
Direct Synthesis of Dimethyl Ether from CO2: Recent Advances in Bifunctional/Hybrid Catalytic Systems




Abstract
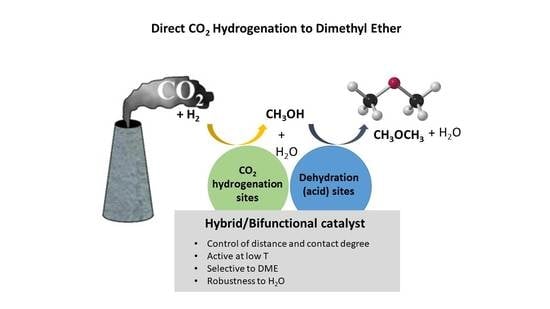
:
1. Introduction
2. Direct Synthesis of DME from CO2: Thermodynamic and Kinetic Considerations
3. Catalytic Functions in Bifunctional/Hybrid Catalysts for Direct Synthesis of DME from CO2
3.1. Catalytic Function for CO2 Hydrogenation to Methanol
3.1.1. Conventional Cu-ZnO Catalysts
3.1.2. Modified Cu-ZnO Catalysts
3.1.3. Alternative Non-Copper Catalysts
3.2. Catalytic Function for the Dehydration of Methanol
3.2.1. Al2O3-Based Catalysts
3.2.2. Zeolite-Based Catalysts
3.2.3. Heteropolyacids (HPAs)-Based Catalysts
3.2.4. Other Acid Catalysts
4. Methods of Preparation of Bifunctional/Hybrid Catalysts for Direct Synthesis of DME from CO2
4.1. Precipitation
4.2. Physical Mixing
4.3. Nanostructuration (Core–Shell Capsule Catalysts)
4.4. Other Methods (CVD and Grafting)
5. Status on Catalyst Development for Direct Synthesis of DME from CO2
6. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- World Energy Outlook 2020, IEA, Paris. Available online: https://www.iea.org/topics/world-energy-outlook (accessed on 13 September 2019).
- Global CO2 Emissions in 2019, IEA 2020, Paris. Available online: https://www.iea.org/articles/global-CO2-emissions-in-2019 (accessed on 10 July 2019).
- Long-term low greenhouse gas emission development strategy of the EU and its Member States. Available online: https://ec.europa.eu/clima/policies/strategies/2050_en (accessed on 12 April 2019).
- CO2 Value Europe. Available online: http://www.co2value.eu/ (accessed on 23 March 2021).
- Styring, P.; Jansen, D.; de Coninck, H.; Reith, H.; Armstrong, K. Carbon Capture and Utilisation in the Green Economy; The Centre for Low Carbon Futures: York, UK, 2011. [Google Scholar]
- Kerry Yu, K.M.; Yu, P.K.M.; Curcic, I.; Gabriel, J.; Tsang, S.C.E. Recent advances in CO2 capture and utilization. ChemSusChem 2008, 1, 893–899. [Google Scholar] [CrossRef]
- Markewitz, P.; Kuckshinrichs, W.; Leitner, W.; Linssen, J.; Zapp, P.; Bongartz, R.; Schreiber, A.; Müller, T.E. Worldwide innovations in the development of carbon capture technologies and the utilization of CO2. Energy Environ. Sci. 2012, 5, 7281–7305. [Google Scholar] [CrossRef] [Green Version]
- Von der Assen, N.; Jung, J.; Bardow, A. Life-cycle assessment of carbon dioxide capture and utilization: Avoiding the pitfalls. Energy Environ. Sci. 2013, 6, 2721–2734. [Google Scholar] [CrossRef]
- Dena Study Integrated Energy Transition, Deutsche Energie-Agentur GmbH (dena), Berlin, Germany. 2018. Available online: https://t.co/8pKe0xiDXz (accessed on 23 March 2021).
- Song, C. Global challenges and strategies for control, conversion and utilization of CO2 for sustainable development involving energy, catalysis, adsorption and chemical processing. Catal. Today 2006, 115, 2–32. [Google Scholar] [CrossRef]
- Neelis, M.; Patel, M.; Blok, K.; Haije, W.; Bach, P. Approximation of theoretical energy-saving potentials for the petrochemical industry using energy balances for 68 key processes. Energy 2007, 32, 1104–1123. [Google Scholar] [CrossRef] [Green Version]
- Marchionna, M.; Patrini, R.; Sanfilippo, D.; Migliavacca, G. Fundamental investigations on di-methyl ether (DME) as LPG substitute or make-up for domestic uses. Fuel Process. Technol. 2008, 89, 1255–1261. [Google Scholar] [CrossRef]
- Fleisch, T.H.; Basu, A.; Sills, R.A. Introduction and advancement of a new clean global fuel: The status of DME developments in China and beyond. J. Nat. Gas Sci. Eng. 2012, 9, 94–107. [Google Scholar] [CrossRef]
- Nasser, G.; Kurniawan, T.; Miyake, K.; Galadima, A.; Hirota, Y.; Nishiyama, N.; Muraza, O. Dimethyl ether to olefins over dealuminated mordenite (MOR) zeolites derived from natural minerals. J. Nat. Gas Sci. Eng. 2016, 28, 566–571. [Google Scholar] [CrossRef]
- Haro, P.; Trippe, F.; Stahl, R.; Henrich, E. Bio-syngas to gasoline and olefins via DME-A comprehensive techno-economic assessment. Appl. Energy 2013, 108, 54–65. [Google Scholar] [CrossRef]
- Liu, Y.; Murata, K.; Inaba, M.; Takahara, I. Synthesis of ethanol from methanol and syngas through an indirect route containing methanol dehydrogenation, DME carbonylation, and methyl acetate hydrogenolysis. Fuel Process. Technol. 2013, 110, 206–213. [Google Scholar] [CrossRef]
- Saravanan, K.; Ham, H.; Tsubaki, N.; Bae, J.W. Recent progress for direct synthesis of dimethyl ether from syngas on the heterogeneous bifunctional hybrid catalysts. Appl. Catal. B Environ. 2017, 217, 494–522. [Google Scholar] [CrossRef]
- Catizzone, E.; Freda, C.; Braccio, G.; Frusteri, F.; Bonura, G. Dimethyl ether as circular hydrogen carrier: Catalytic aspects of hydrogenation/dehydrogenation steps. J. Energy Chem. 2021, 58, 55–77. [Google Scholar] [CrossRef]
- Zannis, T.C.; Hountalas, D.T. DI Diesel Engine Performance and Emissions from the Oxygen Enrichment of Fuels with Various Aromatic Content. Energy Fuels 2004, 18, 659–666. [Google Scholar] [CrossRef]
- Mondal, U.; Yadav, G.D. Perspective of dimethyl ether as fuel: Part I. Catalysis. J. CO2 Util. 2019, 32, 299–320. [Google Scholar] [CrossRef]
- Catizzone, E.; Bonura, G.; Migliori, M.; Frusteri, F.; Giordano, G. CO2 recycling to dimethyl ether: State-of-the-art and perspectives. Molecules 2018, 23, 31. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Yang, G.; Yoneyama, Y.; Tsubaki, N. Catalysis chemistry of dimethyl ether synthesis. ACS Catal. 2014, 4, 3346–3356. [Google Scholar] [CrossRef]
- Azizi, Z.; Rezaeimanesh, M.; Tohidian, T.; Reza Rahimpour, M. Dimethyl ether: A review of technologies and production challenges. Chem. Eng. Process. 2014, 82, 150–172. [Google Scholar] [CrossRef]
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the greener production of formates/formic acid, methanol, and DME by heterogeneously catalyzed CO2 hydrogenation processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Yi, Y.; Wang, L.; Guo, H.; Bogaerts, A. Hydrogenation of carbon dioxide to value-added chemicals by heterogeneous catalysis and plasma catalysis. Catalysts 2019, 9, 275. [Google Scholar] [CrossRef] [Green Version]
- Ye, R.-P.; Ding, J.; Gong, W.; Argyle, M.D.; Zhong, Q.; Wang, Y.; Russel, C.K.; Xu, Z.; Russel, A.G.; Li, Q.; et al. CO2 hydrogenation to-high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10, 5698. [Google Scholar] [CrossRef] [Green Version]
- Borroso-Martín, I.; Infantes-Molina, A.; Fini, F.J.; Ballesteros-Plata, D.; Rodríguez-Castellón, E.; Moretti, E. Silica-related catalysts for CO2 transformation into methanol and dimethyl ether. Catalysts 2020, 10, 1282. [Google Scholar] [CrossRef]
- Huang, M.H.; Lee, H.M.; Liang, K.C.; Tzeng, C.C.; Chen, W.H. An experimental study on single-step dimethyl ether (DME) synthesis from hydrogen and carbon monoxide under various catalysts. Int. J. Hydrog. Energy 2015, 40, 13583–13593. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.L.; Chen, Y.X.; Zhen, J.J.; Li, R.F. Synthesis of dimethyl ether from syngas using a hierarchically porous composite zeolite as the methanol dehydration catalyst. J. Fuel. Chem. Technol. 2013, 41, 873–882. [Google Scholar] [CrossRef]
- Asthana, S.; Samanta, C.; Bhumik, A.; Banerjee, B.; Voolapalli, R.K.; Saha, B. Direct synthesis of dimethyl ether from syngas over Cu-based catalysts: Enhanced selectivity in the presence of MgO. J. Catal. 2016, 334, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Ateka, A.; Pérez-Uriarte, P.; Gamero, M.; Ereña, J.; Aguayo, A.T.; Bilbao, J. A comparative thermodynamic study on the CO2 conversion in the synthesis of methanol and of DME. Energy 2017, 120, 796–804. [Google Scholar] [CrossRef]
- Jia, G.; Tan, Y.; Han, Y. A comparative study on the thermodynamics of dimethyl ether synthesis from CO hydrogenation and CO2 hydrogenation. Ind. Eng. Chem. Res. 2006, 45, 1152–1159. [Google Scholar] [CrossRef]
- Kunkes, E.; Behrens, M. Methanol Chemistry. In Chemical Energy Storage; Walter de Gruyter GmbH: Berlin, Germany, 2013; pp. 413–435. [Google Scholar]
- Sahibzada, M.; Metcalfe, I.S.; Chadwick, D. Methanol synthesis from CO/CO2/H2 at differential and finite conversions. J. Catal. 1998, 174, 111–118. [Google Scholar] [CrossRef]
- Liang, B.; Ma, J.; Su, X.; Yang, C.; Duan, H.; Zhou, H.; Deng, S.; Li, L.; Huang, Y. Investigation on deactivation of Cu/ZnO/Al2O3 catalyst for CO2 hydrogenation to methanol. Ind. Eng. Chem. Res. 2019, 58, 9030–9037. [Google Scholar] [CrossRef] [Green Version]
- Trenco, G.A.; Vidal-Moya, A.; Martinez, A. Study of the interaction between components in hybrid CuZnAl/HZSM-5 catalysts and its impact in the syngas-to –DME reaction. Catal. Today 2012, 179, 43–51. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Neja, S.S.; Blaz, L. Correlation between synthesis pH, structure and Cu/MgO/Al2O3 heterogeneous catalyst activity and selectivity in CO2 hydrogenation to methanol. J. CO2 Util. 2018, 28, 189–199. [Google Scholar] [CrossRef]
- Kunkes, E.L.; Studt, F.; Abild-Pedersen, F.; Schlögl, R.; Behrens, M. Hydrogenation of CO2 to methanol and CO on Cu/ZnO/Al2O3: Is there a common intermediate or not? J. Catal. 2015, 328, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Liao, F.; Huang, Y.; Ge, J.; Zheng, W.; Tedsree, K.; Collier, P.; Hong, X.; Tsang, S.C. Morphol-ogy-dependent interactions of ZnO with Cu nanoparticles at the materials interface in selective hy-drogenation of CO2 to methanol. Angew. Chem. Int. Ed. 2011, 50, 2162–2165. [Google Scholar] [CrossRef]
- Kuld, S.; Thorhauge, M.; Falsig, H.; Elkjær, C.F.; Helveg, S.; Chorkendorff, I.; Sehested, J. Quantifying the promotion of Cu catalysts by ZnO for methanol synthesis. Science 2016, 352, 969–974. [Google Scholar] [CrossRef] [Green Version]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kuhl, S.; Havecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.L.; et al. The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Álvarez Galván, C.; Schumann, J.; Behrens, M.; Fierro, J.L.G.; Schlögl, R.; Frei, E. Reverse water-gas shift reaction at the Cu/ZnO interface: Influence of the Cu/Zn ratio on structure-activity correlations. Appl. Catal. B Environ. 2016, 195, 104–111. [Google Scholar] [CrossRef]
- Tarasov, A.V.; Seitz, F.; Schlögl, R.; Frei, E. In situ quantification of reaction adsorbates in low temperature methanol synthesis on a high-performance Cu/ZnO:Al catalyst. ACS Catal. 2019, 9, 5537–5544. [Google Scholar] [CrossRef]
- Guil-López, R.; Mota, N.; Llorente, J.; Millán, E.; Pawelec, N.; Fierro, J.L.G.; Navarro, R.M. Methanol synthesis from CO2: A review of the latest developments in heterogeneous catalysis. Materials 2019, 12, 3902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.F.; Yang, Y.; Mims, C.; Peden, C.H.F.; Li, J.; Mei, D. Insight into methanol synthesis from CO2 hydrogenation on Cu(1 1 1): Complex reaction network and the effects of H2O. J. Catal. 2011, 281, 199–211. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Yan, B.; Chen, J.G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Zander, S.; Kunkes, E.L.; Schuster, M.E.; Schumann, J.; Weinberg, G.; Teschner, D.; Jacobsen, N.; Schlögl, R.; Behrens, M. The role of the oxide component in the development of copper composite catalysts for methanol synthesis. Angew. Chem. Int. Ed. 2013, 52, 6536–6540. [Google Scholar] [CrossRef]
- Hansen, J.B.; Nielsen, P.E.H. Methanol synthesis. In Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH: Winheim, Germany, 2008; p. 1593. [Google Scholar]
- Behrens, M.; Zander, S.; Kurr, P.; Jacobsen, N.; Senker, J.; Koch, G.; Ressler, T.; Fischer, R.W.; Schlögl, R. Performance Improvement of Nanocatalysts by Promoter-Induced Defects in the Support Material: Methanol Synthesis over Cu/ZnO:Al. J. Am. Chem. Soc. 2013, 135, 6061–6068. [Google Scholar] [CrossRef]
- Schumman, J.; Eichelbaum, M.; Lunkebein, T.; Thomas, N.; Álvarez Galván, M.C.; Schlögl, R.; Behrens, M. Promoting Strong Metal Support Interaction: Doping ZnO for Enhanced Activity of Cu/ZnO:M (M = Al, Ga, Mg) Catalysts. ACS Catal. 2015, 5, 3260–3270. [Google Scholar] [CrossRef]
- Kornas, A.; Grabowski, R.; Śliwa, M.; Samson, K.; Ruggiero-Mikołajczyk, M.; Żelazny, M.A. Dimethyl ether synthesis from CO2 hydrogenation over hybrid catalysts: Effects of preparation methods. React. Kinet. Mech. Catal. 2017, 121, 317–327. [Google Scholar] [CrossRef]
- Li, M.M.J.; Zeng, Z.; Liao, F.; Hong, X.; Tsang, S.C.E. Enhanced CO2 hydrogenation to methanol over CuZn nanoalloy in Ga modified Cu/ZnO catalysts. J. Catal. 2016, 343, 157–167. [Google Scholar] [CrossRef]
- Guil-López, R.; Mota, N.; Llorente, J.; Millán, E.; Pawelec, B.; García, R.; Fierro, J.L.G.; Navarro, R.M. Structure and activity of Cu/ZnO catalysts co-modified with aluminium and gallium for methanol synthesis. Catal. Today 2020, 355, 870–881. [Google Scholar] [CrossRef]
- Chen, W.H.; Lin, B.J.; Lee, H.M.; Huang, M.H. One-step synthesis of dimethyl ether from the gas mixture containing CO2 with high space velocity. Appl. Energy 2012, 98, 92–101. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Li, D.B.; Jiang, D.; Zhu, Y.; Wu, Y.; Zhang, S.J.; Wang, K.J.; Wu, J. Effect of Mn promoter on structure and properties of Mn modified CuO–ZnO–ZrO2/HZSM-5 catalysts for synthesis of dimethyl ether from CO2 hydrogenation. J. Mol. Catal. 2014, 28, 344–350. [Google Scholar]
- Meshkini, F.; Taghizadeh, M.; Bahmani, M. Investigating the effect of metal oxide additives on the properties of Cu/ZnO/Al2O3 catalysts in methanol synthesis from syngas using factorial experimental design. Fuel 2010, 89, 170–175. [Google Scholar] [CrossRef]
- Yang, Y.; White, M.G.; Liu, P. Theoretical study of methanol synthesis from CO2 hydrogenation on metal-doped Cu(111) surfaces. J. Phys. Chem. C 2012, 116, 248–256. [Google Scholar] [CrossRef]
- Liu, L.; Fan, F.; Bai, M.; Xue, F.; Ma, X.; Jiang, Z.; Fang, T. Mechanistic study of methanol synthesis from CO2 hydrogenation on Rh-doped Cu(111) surface. Mol. Catal. 2019, 466, 26–36. [Google Scholar] [CrossRef]
- Pasupulety, N.; Driss, H.; Alhamed, Y.A.; Alzahrani, A.A.; Daous, M.A.; Petrov, L. Influence of preparation method on the catalytic activity of Au/Cu-Zn-Al catalysts for CO2 hydrogenation to methanol. Comptes Rendus Acad. Bulg. Sci. 2015, 68, 1511–1518. [Google Scholar]
- Melián-Cabrera, I.; López Granados, M.; Fierro, J.L.G. Pd-modified Cu–Zn catalysts for methanol synthesis from CO2/H2 mixtures: Catalytic structures and performance. J. Catal. 2002, 210, 285–294. [Google Scholar] [CrossRef]
- Angelo, L.; Kobl, K.; Martínez-Tejada, L.M.; Zimmermann, Y.; Parkhomenko, K.; Roger, A.C. Study of CuZnMOx oxides (M = Al, Zr, Ce, CeZr) for the catalytic hydrogenation of CO2 into methanol. Comptes. Rendus. Chim. 2015, 18, 250–260. [Google Scholar] [CrossRef]
- Dong, X.; Li, F.; Zhao, N.; Xiao, F.; Wang, J.; Tan, Y. CO2 hydrogenation to methanol over Cu/ZnO/ZrO2 catalysts prepared by precipitation-reduction method. Appl. Catal. B Environ. 2016, 191, 8–17. [Google Scholar] [CrossRef]
- Ateka, A.; Sierra, I.; Ereña, J.; Bilbao, J.; Aguayo, A.T. Performance of CuO–ZnO–ZrO2 and CuO–ZnO–MnO as metallic functions and SAPO-18 as acid function of the catalyst for the synthesis of DME co-feeding CO2. Fuel Proc. Technol. 2016, 152, 34–45. [Google Scholar] [CrossRef]
- Frusteri, F.; Cordaro, M.; Cannilla, C.; Bonura, G. Multifunctionality of Cu-ZnO-ZrO2/H-ZSM5 catalysts for the one-step CO2-to-DME hydrogenation reaction. Appl. Catal. B Environ. 2015, 162, 57–65. [Google Scholar] [CrossRef]
- Frusteri, F.; Bonura, G.; Cannilla, C.; Drago Ferrante, G.; Aloise, A.; Catizzone, E.; Migliori, M.; Giordano, G. Stepwise tuning of metal-oxide and acid sites of CuZnZr-MFI hybrid catalysts for the direct DME synthesis by CO2 hydrogenation. Appl. Catal. B Environ. 2015, 176–177, 522–531. [Google Scholar] [CrossRef]
- Mureddu, M.; Ferrara, F.; Pettinau, A. Highly efficient CuO/ZnO/ZrO2@SBA-15 nanocatalysts for methanol synthesis from the catalytic hydrogenation of CO2. Appl. Catal. B Environ. 2019, 258, 117941. [Google Scholar] [CrossRef]
- Prieto, G.; Zečević, J.; Friedrich, H.; de Jong, K.P.; de Jongh, P.E. Towards stable catalysts by controlling collective properties of supported metal nanoparticles. Nat. Mater. 2013, 12, 34–39. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, Y.; Li, J.; Pippel, E.; Yang, H.; Gao, Z.; Qin, Y. High efficiency Cu-ZnO hydrogenation catalyst: The tailoring of Cu-ZnO interface sites by molecular layer deposition. ACS Catal. 2015, 5, 5567–5573. [Google Scholar] [CrossRef]
- Deerattrakul, V.; Puengampholsrisook, P.; Limphirat, W.; Kongkachuichay, P. Characterization of supported Cu-Zn/graphene aerogel catalyst for direct CO2 hydrogenation to methanol: Effect of hydrothermal temperature on graphene aerogel synthesis. Catal. Today 2018, 314, 154–163. [Google Scholar] [CrossRef]
- Zhang, C.; Liao, P.; Wang, H.; Sun, J.; Gao, P. Preparation of novel bimetallic CuZn-BTC coordination polymer nanorod for methanol synthesis from CO2 hydrogenation. Mater. Chem. Phys. 2018, 215, 211–220. [Google Scholar] [CrossRef]
- Xiao, J.; Mao, D.; Guo, X.; Yu, J. Methanol synthesis from CO2 hydrogenation over CuO-ZnO-TiO2 catalysts: The influence of TiO2 content. Energy Technol. 2015, 3, 32–39. [Google Scholar] [CrossRef]
- Xu, J.H.; Su, X.; Liu, X.Y.; Pan, X.L.; Pei, G.X.; Huang, Y.Q.; Wang, X.D.; Zhang, T.; Geng, H.R. Methanol synthesis from CO2 and H2 over Pd/ZnO/Al2O3: Catalyst structure dependence of methanol selectivity. Appl. Catal. A 2016, 514, 51–59. [Google Scholar] [CrossRef]
- Rui, N.; Wang, Z.Y.; Sun, K.H.; Ye, J.Y.; Ge, Q.F.; Liu, C.J. CO2 hydrogenation to methanol over Pd/In2O3: Effects of Pd and oxygen vacancy. Appl. Catal. B Environ. 2017, 218, 488–497. [Google Scholar] [CrossRef]
- Wu, C.Y.; Zhang, P.; Zhang, Z.F.; Zhang, L.J.; Yang, G.Y.; Han, B.X. Efficient hydrogenation of CO2 to methanol over supported subnanometer gold catalysts at low temperature. ChemCatChem 2017, 9, 3691–3696. [Google Scholar] [CrossRef]
- Zhong, J.; Yang, X.; Wu, Z.; Liang, B.; Huang, Y.; Zhanga, T. State of the art and perspectives in heterogeneous catalysis of CO2 hydrogenation to methanol. Chem. Soc. Rev. 2020, 49, 1385–1413. [Google Scholar] [CrossRef] [PubMed]
- Lian, Y.; Fang, T.F.; Zhang, Y.H.; Liu, B.; Li, J.L. Hydrogenation of CO2 to alcohol species over Co@Co3O4/C-N catalysts. J. Catal. 2019, 379, 46–51. [Google Scholar] [CrossRef]
- Li, C.S.; Melaet, G.; Ralston, W.T.; An, K.; Brooks, C.; Ye, Y.F.; Liu, Y.S.; Zhu, J.F.; Guo, J.H.; Alayoglu, S.; et al. High-performance hybrid oxide catalyst of manganese and cobalt for low-pressure methanol synthesis. Nat. Commun. 2015, 6, 6538. [Google Scholar] [CrossRef]
- Hanseul Choi, Sunyoung Oh, Si Bui Trung Tran, Jeong Young Park, Size-controlled model Ni catalysts on Ga2O3 for CO2 hydrogenation to methanol. J. Catal. 2019, 376, 68–76. [CrossRef]
- Sun, K.H.; Fan, Z.G.; Ye, J.Y.; Yan, J.M.; Ge, Q.F.; Li, Y.N.; He, W.J.; Yang, W.; Liu, C.J. Hydrogenation of CO2 to methanol over In2O3 catalyst. J. CO2. Util. 2015, 12, 1–6. [Google Scholar] [CrossRef]
- Martin, O.; Martin, A.J.; Mondelli, C.; Mitchell, S.; Segawa, T.F.; Hauert, R.; Drouilly, C.; Curulla-Ferré, D.; Pérez-Ramírez, J. Indium oxide as a superior catalyst for methanol synthesis by CO2 hydrogenation. Angew. Chem. Int. Ed. 2016, 55, 6261–6265. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.Y.; Cao, C.X.; Chen, T.B.; Ding, X.X.; Huang, H.; Shen, L.; Cao, X.Y.; Zhu, M.H.; Xu, J.; Gao, J.; et al. Unraveling highly tunable selectivity in CO2 hydrogenation over bimetallic In-Zr oxide catalysts. ACS Catal. 2019, 9, 8785–8797. [Google Scholar] [CrossRef]
- Frei, M.S.; Mondelli, C.; Cesarini, A.; Krumeich, F.; Hauert, R.; Stewart, J.A.; Ferre, D.C.; Perez-Ramirez, J. Role of Zirconia in Indium Oxide-Catalyzed CO2 Hydrogenation to Methanol. ACS Catal. 2020, 10, 1133–1145. [Google Scholar] [CrossRef]
- Akkharaphatthawona, N.; Chanlekc, N.; Cheng, C.K.; Chareonpanicha, M.; Limtrakule, J.; Witoon, T. Tuning adsorption properties of GaxIn2−xO3 catalysts for enhancement of methanol synthesis activity from CO2 hydrogenation at high reaction temperature. Appl. Surf. Sci. 2019, 489, 278–286. [Google Scholar] [CrossRef]
- Studt, F.; Sharafutdinov, I.; Abild-Pedersen, F.; Elkjaer, C.F.; Hummelshoj, J.S.; Dahl, S.; Chorkendorff, I.; Norskov, J.K. Discovery of a Ni-Ga catalyst for carbon dioxide reduction to methanol. Nat. Chem. 2014, 6, 320–324. [Google Scholar] [CrossRef]
- Pustovarenko, A.; Dikhtiarenko, A.; Bavykina, A.; Gevers, L.; Ramírez, A.; Russkikh, A.; Telalovic, S.; Aguilar, A.; Hazemann, J.-L.; Ould-Chikh, S.; et al. Metal–organic framework-derived synthesis of cobalt indium catalysts for the hydrogenation of CO2 to methanol. ACS Catal. 2020, 10, 5064–5076. [Google Scholar] [CrossRef]
- Richard, A.R.; Fan, M. The effect of lanthanide promoters on NiInAl/SiO2 catalyst for methanol synthesis. Fuel 2018, 222, 513–522. [Google Scholar] [CrossRef]
- Akarmazyan, S.S.; Panagiotopoulou, P.; Kambolis, A.; Papadopoulou, C.; Kondarides, D.I. Methanol dehydration to dimethyl ether over Al2O3 catalysts. Appl. Catal. B Environ. 2014, 145, 136–148. [Google Scholar] [CrossRef]
- Osman, A.I.; Abu-Dahrieh, J.K.; Rooney, D.W.; Thompson, J.; Halawy, S.A.; Mohamed, M.A. Surface hydrophobicity and acidity effect on alumina catalyst in catalytic methanol dehydration reaction. J. Chem. Technol. Biotechnol. 2017, 92, 2952–2962. [Google Scholar] [CrossRef] [Green Version]
- Raoof, F.; Taghizadeh, M.; Eliassi, A.; Yaripour, F. Effects of temperature and feed composition on catalytic dehydration of methanol to dimethyl ether over γ-alumina. Fuel 2008, 87, 2967–2971. [Google Scholar] [CrossRef]
- Bateni, H.; Able, C. Development of heterogeneous catalysts for dehydration of methanol to dimethyl ether: A review. Catal. Ind. 2019, 11, 7–33. [Google Scholar] [CrossRef]
- Ghorbanpour, A.; Rimer, J.D.; Grabow, L.C. Computational assessment of the dominant factors governing the mechanism of methanol dehydration over H-ZSM-5 with heterogeneous aluminum distribution. ACS Catal. 2016, 6, 2287–2298. [Google Scholar] [CrossRef]
- Jones, J.A.; Iglesia, E. Kinetic, spectroscopic, and theroretical assessment of associative and dissociative metanol dehydration routes in zeolites. Angew. Chem. Int. Ed. 2014, 53, 12177–12181. [Google Scholar] [CrossRef]
- Hosseininejad, S.; Afacan, A.; Hayes, R.E. Catalytic and kinetic study of methanol dehydration to dimethyl ether. Chem. Eng. Res. Des. 2012, 90, 825–833. [Google Scholar] [CrossRef]
- Osman, A.I.; Abu-Dahrieh, J.K. Kinetic investigation of η-Al2O3 catalyst for dimethyl ether production. Catal. Lett. 2018, 148, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Osman, A.I.; Abu-Dahrieh, J.K.; Rooney, D.W.; Halawy, S.A.; Mohamed, M.A.; Abdelkader, A. Effect of precursor on the performance of alumina for the dehydration of methanol to dimethyl ether. Appl. Catal. B 2012, 127, 307–315. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Lee, Y.J.; Bae, J.W.; Potdar, H.S.; Jun, K.W. Synthesis and characterization of a highly active alumina catalyst for methanol dehydration to dimethyl ether. Appl. Catal. A Gen. 2008, 348, 113–120. [Google Scholar] [CrossRef]
- Carvalho, D.F.; Almeida, G.C.; Monteiro, R.S.; Mota, C.J.A. Hydrogenation of CO2 to Methanol and Dimethyl Ether over a Bifunctional Cu·ZnO Catalyst Impregnated on Modified γ-Alumina. Energy Fuels 2020, 34, 7269–7274. [Google Scholar] [CrossRef]
- Yaripour, F.; Shariatinia, Z.; Sahebdelfar, S.; Irandoukht, A. The effects of synthesis operation conditions on the properties of modified-alumina nanocatalysts in methanol dehydration to dimethyl ether using factorial experimental design. Fuel 2015, 139, 40–50. [Google Scholar] [CrossRef]
- Chiang, C.-L.; Lin, K.-S. Preparation and characterization of CuO/Al2O3 catalyst for dimethyl ether production via methanol dehydration. Inter. J. Hydrog. Energy 2017, 42, 23526–23538. [Google Scholar] [CrossRef]
- Armenta, M.A.; Valdez, R.; Quintana, J.M.; Silva-Rodrigo, R.; Cota, L.; Olivas, A. Highly selective CuO/γ-Al2O3 catalyst promoted with hematite for efficient methanol dehydration to dimethyl ether. Inter. J. Hydrog. Energy 2018, 43, 6551–6560. [Google Scholar] [CrossRef]
- Armenta, M.A.; Maytorena, V.M.; Flores–Sánchez, L.A.; Quintana, J.M.; Valdez, R.; Olivas, A. Dimethyl ether production via methanol dehydration using Fe3O4 and CuO over γ-χ- Al2O3 nanocatalysts. Fuel 2020, 280, 118545. [Google Scholar] [CrossRef]
- Catizzone, E.; Aloise, A.; Migliori, M.; Giordano, G. Dimethyl ether synthesis via methanol dehydration: Effect of zeolite structure. Appl. Catal. A Gen. 2015, 502, 215–220. [Google Scholar] [CrossRef]
- Catizzone, E.; Aloise, A.; Migliori, M.; Giordano, G. From 1-D to 3-D zeolite structures: Performance assessment in catalysis of vapour-phase methanol dehydration to DME. Microp. Mesopo. Mat. 2017, 243, 102–111. [Google Scholar] [CrossRef]
- Frusteri, F.; Migliori, M.; Cannilla, C.; Frusteri, L.; Catizzone, E.; Aloise, A.; Giordano, G.; Bonura, G. Direct CO2-to-DME hydrogenation reaction: New evidences of a superior behavior of FER-based hybrid systems to obtain high DME yield. J. CO2 Util. 2017, 18, 353–361. [Google Scholar] [CrossRef]
- Dębek, R.; Ribeiro, M.F.G.; Fernandes, A.; Motak, M. Dehydration of methanol to dimethyl ether over modified vermiculites. C. R. Chim. 2015, 18, 1211e22. [Google Scholar] [CrossRef]
- Cai, M.; Palčić, A.; Subramanian, V.; Moldovan, S.; Ersen, O.; Valtchev, V.; Ordomsky, V.V.; Khodakov, A.Y. Direct Dimethyl Ether Synthesis from Syngas on Copper-Zeolite Hybrid Catalysts with a Wide Range of Zeolite Particle Sizes. J. Catal. 2016, 338, 227. [Google Scholar] [CrossRef] [Green Version]
- Aboul-Fotouh, S.M.K.; Ali, L.I.; Naghmash, M.A.; Aboul-Gheit, N.A.K. Effect of the Si/Al ratio of HZSM-5 zeolite on the production of dimethyl ether before and after ultrasonication. J. Fuel Chem. Technol. 2017, 45, 581–588. [Google Scholar] [CrossRef]
- Aboul-Fotouh, S.M.K.; Aboul-Gheit, N.A.K.; Hassan, M.M.I. Conversion of methanol using modified H-MOR zeolite catalysts. Chin. J. Catal. 2011, 32, 412–417. [Google Scholar] [CrossRef]
- Aboul-Fotouh, S.M.K.; Aboul-Gheit, N.A.K.; Naghmash, M.A. Dimethylether production on zeolite catalysts activated by Cl-, F- and/or ultrasonication. J. Fuel Chem. Technol. 2016, 44, 428–436. [Google Scholar] [CrossRef]
- Aloise, A.; Marino, A.; Dalena, F.; Giorgianni, G.; Migliori, M.; Frusteri, L.; Cannilla, C.; Bonura, G.; Frusteri, F.; Giordano, G. Desilicated ZSM-5 zeolite: Catalytic performances assessment in methanol to DME dehydration. Micropor. Mesopor. Mat. 2020, 302, 110198. [Google Scholar] [CrossRef]
- Dennis-Smither, B.J.; Yang, Z.; Buda, C.; Liu, X.; Sainty, N.; Tanb, X.; Sunley, G.J. Getting zeolite catalysts to play your tune: Methyl carboxylate esters as switchable promoters for methanol dehydration to DME. Chem. Commun. 2019, 55, 13804. [Google Scholar] [CrossRef] [PubMed]
- Magzoub, F.; Li, X.; Lawson, S.; Rezaei, F.; Rownaghi, A.A. 3D-printed HZSM-5 and 3D-HZM5@SAPO-34 structured monoliths with controlled acidity and porosity for conversion of methanol to dimethyl ether. Fuel 2020, 280, 118628. [Google Scholar] [CrossRef]
- Catizzone, E.; Bonura, G.; Migliori, M.; Braccio, G.; Frusteri, F.; Giordano, G. Direct CO2-to-dimethyl ether hydrogenation over CuZnZr/zeolite hybrid catalyst: New evidence on the interaction between acid and metal sites. Ann. Chim. Sci. Mat. 2019, 43, 141–149. [Google Scholar] [CrossRef]
- Solyman, S.M.; Aboul-Gheit, N.A.K.; Sadek, M.; Tawfik, F.M.; Ahmed, H.A. The effect of physical and chemical treatment on nano-zeolite characterization and their performance in dimethyl ether preparation. Egypt. J. Pet. 2015, 24, 289–297. [Google Scholar] [CrossRef] [Green Version]
- Micek-Ilnicka, A. The role of water in the catalysis on solid heteropolyacids. J. Mol. Catal. A Chem. 2009, 308, 1–14. [Google Scholar] [CrossRef]
- Schnee, J.; Gaigneaux, E.M. Elucidating and exploiting the chemistry of keggin heteropolyacids in the methanol-to-dme conversion: Enabling the bulk reaction thanks to operando raman. Catal. Sci. Technol. 2017, 7, 817–830. [Google Scholar] [CrossRef]
- Schnee, J.; Eggermont, A.; Gaigneaux, E.M. Boron nitride: A support for highly active heteropolyacids in the methanol-to-dme reaction. ACS Catal. 2017, 7, 4011–4017. [Google Scholar] [CrossRef]
- Ladera, R.M.; Ojeda, M.; Fierro, J.L.G.; Rojas, S. Tio2-supported heteropoly acid catalysts for dehydration of methanol to dimethyl ether: Relevance of dispersion and support interaction. Catal. Sci. Technol. 2015, 5, 484–491. [Google Scholar] [CrossRef]
- García-López, E.I.; Marcì, G.; Krivtsov, I.; Casado Espina, J.; Liotta, L.F.; Serrano, A. Local structure of supported keggin and wells–dawson heteropolyacids and its influence on the catalytic activity. J. Phys. Chem. 2019, 123, 19513–19527. [Google Scholar] [CrossRef]
- Alharbi, W.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Dehydration of methanol to dimethyl ether over heteropoly acid catalysts: The relationship between reaction rate and catalyst acid strength. ACS Catal. 2015, 5, 7186–7193. [Google Scholar] [CrossRef]
- Peinado, C.; Liuzzi, D.; Ladera-Gallardo, R.M.; Retuerto, N.; Ojeda, M.; Peña, M.A.; Rojas, S. Effects of support and reaction pressure for the synthesis of dimethyl ether over heteropolyacid catalysts. Sci. Rep. 2020, 10, 8551. [Google Scholar] [CrossRef] [PubMed]
- Millán, E.; Mota, N.; Guil-López, R.; Pawelec, B.; Fierro, J.L.G.; Navarro, R.M. Bifunctional Hybrid Catalysts Based on Supported H3PW12O40 and Cu-ZnO(Al): Effect of Heteropolyacid Loading on Hybrid Structure and Catalytic Activity. Catalysts 2020, 10, 1071. [Google Scholar] [CrossRef]
- Ladera, R.M.; Fierro, J.L.G.; Ojeda, M.; Rojas, S. TiO2-supported heteropoly acids for low-temperature synthesis of dimethyl ether from methanol. J. Catal. 2014, 312, 195–203. [Google Scholar] [CrossRef]
- Kornas, A.; Śliwa, M.; Ruggiero-Mikołajczyk, M.; Samson, K.; Podobiński, J.; Karcz, R.; Duraczyńska, D.; Rutkowska-Zbik, D.; Grabowski, R. Direct hydrogenation of CO2 to dimethyl ether (DME) over hybrid catalysts containing CuO/ZrO2 as a metallic function and heteropolyacids as an acidic function. Reac. Kinet. Mech. Cat. 2020, 130, 179–194. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Sun, D.; Wang, S.; Xiao, M.; Sun, L.; Meng, Y. Heteropolyacid Salt Catalysts for Methanol Conversion to Hydrocarbons and Dimethyl Ether: Effect of Reaction Temperature. Catalysts 2019, 9, 320. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Li, X.; Xu, Y.; Dong, Y.; Lai, W.; Fang, W.; Yi, X. Fabrication of nano-sized SAPO-11 crystals with enhanced dehydration of methanol to dimethyl ether. Catal. Commun. 2018, 103, 1–4. [Google Scholar] [CrossRef]
- Sánchez-Contador, M.; Ateka, A.; Aguayo, A.T.; Bilbao, J. Behavior of SAPO-11 as acid function in the direct synthesis of dimethyl ether from syngas and CO2. J. Ind. Eng. Chem. 2018, 63, 245–254. [Google Scholar] [CrossRef]
- Pranee, W.; Neramittagapong, S.; Assawasaengrat, P.; Neramittagapong, A. Methanol dehydration to dimethyl ether over strong-acid-modified diatomite catalysts. Energy Sources Part A Recovery Util. Environ. Eff. 2016, 38, 3109–3115. [Google Scholar] [CrossRef]
- Temvuttirojn, C.; Chuasomboon, N.; Numpilai, T.; Faungnawakij, K.; Chareonpanich, M.; Limtrakul, J.; Witoon, T. Development of SO42−–ZrO2 acid catalysts admixed with a CuO-ZnO-ZrO2 catalyst for CO2 hydrogenation to dimethyl ether. Fuel 2019, 241, 695–703. [Google Scholar] [CrossRef]
- Witoon, T.; Permsirivanich, T.; Kanjanasoontorn, N.; Akkaraphataworn, C.; Seubsai, A.; Faungnawakij, K.; Warakulwit, C.; Chareonpanich, M.; Limtrakul, J. Direct synthesis of dimethyl ether from CO2 hydrogenation over cu-ZnO-ZrO2/SO42--ZrO2 hybrid catalysts: Effects of sulfur-to-zirconia ratios. Catal. Sci. Technol. 2015, 5, 2347–2357. [Google Scholar] [CrossRef]
- Suwannapichat, Y.; Numpilai, T.; Chanlek, N.; Faungnawakij, K.; Chareonpanich, M.; Witoon, T. Direct synthesis of dimethyl ether from CO2 hydrogenation over novel hybrid catalysts containing a Cu-ZnO-ZrO2 catalyst admixed with WOx/Al2O3 catalysts: Effects of pore size of Al2O3 support and W loading content. Energy Convers. Manag. 2018, 159, 20–29. [Google Scholar] [CrossRef]
- Witoon, T.; Kachaban, N.; Donphai, W.; Kidkhunthod, P.; Faungnawakij, K.; Chareonpanich, M.; Limtrakul, J. Tuning of catalytic CO2 hydrogenation by changing composition of CuO-ZnO-ZrO2 catalysts. Energy Convers. Manag. 2016, 118, 21–31. [Google Scholar] [CrossRef]
- Macht, J.; Baertsch, C.D.; May-Lozano, M.; Soled, S.L.; Wang, Y.; Iglesia, E. Support effects on Brønsted acid site densites and alcohol dehydration turnover rates on tungsten oxide domians. J. Catal. 2004, 227, 479–491. [Google Scholar] [CrossRef]
- Ladera, R.M.; Finocchio, E.; Rojas, S.; Busca, G.; Fierro, J.L.G.; Ojeda, M. Supported WOx-based catalysts for methanol dehydration to dimethyl ether. Fuel 2013, 113, 1–9. [Google Scholar] [CrossRef]
- Ladera, R.M.; Finocchio, E.; Rojas, S.; Fierro, J.L.G.; Ojeda, M. Supported niobium catalysts for methanol dehydration to dimethyl ether: FTIR studies of acid properties. Catal. Today 2012, 192, 136–143. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Spadaro, L.; Cannilla, C.; Arena, F.; Frusteri, F. Hybrid Cu-ZnO-ZrO2/H-ZSM-5 system for the direct synthesis of DME by CO2 hydrogenation. Appl. Catal. B Environ. 2013, 140–141, 16–24. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Cannilla, C.; Arena, F.; Frusteri, F. The changing nature of the active site of Cu-Zn-Zr catalysts for the CO2 hydrogenation reaction to methanol. Appl. Catal. B Environ. 2014, 152–153, 152–161. [Google Scholar] [CrossRef]
- Bonura, G.; Frusteri, F.; Cannilla, C.; Drago Ferrante, G.; Aloise, A.; Catizzone, E.; Migliori, M.; Giordano, G. Catalytic features of CuZnZr–zeolite hybrid systems for the direct CO2-to-DME hydrogenation reaction. Catal. Today 2016, 277, 48–54. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Ding, F.; Wang, K.; Wang, X.; Ren, B.; Wu, J. Synthesis of DME by CO2 hydrogenation over La2O3-modified CuO-ZnO-ZrO2/HZSM-5 catalysts. Chem. Ind. Chem. Eng. Q. 2017, 23, 49–56. [Google Scholar] [CrossRef]
- Guo, X.; Mao, D.; Lu, G.; Wang, S.; Wu, G. The influence of La doping on the catalytic behavior of Cu/ZrO2 for methanol synthesis from CO2 hydrogenation. J. Mol. Catal. A Chem. 2011, 345, 60–68. [Google Scholar] [CrossRef]
- Jeong, C.; Park, J.; Kim, J.; Baik, J.H.; Suh, Y.-W. Effects, of Al3+ precipitation onto primitive amorphous Cu-Zn precipitate on methanol synthesis over Cu/ZnO/Al2O3 catalyst. Korean J. Chem. Eng. 2019, 36, 191–196. [Google Scholar] [CrossRef]
- Jiang, Q.; Liu, Y.; Dintzer, T.; Luo, J.; Parkhomenko, K.; Roger, A. Tuning the highly dispersed metallic cu species via manipulating Brønsted acid sites of mesoporous aluminosilicate support for CO2 hydrogenation reactions. Appl. Catal. B Environ. 2020, 269, 118804. [Google Scholar] [CrossRef]
- Lu, S.; Si, B.; Chang, Q.; Liu, B.; Liu, E.; Fan, J. Modification of the CuO-ZnO-Al2O3/HZSM-5 bifunctional catalyst with promoters and their catalytic performance research. Huaxue Gongcheng/Chem. Eng. 2016, 44, 42–47. [Google Scholar] [CrossRef]
- Qin, Z.-Z.; Zhou, X.-H.; Su, T.-M.; Jiang, Y.-X.; Ji, H.-B. Hydrogenation of CO2 to dimethyl ether on La-, Ce-modified Cu-Fe/HZSM-5 catalysts. Catal. Commun. 2016, 75, 78–82. [Google Scholar] [CrossRef]
- Bao, J.; Tsubaki, N. Design and synthesis of powerful capsule catalysts aimed at applications in C1 chemistry and biomass conversion. Chem. Rec. 2018, 18, 4–19. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.H.; Ma, Q.X.; Zhao, T.S.; Bao, J.; Tsubaki, N. Recent advances in multifunctional capsule catalysts in heterogeneous catalysis. Chin. J. Chem. Phys. 2018, 31, 393–403. [Google Scholar] [CrossRef]
- Yang, G.; Tsubaki, N.; Shamoto, J.; Yoneyama, Y.; Zhang, Y. Confinement effect and synergistic function of H-ZSM-5/Cu-ZnO-Al2O3 capsule catalyst for one-step controlled synthesis. J. Am. Chem. Soc. 2010, 132, 8129–8136. [Google Scholar] [CrossRef]
- Yang, G.; Wang, D.; Yoneyama, Y.; Tan, Y.; Tsubaki, N. Facile synthesis of H-type zeolite shell on a silica substrate for tandem catalysis. Chem. Commun. 2012, 48, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Phienluphon, R.; Pinkaew, K.; Yang, G.; Li, J.; Wei, Q.; Yoneyama, Y.; Vitidsant, T.; Tsubaki, N. Designing core (Cu/ZnO/Al2O3)-shell (SAPO-11) zeolite capsule catalyst with a facile physical way for dimethyl ether direct synthesis from syngas. Chem. Eng. J. 2015, 270, 605–611. [Google Scholar] [CrossRef]
- Tan, L.; Zhang, P.; Suzuki, Y.; Li, H.; Guo, L.; Yoneyama, Y.; Chen, J.; Peng, X.; Tsubaki, N. Bifunctional capsule catalyst of Al2O3@Cu with strengthened dehydration reaction field for direct synthesis of dimethyl ether from syngas. Ind. Eng. Chem. Res. 2019, 58, 51, 22905–22911. [Google Scholar] [CrossRef]
- Atakan, A.; Keraudy, J.; Mäkie, P.; Hulteberg, C.; Björk, E.M.; Odén, M. Impact of the morphological and chemical properties of copper-zirconium-SBA-15 catalysts on the conversion and selectivity in carbon dioxide hydrogenation. J. Colloid Interface Sci. 2019, 546, 163–173. [Google Scholar] [CrossRef]
- Bahruji, H.; Amstrong, R.D.; Ruiz Esquius, J.; Jones, W.; Bowker, M.; Hutchings, G.J. Hydrogenation of CO2 to Dimethyl Ether over Brønsted Acidic PdZn Catalysts. Ind. Eng. Chem. Res. 2018, 57, 6821–6829. [Google Scholar] [CrossRef]
- Ahoba-Sam, C.; Borfecchia, E.; Lazzarini, A.; Bugaev, A.; Adamu Isah, A.; Taoufik, M.; Bordiga, S.; Olsbye, U. On the conversion of CO2 to value added products over composite PdZn and H-ZSM-5 catalysts: Excess Zn over Pd, a compromise or a penalty? Catal. Sci. Technol. 2020, 10, 4373–4385. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Li, D.B.; Zhang, S.J.; Wang, K.J.; Wu, J. CO2 hydrogenation to dimethyl ether over CuO–ZnO–Al2O3/HZSM-5 prepared by combustion route. RSC Adv. 2014, 4, 16391–16396. [Google Scholar] [CrossRef]
- Ren, S.; Fan, X.; Shang, Z.; Shoemaker, W.R.; Ma, L.; Wu, T.; Li, S.; Klinghoffer, N.B.; Yu, M.; Liang, X. Enhanced catalytic performance of Zr modified CuO/ZnO/Al2O3 catalyst for methanol and DME synthesis via CO2 hydrogenation. J. CO2 Util. 2020, 36, 82–95. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, Y.; Du, J.; Li, C.; Wang, K.; Liu, L.; Yu, X.; Wang, K.; Li, N. The influence of composition on the functionality of hybrid CuO–ZnO–Al2O3/HZSM-5 for the synthesis of DME from CO2 hydrogenation. RSC Adv. 2018, 8, 30387–30395. [Google Scholar] [CrossRef] [Green Version]
- Li, L.Y.; Mao, D.S.; Xiao, J.; Li, L.; Guo, X.M.; Yu, J. Facile preparation of highly efficient CuO-ZnO-ZrO2/HZSM-5 bifunctional catalyst for one-step CO2 hydrogenation to dimethyl ether: Influence of calcination temperature. Chem. Eng. Res. Des. 2016, 111, 100–108. [Google Scholar] [CrossRef]
- Bonura, G.; Cannilla, C.; Frusteri, L.; Frusteri, F. The influence of different promoter oxides on the functionality of hybrid CuZn-ferrierite systems for the production of DME from CO2-H2 mixtures. Appl. Catal. A Gen. 2017, 544, 21–29. [Google Scholar] [CrossRef]
- Miletto, I.; Catizzone, E.; Bonura, G.; Ivaldi, C.; Migliori, M.; Gianotti, E.; Marchese, L.; Frusteri, F.; Giordano, G. In situ FT-IR characterization of CuZnZr/ferrierite hybrid catalysts for one-pot CO2-to-DME conversion. Materials 2018, 11, 2275. [Google Scholar] [CrossRef] [PubMed] [Green Version]







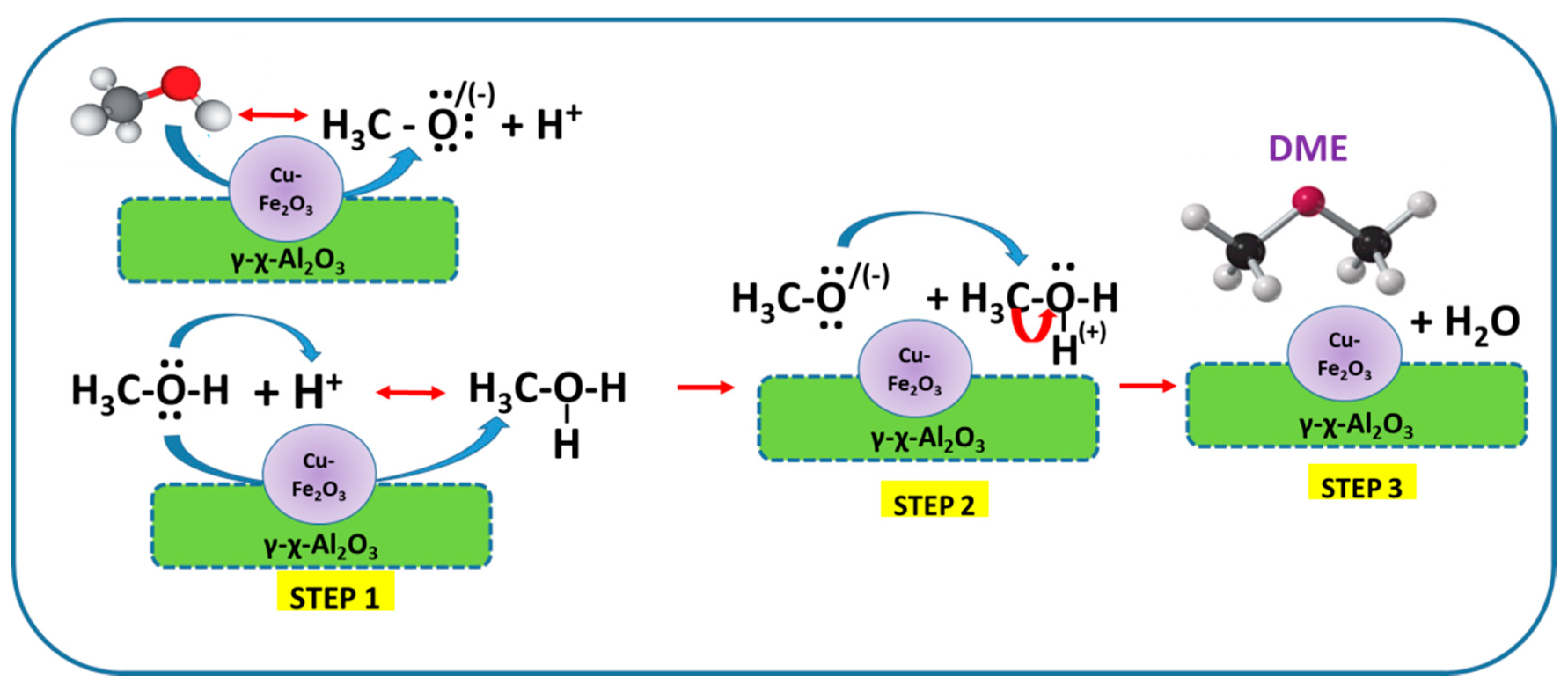








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DME Property | |
|---|---|
| Carbon content (wt%) | 52.2 |
| Boiling point (°C 1 atm) | −24.8 |
| LHV (kJ/mol) | −1328.4 |
| Vapor pressure (20 °C) | 5.1 |
| Energy (MJ/L) | 20.6 |
| Catalyst a | Preparation Method d | GHSV (mL g−1 h−1) | T (°) | P (bar) | XCO2 (%) | SDME (%) | Yield DME (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| CuO-ZnO-Al2O3/ZSM-5 | (A) | 1525 | 260 | 42 | 30.5 | 72.0 | 21.9 | [155] |
| CuO-ZnO-ZrO2-La2O3(1%)/ZSM-5 | (B) | 4200 | 270 | 30 | 34.3 | 57.3 | 19.6 | [139] |
| CuO-ZnO-Al2O3/ZSM-5 | (B) | 1500 | 260 | 30 | 27.3 | 67.1 | 18.3 | [156] |
| CuO-ZnO-Al2O3/ZSM-5 | (C) b | 3600 | 250 | 30 | 21.3 | 63.4 | 13.5 | [157] |
| CuO-ZnO-Al2O3/ZSM-5 | (D) | 4200 | 270 | 30 | 30.6 | 49.2 | 15.1 | [154] |
| CuO-ZnO-Al2O3/ZSM-5 | (B) | 8800 | 260 | 30 | 21 | 35 | 7.4 | [158] |
| CuO-ZnO-ZrO2/FER | (E) | 8800 | 280 | 50 | 29 | 62 | 18.0 | [138] |
| CuO-ZnO-ZrO2/MOR | (E) | 8800 | 280 | 50 | 26 | 51 | 13.0 | [138] |
| CuO-ZnO-ZrO2/ZSM-5 | (B) | 8800 | 260 | 50 | 21 | 41 | 8.6 | [104] |
| CuO-ZnO-Ga2O3/ZSM-5 | (B) | 8800 | 260 | 30 | 21 | 36 | 7.6 | [158] |
| CuO-ZnO-La2O3/FER | (B) | 8800 | 260 | 30 | 18 | 34 | 6.1 | [158] |
| CuO-ZnO-CeO2/FER | (B) | 8800 | 260 | 30 | 14 | 36 | 5.0 | [158] |
| CuO-ZrO2 + K10 c +3% Mn | (F) | 1800 | 260 | 40 | 7 | 22 | 1.5 | [51] |
| CuO-ZrO2 + K10 c + 3% Ga | (F) | 1800 | 260 | 40 | 7 | 20 | 1.4 | [51] |
| Cu-Fe-La/ZSM-5 | (G) | 1500 | 260 | 30 | 17.2 | 51.3 | 8.8 | [144] |
| Cu-Fe-Ce/ZSM-5 | (G) | 1500 | 260 | 30 | 18.1 | 52.0 | 9.4 | [144] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mota, N.; Millán Ordoñez, E.; Pawelec, B.; Fierro, J.L.G.; Navarro, R.M. Direct Synthesis of Dimethyl Ether from CO2: Recent Advances in Bifunctional/Hybrid Catalytic Systems. Catalysts 2021, 11, 411. https://doi.org/10.3390/catal11040411
Mota N, Millán Ordoñez E, Pawelec B, Fierro JLG, Navarro RM. Direct Synthesis of Dimethyl Ether from CO2: Recent Advances in Bifunctional/Hybrid Catalytic Systems. Catalysts. 2021; 11(4):411. https://doi.org/10.3390/catal11040411
Chicago/Turabian StyleMota, Noelia, Elena Millán Ordoñez, Bárbara Pawelec, José Luis G. Fierro, and Rufino M. Navarro. 2021. "Direct Synthesis of Dimethyl Ether from CO2: Recent Advances in Bifunctional/Hybrid Catalytic Systems" Catalysts 11, no. 4: 411. https://doi.org/10.3390/catal11040411
APA StyleMota, N., Millán Ordoñez, E., Pawelec, B., Fierro, J. L. G., & Navarro, R. M. (2021). Direct Synthesis of Dimethyl Ether from CO2: Recent Advances in Bifunctional/Hybrid Catalytic Systems. Catalysts, 11(4), 411. https://doi.org/10.3390/catal11040411