
Layer-Like Zeolite X as Catalyst in a Knoevenagel Condensation: The Effect of Different Preparation Pathways and Cation Exchange

Abstract
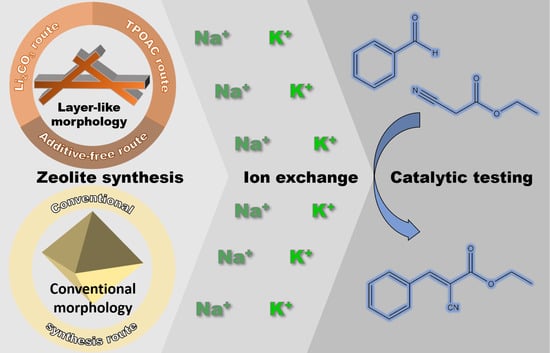
1. Introduction
2. Results and Discussion
2.1. Material Characterization before and after Ion Exchange
2.1.1. Parent NaX Samples
2.1.2. Na/KX Ion Exchange: Effect on Structure, Texture, and Morphology
2.2. Catalytic Behaviour in Knoevenagel Condensation
2.2.1. Catalytic Activity of the Parent NaX Samples
2.2.2. Catalytic Activity after Ion Exchange (NaKX and KX)
3. Materials and Methods
3.1. Zeolite Synthesis
3.2. Ion Exchange
3.3. Catalytic Testing
3.4. Characterisation Methods
4. Conclusions
- Reaction rate and conversion in Knoevenagel condensation of benzaldehyde with ethyl cyanoacetate over layer-like zeolite X exceed that over conventional zeolite X.
- Depending on the zeolite crystal morphology, the charge balancing cation (Na+ and/or K+) affected the catalytic activity differently. In most cases, zeolite KX with layer-like morphology was found to be the most active catalyst, reaching around 80% conversion after 6 h.
- The crystal structure of zeolite X remained widely intact upon ion exchange Na+ → K+.
- Full ion exchange for potassium can be reached after six consecutive ion-exchange steps.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Opanasenko, M.; Dhakshinamoorthy, A.; Shamzhy, M.; Nachtigall, P.; Horáček, M.; Garcia, H.; Čejka, J. Comparison of the catalytic activity of MOFs and zeolites in Knoevenagel condensation. Catal. Sci. Technol. 2013, 3, 500–507. [Google Scholar] [CrossRef]
- Joshi, U.; Joshi, P.; Tamhankar, S.; Joshi, V.; Rode, C.; Shiralkar, V. Effect of nonframework cations and crystallinity on the basicity of NaX zeolites. Appl. Catal. A Gen. 2003, 239, 209–220. [Google Scholar] [CrossRef]
- Saravanamurugan, S.; Palanichamy, M.; Hartmann, M.; Murugesan, V. Knoevenagel condensation over β and Y zeolites in liquid phase under solvent free conditions. Appl. Catal. A Gen. 2006, 298, 8–15. [Google Scholar] [CrossRef]
- Corma, A.; Fornes, V.; Martín-Aranda, R.; Garcia, H.; Primo, J. Zeolites as base catalysts: Condensation of aldehydes with derivatives of malonic esters. Appl. Catal. 1990, 59, 237–248. [Google Scholar] [CrossRef]
- Corma, A.; Fornés, V.; Martín-Aranda, R.; Rey, F. Determination of base properties of hydrotalcites: Condensation of benzaldehyde with ethyl acetoacetate. J. Catal. 1992, 134, 58–65. [Google Scholar] [CrossRef]
- Verboekend, D.; Nuttens, N.; Locus, R.; Van Aelst, J.; Verolme, P.; Groen, J.C.; Perezramirez, J.; Sels, B.F. Synthesis, characterisation, and catalytic evaluation of hierarchical faujasite zeolites: Milestones, challenges, and future directions. Chem. Soc. Rev. 2015, 45, 3331–3352. [Google Scholar] [CrossRef]
- Verboekend, D.; Keller, T.C.; Mitchell, S.; Pérez-Ramírez, J. Hierarchical FAU- and LTA-type zeolites by post-synthetic design: A new generation of highly efficient base catalysts. Adv. Funct. Mater. 2012, 23, 1923–1934. [Google Scholar] [CrossRef]
- Reiprich, B.; Weissenberger, T.; Schwieger, W.; Inayat, A. Layer-like FAU-type zeolites: A comparative view on different preparation routes. Front. Chem. Sci. Eng. 2020, 52, 1131. [Google Scholar] [CrossRef]
- Inayat, A.; Schneider, C.; Schwieger, W. Organic-free synthesis of layer-like FAU-type zeolites. Chem. Commun. 2014, 51, 279–281. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Inayat, A.; Knoke, I.; Spiecker, E.; Schwieger, W. Assemblies of mesoporous FAU-type zeolite nanosheets. Angew. Chem. Int. Ed. 2012, 51, 1962–1965. [Google Scholar] [CrossRef]
- Khaleel, M.; Wagner, A.J.; Mkhoyan, K.A.; Tsapatsis, M. On the rotational intergrowth of hierarchical FAU/EMT zeolites. Angew. Chem. Int. Ed. 2014, 53, 9456–9461. [Google Scholar] [CrossRef]
- Medeiros-Costa, I.C.; Laroche, C.; Pérez-Pellitero, J.; Coasne, B. Characterization of hierarchical zeolites: Combining adsorption/intrusion, electron microscopy, diffraction and spectroscopic techniques. Microporous Mesoporous Mater. 2019, 287, 167–176. [Google Scholar] [CrossRef]
- Schwieger, W.; Machoke, A.G.; Weissenberger, T.; Inayat, A.; Selvam, T.; Klumpp, M.; Inayat, A. Hierarchy concepts: Classification and preparation strategies for zeolite containing materials with hierarchical porosity. Chem. Soc. Rev. 2016, 45, 3353–3376. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Machoke, A.G.; Schwieger, W. Catalytic test reactions for the evaluation of hierarchical zeolites. Chem. Soc. Rev. 2016, 45, 3313–3330. [Google Scholar] [CrossRef]
- Keller, T.C.; Isabettini, S.; Verboekend, D.; Rodrigues, E.G.; Perez-Ramirez, J. Hierarchical high-silica zeolites as superior base catalysts. Chem. Sci. 2014, 5, 677–684. [Google Scholar] [CrossRef]
- Keller, T.C.; Arras, J.; Wershofen, S.; Pérez-Ramírez, J. Design of hierarchical zeolite catalysts for the manufacture of polyurethane intermediates. ACS Catal. 2015, 5, 734–743. [Google Scholar] [CrossRef]
- Yutthalekha, T.; Wattanakit, C.; Warakulwit, C.; Wannapakdee, W.; Rodponthukwaji, K.; Witoon, T.; Limtrakul, J. Hierarchical FAU-type zeolite nanosheets as green and sustainable catalysts for benzylation of toluene. J. Clean. Prod. 2017, 142, 1244–1251. [Google Scholar] [CrossRef]
- Yutthalekha, T.; Suttipat, D.; Salakhum, S.; Thivasasith, A.; Nokbin, S.; Limtrakul, J.; Wattanakit, C. Aldol condensation of biomass-derived platform molecules over amine-grafted hierarchical FAU-type zeolite nanosheets (Zeolean) featuring basic sites. Chem. Commun. 2017, 53, 12185–12188. [Google Scholar] [CrossRef]
- Lechert, H.; Kacirek, H. The kinetics of nucleation of X zeolites. Zeolites 1993, 13, 192–200. [Google Scholar] [CrossRef]
- Ferdov, S. Conventional synthesis of layer-like zeolites with faujasite (FAU) structure and their pathway of crystallization. Microporous Mesoporous Mater. 2020, 303, 110263. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Haouas, M.; Taulelle, F.; Martineau, C. Recent advances in application of 27 Al NMR spectroscopy to materials science. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 94–95, 11–36. [Google Scholar] [CrossRef] [PubMed]
- Demtröder, W. Experimentalphysik, 5, neu Bearbeitete und Aktualisierte Auflage; Springer: Berlin/Heidelberg, Germany, 2016; ISBN 978-3662490938. [Google Scholar]
- Bordiga, S.; Lamberti, C.; Bonino, F.; Travert, A.; Thibault-Starzyk, F. Probing zeolites by vibrational spectroscopies. Chem. Soc. Rev. 2015, 44, 7262–7341. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N2 Physisorption Results | ICP-OES Results | |||||||
|---|---|---|---|---|---|---|---|---|
| Aspec\m²g−1 | Aext\m²g−1 | Vmicro\cm3g−1 | Vmeso\cm3g−1 | Si Al−1 \- | Na Al−1 \- | K Al−1 \- | Li Al−1 \- | |
| X-Conv | ||||||||
| As-synthesised | 923 | 110 | 0.308 | 0.093 | 1.45 | 0.87 | - | - |
| 1st ion exchange | 789 | 97 | 0.261 | 0.076 | 1.50 | 0.33 | 0.53 | - |
| 6th ion exchange | 781 | 98 | 0.258 | 0.077 | 1.33 | 0.01 | 0.95 | - |
| (change compared to as-synthesised) | −15% | -11% | −16% | −17% | ||||
| X-Addfree | ||||||||
| As-synthesised | 821 | 153 | 0.258 | 0.170 | 1.47 | 0.98 | - | - |
| 1st ion exchange | 675 | 142 | 0.206 | 0.189 | 1.29 | 0.38 | 0.56 | - |
| 6th ion exchange | 643 | 162 | 0.188 | 0.222 | 1.30 | 0.01 | 0.96 | - |
| (change compared to as-synthesised) | −22% | +6% | −27% | +31% | ||||
| X-Li2CO3 | ||||||||
| As-synthesised | 774 | 131 | 0.248 | 0.141 | 1.47 | 0.92 | - | 0.11 |
| 1st ion exchange | 663 | 132 | 0.205 | 0.152 | 1.53 | 0.32 | 0.54 | 0.03 |
| 6th ion exchange | 641 | 122 | 0.199 | 0.158 | 1.30 | 0.01 | 1.02 | 0.00 |
| (change compared to as-synthesised) | −17% | −7% | −20% | +12% | ||||
| X-TPOAC (calcined) | ||||||||
| calcined | 611 | 173 | 0.173 | 0.240 | 1.44 | 0.89 | - | - |
| 1st ion exchange | 520 | 155 | 0.145 | 0.216 | 1.51 | 0.34 | 0.43 | - |
| 6th ion exchange | 512 | 151 | 0.144 | 0.219 | 1.41 | 0.05 | 0.81 | - |
| (change compared to as-synthesised) | −16% | −13% | −17% | −9% | ||||
| NaX-Conv | NaX-Li2CO3 | NaX-Addfree | NaX-TPOAC | |
|---|---|---|---|---|
| Plate thickness\nm | - | 183 ± 34 | 126 ± 22 | 96 ± 19 |
| Particle diameter\μm | 2.56 ± 0.42 | 3.56 ± 0.39 | 1.60 ± 0.22 | 5.90 ± 1.32 |
| Parent (NaX) /(mol·L−1·min−1) | 1st Ion Exchange (Na/KX) /(mol·L−1·min−1) | 6th Ion Exchange (KX) /(mol·L−1·min−1) | |
|---|---|---|---|
| X-Conv | 0.013 | 0.031 (60% K) | 0.054 |
| X-Li2CO3 | 0.025 | 0.051 (60% K) | 0.087 |
| X-Addfree | 0.045 | 0.047 (55% K) | 0.133 |
| X-TPOAC (calcined) | 0.092 | 0.050 (50% K) | 0.069 |
| 1.0 Al203: 3.0 SiO2: 180 H20: | Crystallisation Temperature/ Duration | |||
|---|---|---|---|---|
| Na2O | Li2CO3 | TPOAC MeOH | ||
| NaX-Conv [8] | 4.0 | - | - - | 75 °C/4 d |
| NaX-Addfree [8] | 4.0 | - | - - | 50 °C/3 d |
| NaX-Li2CO3 [9] | 3.7 | 0.6 | - - | 75 °C/4 d |
| NaX-TPOAC [10] | 3.5 | - | 0.06 0.62 | 75 °C/5 d |
| Sample | Sodium Silicate Solution | Sodium Aluminate Solution | NaOH | Deionized Water | Li2CO3 | TPOAC Solution |
|---|---|---|---|---|---|---|
| NaX-Conv [8] | 47.15 | 37.03 | 8.20 | 175.50 | - | - |
| NaX-Addfree [8] | 47.15 | 37.03 | 8.20 | 175.50 | - | - |
| NaX-Li2CO3 [9] | 47.15 | 37.03 | 6.47 | 175.90 | 12.59 | - |
| NaX-TPOAC [10] | 47.15 | 37.03 | 5.32 | 176.18 | - | 5.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grass, J.-P.; Klühspies, K.; Reiprich, B.; Schwieger, W.; Inayat, A. Layer-Like Zeolite X as Catalyst in a Knoevenagel Condensation: The Effect of Different Preparation Pathways and Cation Exchange. Catalysts 2021, 11, 474. https://doi.org/10.3390/catal11040474
Grass J-P, Klühspies K, Reiprich B, Schwieger W, Inayat A. Layer-Like Zeolite X as Catalyst in a Knoevenagel Condensation: The Effect of Different Preparation Pathways and Cation Exchange. Catalysts. 2021; 11(4):474. https://doi.org/10.3390/catal11040474
Chicago/Turabian StyleGrass, Jan-Paul, Katharina Klühspies, Bastian Reiprich, Wilhelm Schwieger, and Alexandra Inayat. 2021. "Layer-Like Zeolite X as Catalyst in a Knoevenagel Condensation: The Effect of Different Preparation Pathways and Cation Exchange" Catalysts 11, no. 4: 474. https://doi.org/10.3390/catal11040474
APA StyleGrass, J.-P., Klühspies, K., Reiprich, B., Schwieger, W., & Inayat, A. (2021). Layer-Like Zeolite X as Catalyst in a Knoevenagel Condensation: The Effect of Different Preparation Pathways and Cation Exchange. Catalysts, 11(4), 474. https://doi.org/10.3390/catal11040474