1. Introduction
The vast majority of catalysts used on industrial scale are heterogenous catalyst with a high surface-to-volume ratio to increase the number of active sites. Very often, these catalysts are microcrystalline or even amorphous. This makes it difficult to gain knowledge on the electronic and structural properties of the active sites, a prerequisite to improve the performance of the catalyst.
In the past two decades, metal fluoride catalysts, especially high-surface aluminium fluoride, HS-AlF
and amorphous ACF (aluminiumchlorofluoride, AlCl
F
,
x = 0.05−0.3), have shown high activity in several reactions such as C-H and C-F bond activation or hydroarylation reactions [
1,
2,
3,
4,
5,
6]. In selected reactions, these catalysts show comparable activity with the homogeneous catalyst SbF
[
2]. To get more insight into the chemistry of these heterogenous catalysts, the Lewis acidity of the active sites, which describes their potential to bind to electron rich compounds, can be compared [
7].
These metal fluoride catalysts show their catalytic behaviours only if they are synthesised as nanocrystalline material or like ACF only in its amorphous form. Therefore, it is far from trivial to get experimental characterization of these materials. One way to quantify the Lewis acidity is the adsorption of small molecules like CO and NH
, where their lone pairs can bind to the positively charged catalytic centres. The adsorption of carbon monoxide (CO) and ammonia (NH
) as probe molecules is an essential tool in the characterization of a surfaces’ local properties [
8,
9,
10,
11,
12,
13,
14,
15]. Due to their adsorption at the active sites, the corresponding frequency shifts allow an estimation of Lewis-acidities, both experimentally and computationally [
8,
9,
10,
11,
12,
13,
14,
15]. For example, a large blue shift in the CO stretching vibration is associated with high Lewis acidity of an active, so-called coordinatively unsaturated site (CUS). Different frequency shifts can be associated with coordination numbers of the CUS on the surfaces [
16].
In different experimental investigations, the Lewis acidity of milled
-AlF
,
-AlF
, high-surface AlF
(HS-AlF
) and ACF were previously determined by the adsorption of CO and the corresponding IR spectra (see
Table 1) [
2,
3,
17,
18]. Additionally Bailey et al. [
19,
20] performed theoretical studies at four- and fivefold coordinated aluminium centres of the
-AlF
(001),
-AlF
-(0001) and
-AlF
-(01
2) surfaces with adsorbed CO, which show blue shifts comparable to the available experimental values. The measured frequencies were used as a basis for the classification of the Lewis acidic centres, by comparison of the frequency shift with the experimental CO frequency in the free molecule (2143 cm
) [
3]. Whereas weakly physiosorbed CO shows nearly no shift (smaller than 10 cm
), very strong Lewis acidic centres can result in a blue shift up to ~80 cm
[
3]. In this work, six different types of acidic centres were proposed, the most common ones being four- and fivefold coordinated aluminium centres, with fourfold coordinated ones assumed to be less Lewis-acidic than fivefold coordinated centres. This on the first glance counterintuitive behaviour is explained by the low accessibility of the fourfold active centre [
14], due to the tetrahedral coordination environment. There are three types of fivefold coordinated aluminium centres on AlF
-surfaces found, which differ by the number of terminal fluorine atoms [
14]. Krahl and Kemnitz proposed that due to the higher electron withdrawing effect of bridging fluorine atoms, the electron pair acceptor strength of the aluminium centre decreases with the number of terminal fluorine atoms [
14].
In the dissertation by Bailey [
20], few surface structures of
- and
-AlF
were characterized via the adsorption of CO at the model surfaces. In this work, the adsorption of CO at the fourfold coordinated aluminium centers on the
-AlF
-(0001) surface within a 2 × 1 cell and on the
-AlF
-(01
2) surface with a 1 × 1 and a
×
surface cell were investigated under the usage of the Crystal code with the B3LYP functional and the corresponding basis sets. The
-AlF
-(0001) surface model shows adsorption energies between −0.16 to −0.30 eV with blue shifts of the CO stretch vibration of 78 and 88 cm
. For the
-AlF
-(01
2) surfaces, adsorption energies between −0.52 eV for the 1 × 1 and −0.21 eV for the
×
surface were found together with blue shifts of the CO stretch vibration between 72 and 87 cm
. For both surface models, the theoretical results are in the range of the experimental data [
20]. Our previous investigations [
21] have shown that the (01
2) surface is quite high in energy and only another layer of fluorides and surface reconstruction stabilizes the
-AlF
-(01
2) surface. Locally, the structural motifs found by Bailey [
20] and ours agree well, as proven by similar frequency shift obtained for CO. Contrary to the model of Bailey et al., the (0001) surface model used in this work shows threefold coordinated aluminium centers due to the usage of a different theoretical set-up and no additional fluorides [
21].
For NH
as the probe molecule, there are three different vibrational frequencies that are affected by adsorption; the symmetric and asymmetric stretch modes in the range of 3300 cm
and a bending mode in the range of 1630 cm
. Different chemisorption modes for NH
on MgO [
8,
9] and ZnO [
11] surfaces were characterized, but not much is known for the adsorption of NH
on metal fluoride surfaces. On these oxides, the adsorption of NH
on Lewis acidic CUS resulted in a blue shift of the asymmetric bending mode. This mode is most sensitive to the structural surrounding of the surface atoms, as it is highly affected by the formation of hydrogen bonds to the surface ions [
9].
Furthermore, vibrational spectroscopy is a valuable experimental tool to characterize surfaces, and not much can be directly deduced on the local surrounding and possible secondary interactions to the anions. In this sense, first-principle investigations of different surface models of the heterogeous catalyst can help to elucidate the local properties.
Currently, there are only a few publications by Bailey et al. that characterize NH
adsorption on metal fluorides [
15]. In related studies published by the same authors, the interactions with
- and
-AlF
-surfaces were investigated with the focus on the adsorption of HF [
22], H
O [
22] and CCl
F
[
23]. It can be assumed from the modelled structures by Bailey et al., that the binding motif of NH
is similar to the “upright“ conformation of NH
on MgO surfaces [
9], where the hydrogens can interact with the neighbouring terminal fluorine atoms of the surface. In the existing literature, only structural properties have been determined, and no frequencies for NH
on AlF
surfaces have been provided.
This paper covers studies on the adsorption of CO and NH
on different surface models of
-AlF
and Cl-doped
-AlF
under the usage of periodic DFT calculations, applying the PBE functional with D3-BJ dispersion correction. In earlier studies [
21], different surfaces, which appear in the nano-crystal of the
-AlF
, were characterized in detail and stable chloride-doped variants were proposed. The structural changes due to adsorption and the frequency shifts of the probe molecules are compared with the experimental results [
3,
17,
18] of the corresponding fluorides. For the analysis of the electronic structure, the Bader charges for all surface models and density of states for selected ones are added to the
Supplementary Materials. In none of the cases were interesting electronic contributions to binding found, therefore a more detailed analysis of the electronic structure with other methods like ELF or bond analysis on surfaces is not necessary [
24]. Both are not straightforward methods with a plane-wave code used in this work.
The paper is organized as follows. After providing the computational details in
Section 2, we describe the models in
Section 3. The results are presented, discussed and compared with the experimental data in
Section 4. The conclusion follows in
Section 5.
4. Results
In [
21], we found, with the same computational methods, that
-AlF
is characterized by five different surfaces in vacuum. We could also show that replacing terminal and bridging fluorides by chloride results in no significant change in the relative stability of the surfaces. Therefore, the adsorption of the probe molecules was performed on the same surface cuts for both systems.
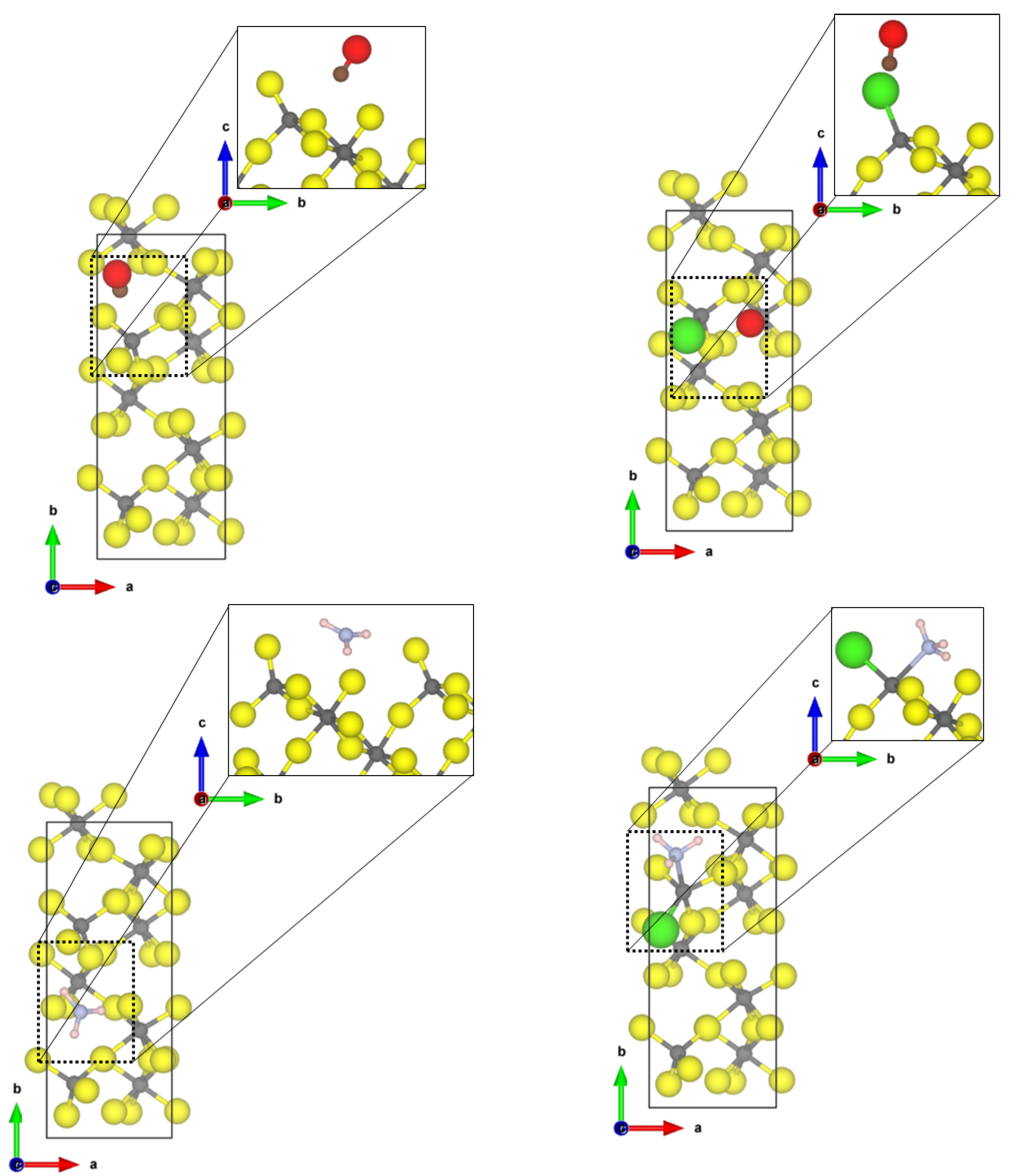
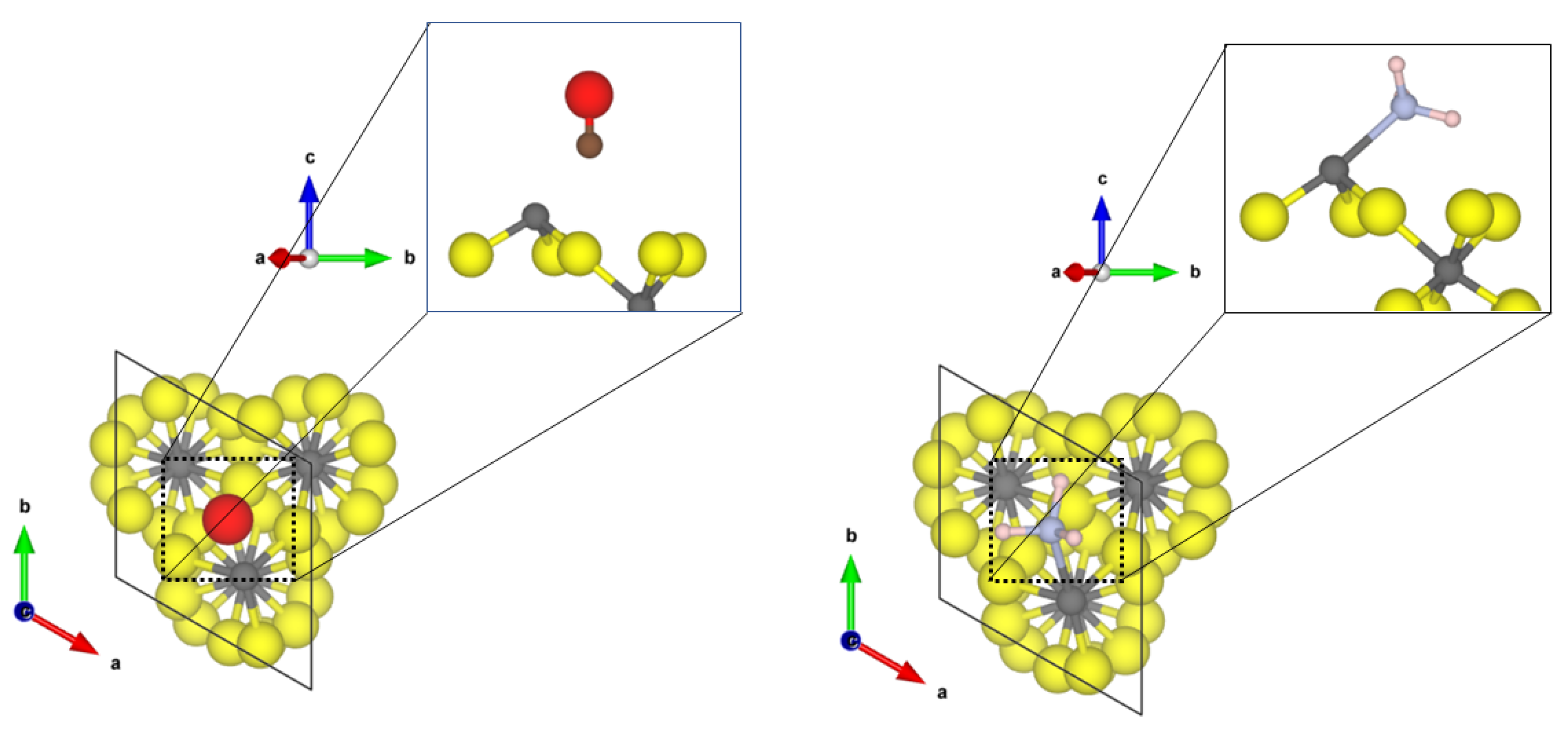
The most stable and most exposed (35–40%) (01
0) surface shows two tetrahedral coordinated aluminium sites. For the Cl-doped surface model, only one terminal fluoride was replaced by a chloride ion. The obtained structures of the probe molecules adsorbed to the
-AlF
- and the Cl-doped
-AlF
-(01
0) surface are depicted in
Figure 1.
For CO, the Cl-doping affects the binding significantly. On the -AlF-(010) surface, CO is physisorbed to the fourfold coordinated aluminium centre with a Al-C distance of 2.67 Å and an adsorption energy of −0.31 eV. The fluoride ions are far away from the adsorbed molecule; therefore, it is assumed that they have no influence on the binding. For the adsorbed CO, the bond distance with 1.134 Å is only 0.005 Å shorter as in the free molecule, but a medium-sized blue shift of the CO stretching frequency to 2165 cm was determined.
In contrast, for the Cl-doped -AlF-(010) surface, the CO adsorbs symmetrically between two terminal bound chlorine atoms with a Cl-C distance of 2.69 Å and an adsorption energy of −0.26 eV. The intramolecular distance in CO is 1.138 Å, which is in agreement with the assumed accuracy of the values for the free molecule. The larger Cl atoms hamper the approach of the CO to the aluminium site. The interaction of the CO molecule with the Cl atoms on this surface causes a slight red shift of the CO frequency to 2114 cm.
The NH
molecule coordinates to three terminal fluorine atoms, with H⋯F hydrogen bond distances of 1.67 Å, 2.06 Å and 2.49 Å and the corresponding N-H bonds are changed by +0.014 Å, −0.004 Å and −0.007 Å, respectively. Comparing these distances to the intermolecular F-H⋯F-H distance of solid HF (1.53 Å [
45]), we suggest that at least one hydrogen bond of medium strength is formed. The NH
molecule itself adopts a nearly planar conformation. For this interaction, an adsorption energy of −1.29 eV was found, which is characteristic for a chemisorption of the molecule; however, no covalent bonds are formed. Therefore, it can be assumed that each hydrogen bond add −0.43 eV, respectively, to the overall adsorption energy of the molecule. This is within the range of medium-sized hydrogen bonds in small molecules [
46]. Unfortunately, it was not possible to converge the structure with higher accuracy, a necessity for determining the vibrational frequencies. Different approaches, including the electronic minimization algorithms [
47,
48,
49], ionic relaxation algorithms [
50] and different starting points were used, however, with no success. These difficulties in reaching the convergence can be attributed to a very shallow potential energy surface, and for accurate results, the quantum nature of the hydrogens also has to be considered, which is far beyond the scope of this publication.
Whilst the CO on the Cl-doped -AlF-(010) surface cannot approach the CUS, NH coordinates to the fourfold coordinated Al with a N-Al distance of 2.06 Å. One of the three hydrogen atoms forms a medium-strength hydrogen bond with a distance of 1.77 Å to a terminal fluoride of the surface, which elongates the NH bond by 0.002 Å compared to the free ammonia. The covalent bond formed between Al-N is strong enough (adsorption energy of −1.49 eV) to distort the tetrahedral enviroment to a trigonal pyramidal one. Surprisingly, the asymmetric bending frequency is only affected slightly with a small blue shift of 4 cm.
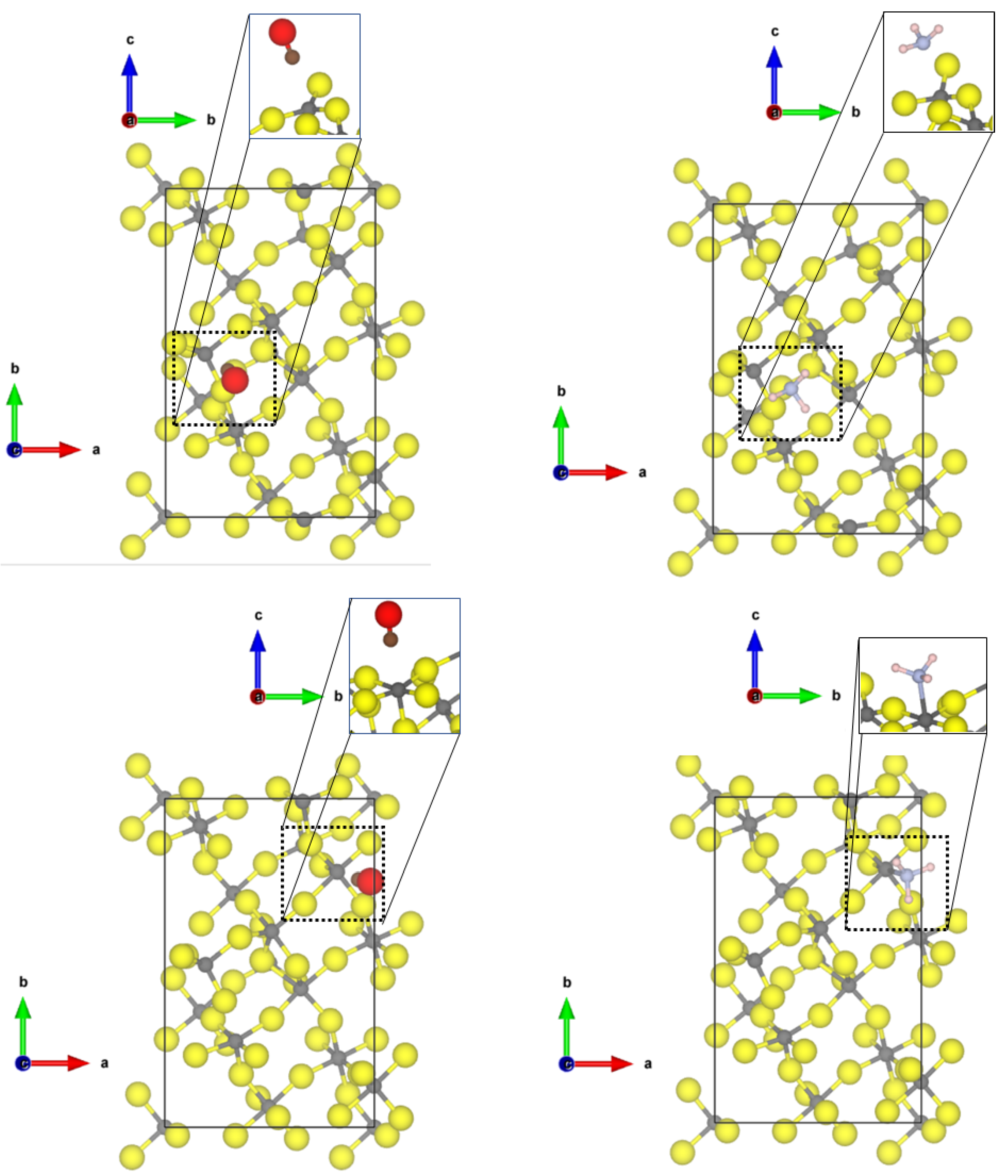
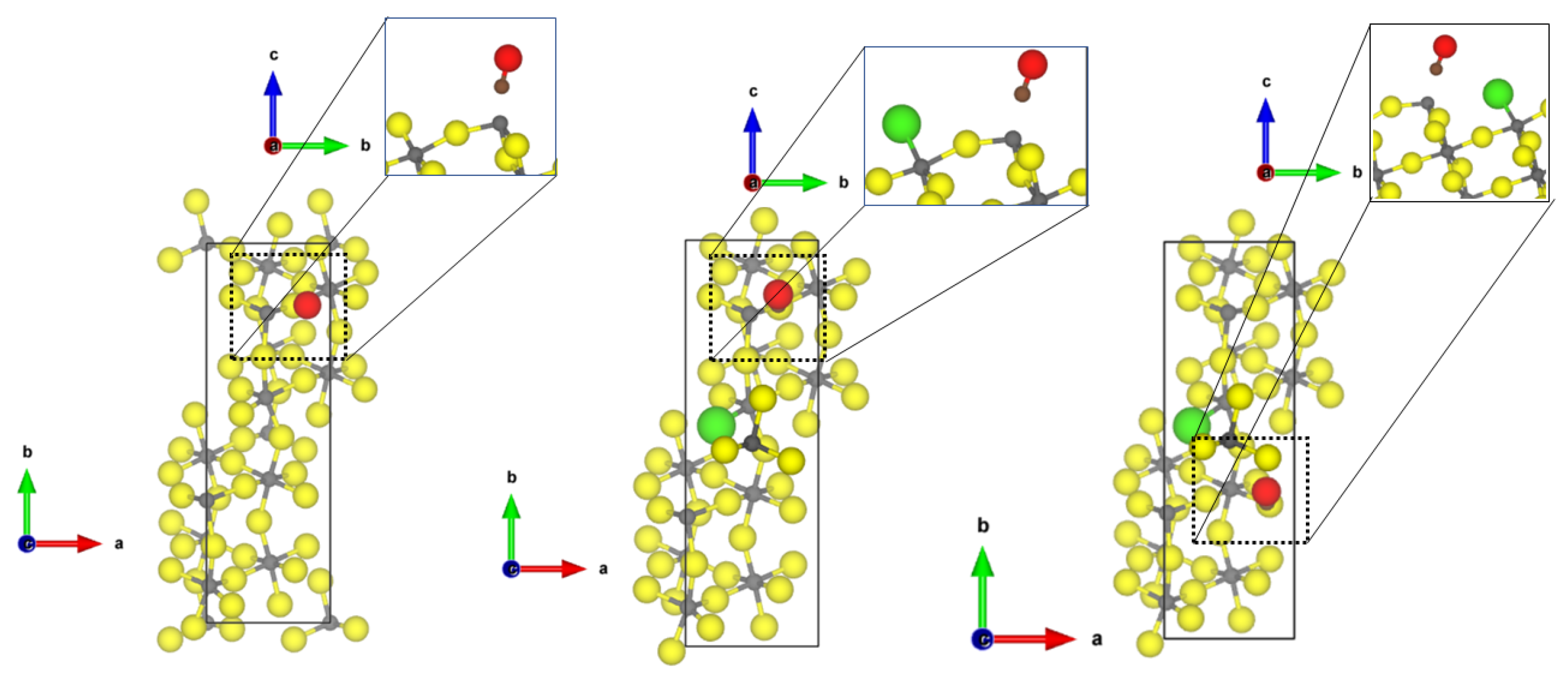
Investigating now the second most abandoned (11
2) surfaces, there are two differently coordinated aluminium sites (see
Figure 2) at which CO can adsorb.
At the tetrahedral site, CO is bound with very similar adsorption energy (−0.30 eV, physisorption) as to the tetrahedral coordinated -AlF-(010) active site; however, a shorter Al-C distance of 2.40 Å and a larger blue shift to 2187 cm was found. The adsorption of CO to the five-fold coordinated CUS is stronger compared to the fourfold coordinated one (adsorption energy of −0.60 eV), and a shorter Al-C distance of 2.24 Å was found, but the frequency is only shifted to 2158 cm. In this case, there is no direct relation between adsorption strength and frequency shift. Despite the fact that the adsorption energy on the 5-fold CUS is doubled, the frequency shift is only half of the one on the fourfold CUS.
NH
also adsorbs on both available CUS of the
-AlF
-(11
2) surface. When bound to the fourfold CUS, the adsorption energy is in the range of physisorption (−0.48 eV) with a quite long Al-N distance (2.77 Å, see
Figure 2) and a frequency shift of 7 cm
. The missing possibility of forming hydrogen bonds to the fluorides in this upright configuration (as found for the structure by Allouche et al. on MgO [
9]) prevents a stronger binding. Therefore, all N-H distances are similar as in the free molecule, and the bending vibration is not affected. The situation is totally different at the 5-fold CUS. The NH
chemisorbs with an adsorption energy of −1.76 eV and an Al-N distance of 2.03 Å. Only weak hydrogen bonds are formed (H⋯F distances of 2.28 and 2.39 Å), and the bending frequency undergoes very little change (frequency shift of 8 cm
). The strong binding solely emerges from the chemical bond between Al-N. The determined Al-N bond length is only 0.1 Å longer than the experimental value found in crystalline AlN (1.89 Å [
51]).
At the Cl-doped
-AlF
-(11
2), the CO molecule interacts with the tetragonal CUS with the terminal Cl atom (see
Figure 3). The intramolecular CO distance undergoes very little change (1.137 Å), and an Al-C distance of 3.26 Å can be found. Due to the long Al-C distance, the adsorption energy is low (−0.22 eV). Cl-doping at the 5-fold CUS was found to be energetically not favourable [
21]; therefore, no differences for the adsorption of the molecules are found compared to the equivalent CUS on
-AlF
-(11
2) (see
Figure 2).
For NH at the tetragonal CUS bound to the Cl-doped -AlF-(112), a weak chemisorption with an adsorption energy of −0.75 eV and an Al-N distance of 2.61 Å is found. The molecule shows H⋯F/Cl distances between 2.7 Å and 2.9 Å. The conformation of the molecule is also comparable to the “upright” case found for the (010) surface. Unfortunately, the structures of CO and NH on the Cl-doped -AlF-(112) surface could not be optimized to the degree of accuracy necessary for the frequency analysis.
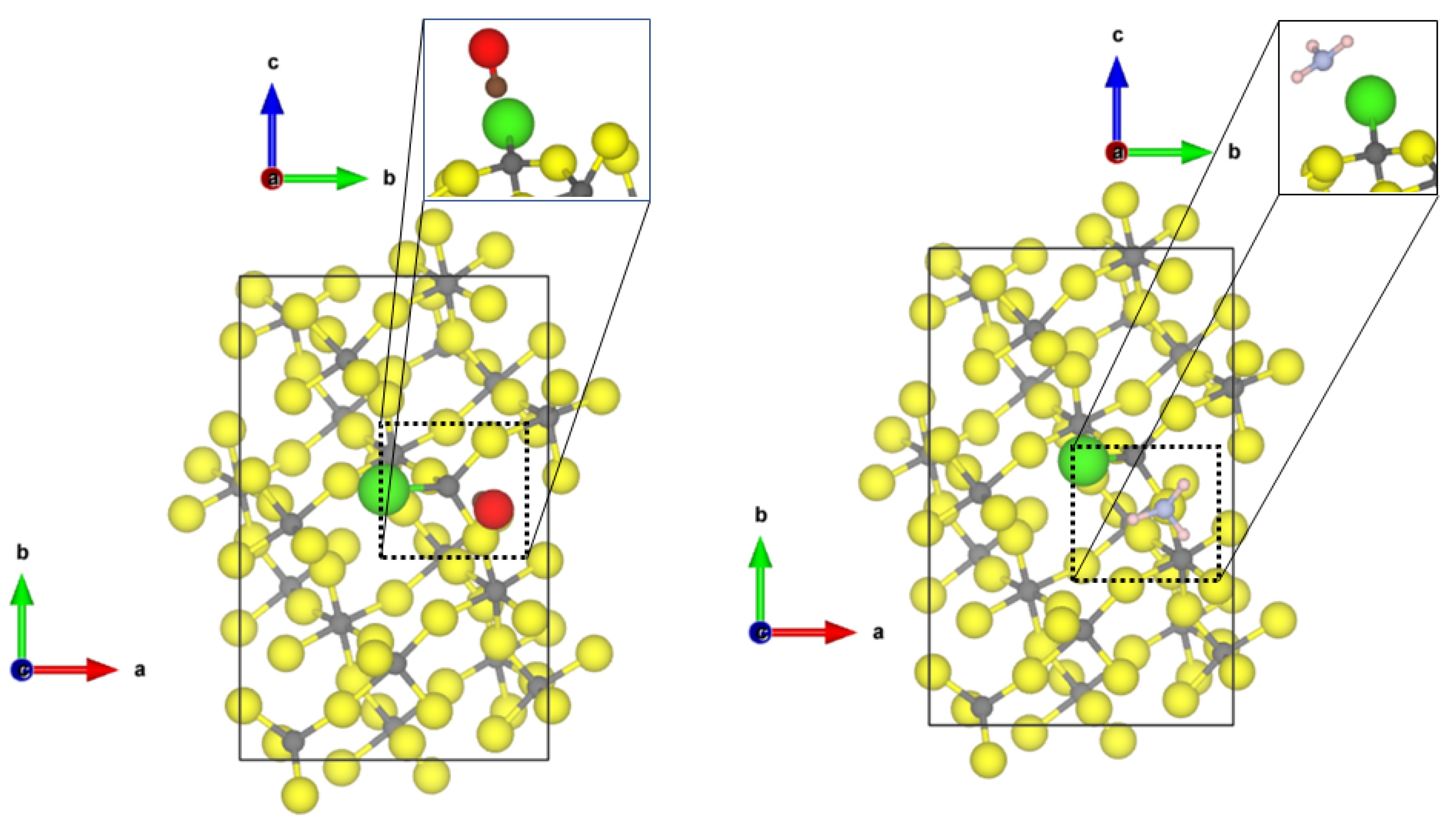
The third most abandoned surface in
-AlF
is the (11
0) surface; however, by Cl-doping its surface energy increases. This surface shows two 5-fold CUS with a square-pyramidal structure. Both CO and NH
adsorb at the CUS, independently of terminal or bridging substitution of fluorine with chlorine atoms (see
Figure 4). For CO, a weak chemisorption (adsorption energy in the range of −0.7 eV) and Al-C distances of 2.19 Å were found. In all three cases, the intramolecular CO distance is around 1.132 Å and the frequencies were in the range of 2190 to 2205 cm
(blue shifts of 64 to 79 cm
).
NH shows a weak chemisorption on the (110) surfaces (adsorption energy in the range of 0.9 eV), independently of the Cl-substitution. In all three cases, the Al-N bond length is about 2.3 Å. Alongside the weak chemical bond to Al, NH forms a strong hydrogen bond to one fluoride at the surface (distance 1.74 Å) and another weak hydrogen bond with the distance of 2.14 Å. Although all NH distances were not changed by adsorption, owing to the quite strong hydrogen bond, the asymmetric bending mode is strongly affected with a blue shift in the range of 20 cm.
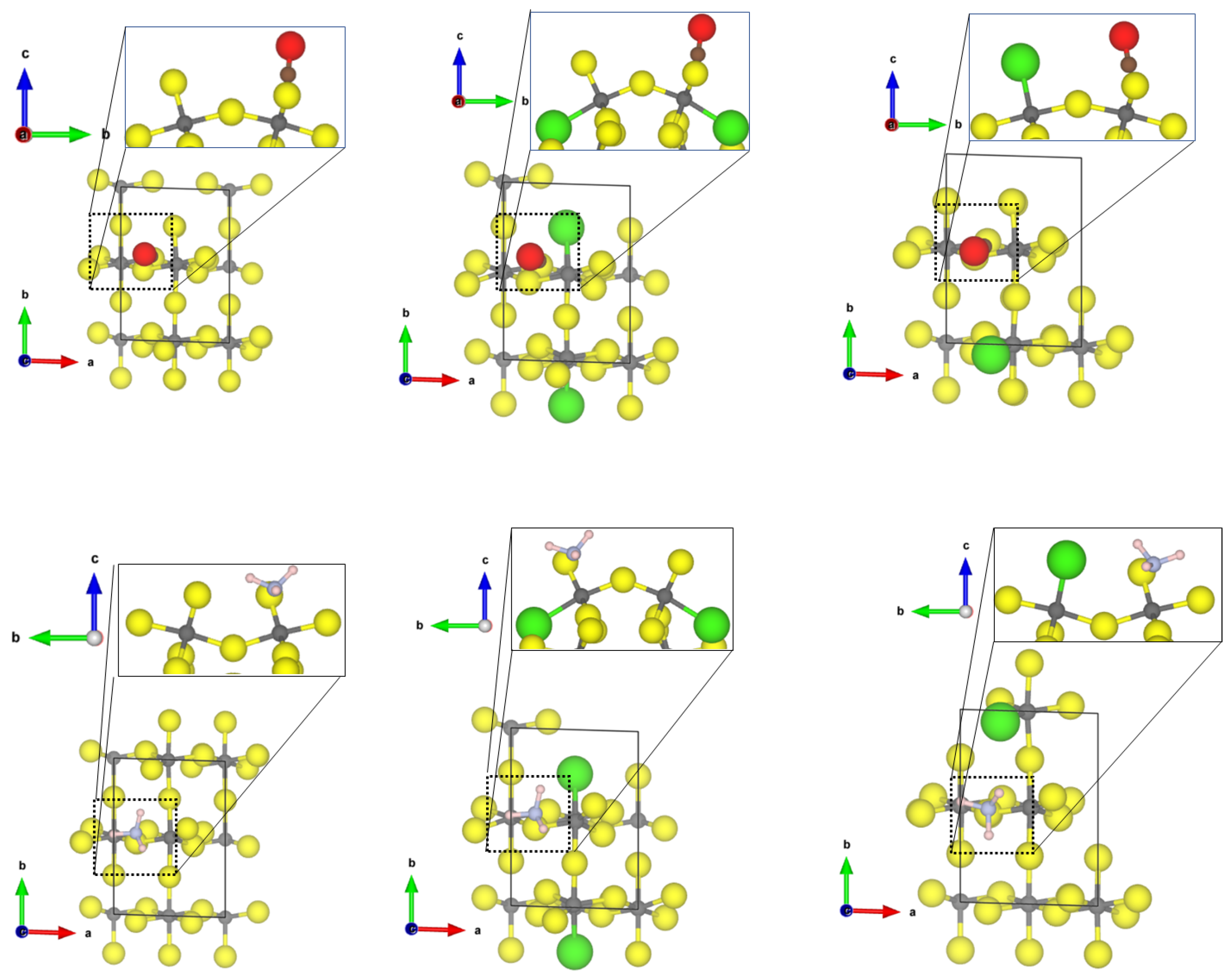
For the less abundant (0001) and (10
2) surfaces, a highly undercoordinated threefold CUS structure occurs. This surface most likely only exists in high vacuum; however, it is interesting from a theoretical point of view. The binding structures of CO and NH
on the
-AlF
(0001) surface are shown in
Figure 5.
Compared to the adsorption of CO to the fourfold and 5-fold CUS of the previous surfaces, a significant elongation of the CO bond length to 1.165 Å and a drastic red shift of the frequency by roughly 200 cm upon coordination at the -AlF-(0001) surface was found. This is an indication that an Al-C chemical bond is formed. The adsorption energy of CO on the threefold CUS (−1.01 eV) is somewhat higher than for the fourfold or fivefold ones, however, not as significant as one would expect from the large red shift. A significant amount of energy is necessary to elongate the CO bond, as the adsorption energy is referred to the free CO in equilibrium. By using the elongated CO bond as reference for the adsorption at the threefold CUS, the corresponding adsorption energy is lowered by 1.04 eV to −2.05 eV.
Furthermore, NH adsorbs strongly on the threefold CUS of the (0001) surface, resulting in a short Al-N distance of 2.05 Å and a strong red shift (58 cm) of the asymmetric bending mode. The adsorption energy of −1.25 eV (chemisorption) is not enhanced compared to fourfold and fivefold CUS. This is due to the missing hydrogen bonds to the fluorides (nearest H⋯F distance is 2.35 A) on the (0001) surface.
While the high surface energy prevents the (0001) surface from occuring in a Cl-doped
-AlF
crystal, the (10
2) surface is stabilized by Cl-substitution [
21] Therefore, only the latter surface is used for investigation of the effect of Cl substitution on the binding properties of CO and NH
. Analysis of the threefold CUS for the adsorption of CO will be described here only since it adsorbs on the 5-fold CUS of the
-AlF
-(10
2) surface similar to the already discussed structures for the (11
0) and (11
2) surface (see
Figure S1 in the Supplementary informations for the optimized structure).
The adsorption of CO and NH
at the threefold CUS of
-AlF
-(10
2) surface is very similar to the one found for the
-AlF
-(0001) surface with a slightly larger Al-C distance and a weaker adsorption energy; however, the Cl-substitution has an effect on the binding. For the CO adsorption, two energetically similar adsorption sites were found, one similar to the case of the undoped surface (see
Figure 6 middle) and one where the CO coordinates between the threefold CUS and the terminal chloride (see
Figure 6 right). For these adsorption structures, the energies were determined to be between −0.54 for the
-AlF
-(10
2) surface and to −0.93 eV for the Cl-doped
-AlF
-(10
2) surface. Surprisingly, both show almost the same strong red shift (~200 cm
), although the structural binding motif is quite different.
The Cl substitution also has a beneficial effect on the NH
adsorption by lowering the adsorption energy by 0.4 eV and decreasing the Al-N bond length to 1.97 Å. However, in this case, the Cl is not involved in the binding. As previously stated, these changes are not correlated to a change in the frequency shift. In all cases, the NH
asymmetric binding mode is strongly red-shifted by about 45 cm
. The optimized structures of NH
on the (10
2) surfaces can be found in the
Supplementary Materials (
Figures S2 and S3 for optimized structures).
5. Discussion
For comparison, the results for all adsorption sites on different surfaces are summarized in
Table 2 for CO and
Table 3 for NH
.
It can be observed that the aluminium centres with less sterical hindrance, such as threefold and fivefold coordinated centres, show usual more negative adsorption energies than fourfold coordinated centres. The highly symmetrical tetrahedral charge distribution around the aluminium site disfavours binding. Krahl et al. [
14] has attributed this effect to a better electrostatic repulsions on the probe molecules. In the case of the Cl-doped
-AlF
-(01
0) surface, the interaction formed with the NH
molecule is strong enough to undergo a reconstruction of the tetrahedron, and a trigonal-bipyramidal structure is formed, which is stabilized with additional hydrogen bonds to the surface fluorides. This stabilizing effect cannot occur in the case of the CO adsorption due to missing hydrogen atoms.
Concerning the influence of Cl-doping (comparing Cl-doped -AlF and corresponding -AlF aluminium sites) only small to negligible effects can be found for the adsorption of CO. It can be seen that the shielding of the surface by chlorine atoms is higher than the shielding by the fluorine atoms due to the more extended electron density, and therefore the binding is slightly reduced for the Cl-doped -AlF sites. For NH, however, the effect is weakened by the possibility of the molecule to form additional hydrogen bonds to the surface, which results in a additional stabilitation.
Regarding the frequency shift of the probe molecules adsorbed to the different coordinated aluminium centres, the molecules adsorbed on the unsaturated threefold coordinated aluminium centres show a large red shift. Due to the high binding affinity of the threefold coordinated aluminium centers a saturation with oxygen, water or other compounds of the realistic catalytic mixture would occur. Additionally, the abundance of the surfaces, where the threefold CUS occur, is below 12% for
-AlF
and 3% for Cl-doped
-AlF
[
21]. Therefore the results would be only apply for special designed high-vacuum surface science experiments.
In constrast, the fourfold and fivefold coordinated aluminium centres are very stable and occur on many different surface cuts with high probability. For the CO stretching vibration, a significant blue shift, from 32 cm
to 79 cm
, was observed. Furthermore, the NH
bending vibration is blue-shifted by 4 cm
to 22 cm
. The CO blue shifts are in good agreement with the experimental results [
2,
3,
17,
18]. Additionally the adsorption energies for CO at the fourfold coordinated centres are in good agreement with the theoretical results of Bailey et al. [
20]. According to the classification of Krahl et al. [
3], weak, medium and strong Lewis acidic centres can be also found within our models.
-AlF
-(11
0) and the Cl-doped variants show strong Lewis acidic centres with frequency shifts between 64 and 79 cm
(see
Table 2). With the same abundance, we find medium Lewis acidic centres on the
-AlF
-(11
2) and
-AlF
-(10
2) (frequency shifts between 52 and 62 cm
) and surfaces with frequency shifts of 32 cm
and 39 cm
, which correspond to weak Lewis acidic centres.
Cl-doping only yields small changes in the frequency shifts of the CO molecules. Therefore, it is likely that the same structural properties of the active sites in
-AlF
and Cl-doped
-AlF
occur. Because no electronic influence and nearly no effect on the local structure of the Cl-doping at
-AlF
surfaces on the adsorption of the probe molecules can be found, we assume that the experimentally measured activity in the ACF is due to its higher amorphicity and therefore due to a higher number of active sites. Besides the structural and vibrational analysis of the surface models, Bader charges and projected density of states for the chlorinated
-AlF
-(01
0) surface are also provided in the
Supplementary Materials.
Additionally, the results of the investigations agree well with the statement of Krahl et al. [
14], that the coordination number of the metal atom of a catalytic active site alone is not enough to determine the Lewis acid strength. For example, the fivefold coordinated aluminium centres show CO frequency shifts in a range of 2158 cm
to 2205 cm
, including weak, medium and strong Lewis acidic centres. The main difference between these active centres, shown by our calculations, is the explicit surrounding of the aluminium sites with different numbers of
or
-F/Cl atoms.
Furthermore, for NH
, different adsorption energies can be found for aluminium centres with the same coordination numbers. A range of adsorption energies from −0.48 to −1.76 eV was found independent of whether NH
binds to five-, four- and threefold coordinated aluminium centres. In comparison to the results of Bailey et al. [
15], our calculations show adsorption energies that are somewhat decreased for the fivefold CUS. Due to the fact that the model of Bailey et al. does not include fourfold coordinated sites for the adsorption of ammonia, no comparison to this study can be made. Additionally, the H⋯F distances of NH
to surface fluorines are within the range determined by Bailey et al. [
15]. The maximum adsorption energy of −2.0 eV determined by Bailey et al. [
15] cannot be observed within our calculations, this can be due to different surface models used or different functionals. Nevertheless, the two computational studies agree well.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}