
Coordination Properties of Non-Rigid Phosphinoyldithioformate Complexes of the [Mo2O2(µ-S)2]2+ Cation in Catalytic Sulfur Transfer Reactions with Thiiranes

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Syntheses
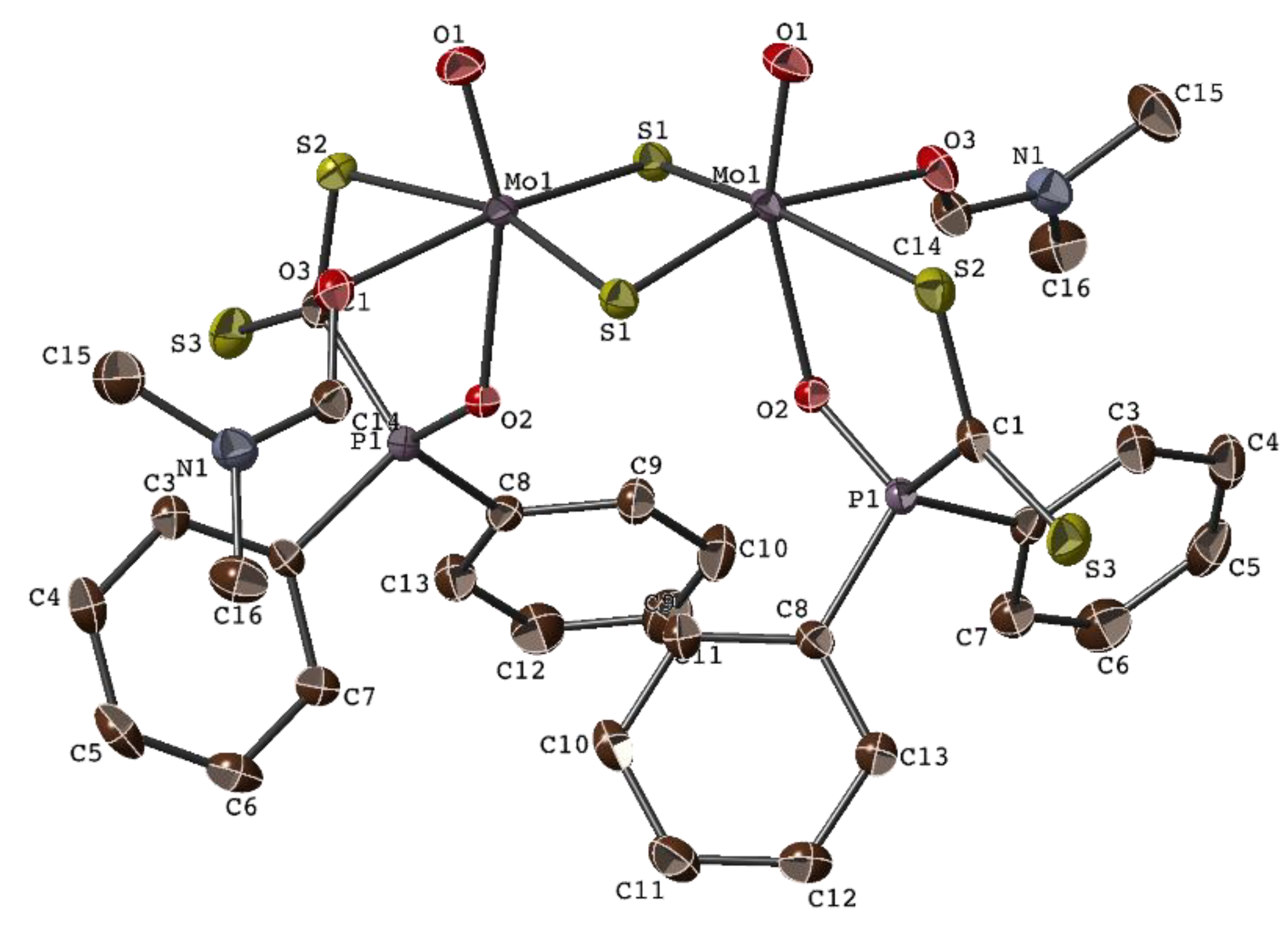
2.2. Molecular Structure of [Mo2O2(µ-S)2{R2P(O)CS2}2(DMF)2], 1
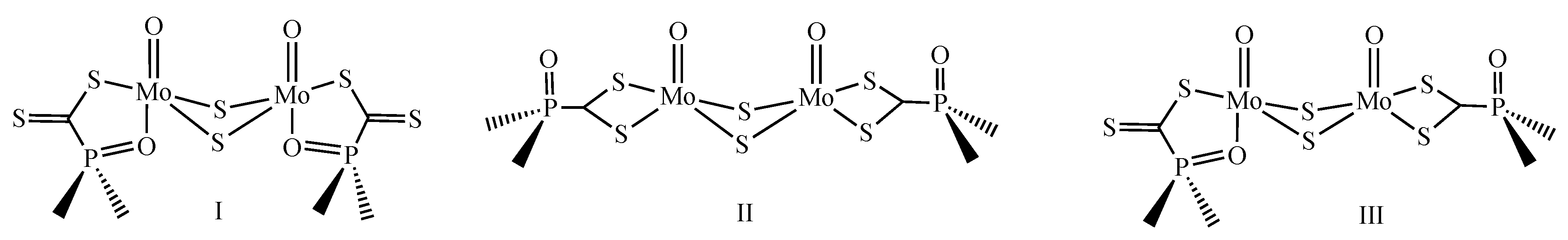
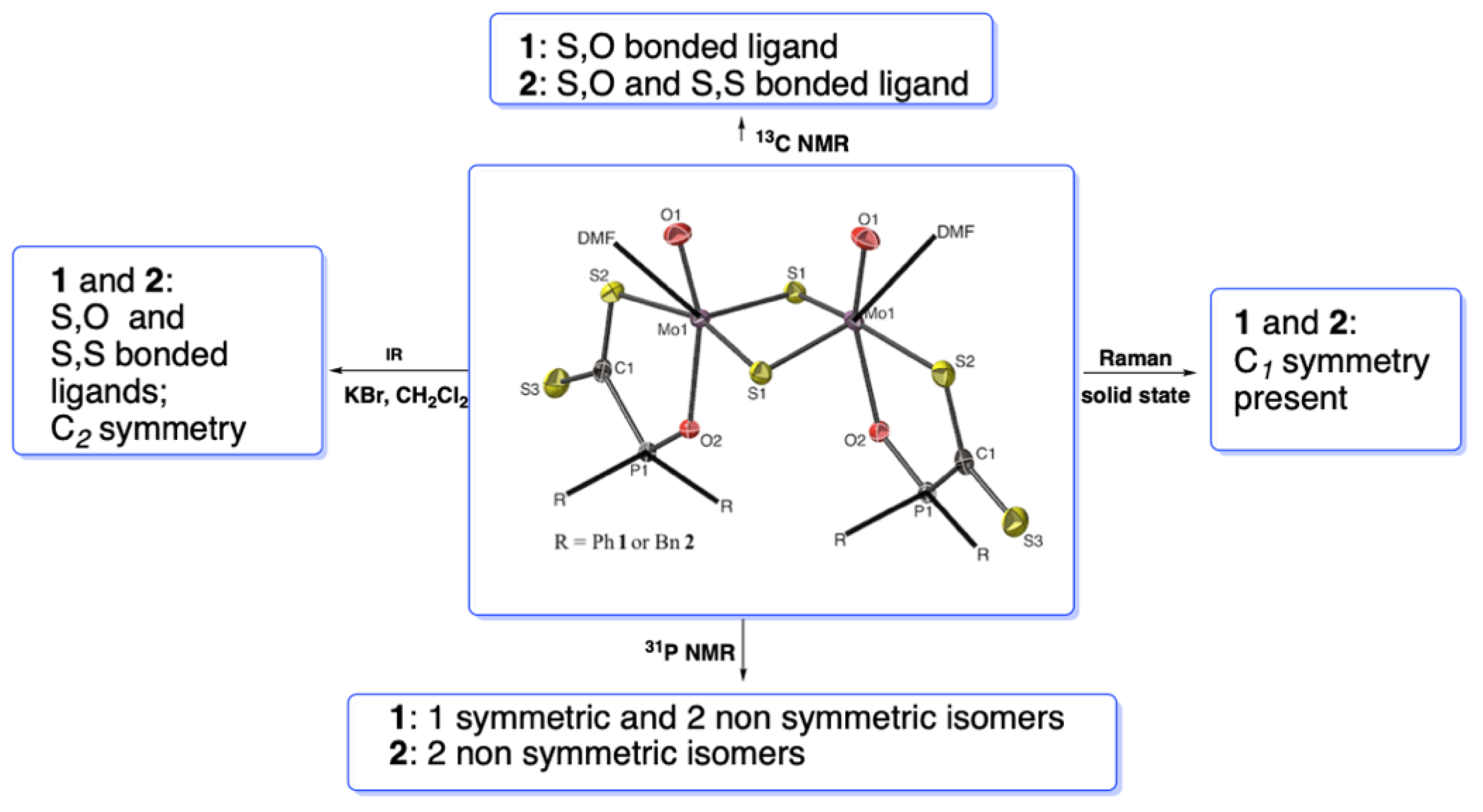
2.3. IR and Raman Spectroscopy
2.4. NMR Spectroscopy
2.5. Variable 31P NMR study of 1 and 2; Non Rigid Behavior
3. Catalytic Experiments on the NMR Scale
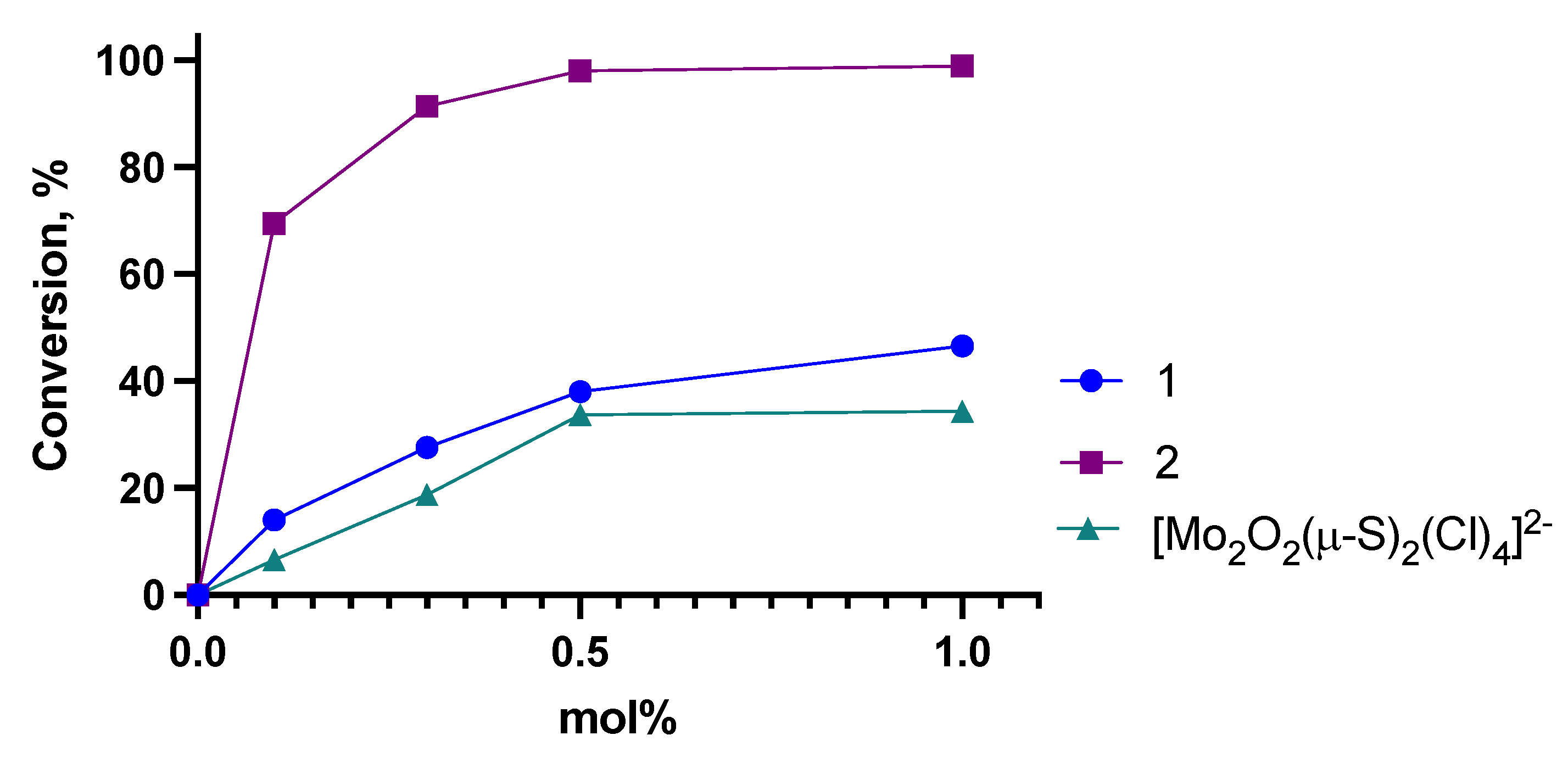
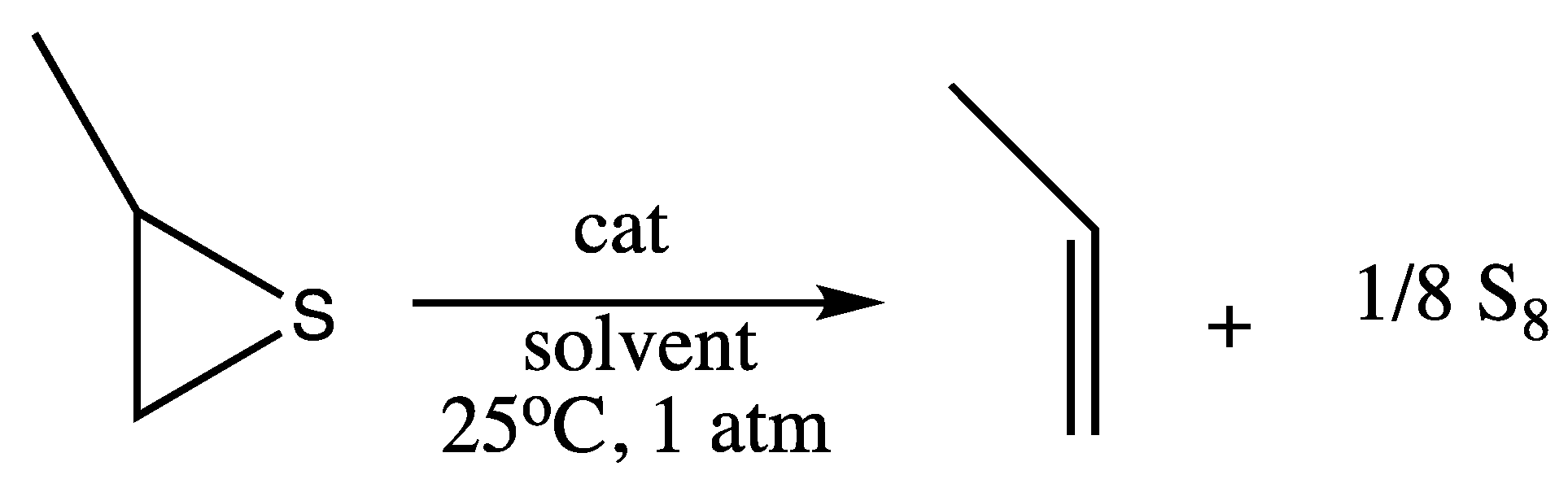
3.1. SAT Reaction of Cyclohexene Sulfide
3.2. SAT Reaction of Propylene Sulfide
3.3. Proposed Mechanism of the Catalytic SAT Reaction
4. Materials and Methods
4.1. General Consideration
4.2. Syntheses
4.3. Catalytic Experiments
4.4. X-ray Crystallography
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rakowski Dubois, M. Catalytic Applications of Transition-Metal Complexes with Sulfide Ligands. Chem. Rev. 1989, 89, 1–9. [Google Scholar] [CrossRef]
- Topsöe, H.; Clausen, B.S. Importance of Co-Mo-S Type Structures in Hydrodesulfurization. Catal. Rev. 2007, 26, 395–420. [Google Scholar] [CrossRef]
- Adam, W.; Bargon, R.M. Synthesis of Thiiranes by Direct Sulfur Transfer: The Challenge of Developing Effective Sulfur Donors and Metal Catalysts. Chem. Rev. 2004, 104, 251–262. [Google Scholar] [CrossRef]
- Garrett, B.R.; Click, K.A.; Durr, C.B.; Hadad, C.M.; Wu, Y. [MoO(S2)2L]1- (L = picolinate or pyrimidine-2-carboxylate) Complexes as MoSx Inspired Electrocatalysts for Hydrogen Production in Aqueous Solution. J. Am. Chem. Soc. 2016, 138, 13726–13731. [Google Scholar] [CrossRef] [PubMed]
- Young, C.G. Facets of early transition metal-sulfur chemistry: Metal-sulfur ligand redox, induced internal electron transfer, and the reactions of metal-sulfur complexes with alkynes. J. Inorg. Biochem. 2007, 101, 1562–1585. [Google Scholar] [CrossRef]
- Donahue, J.P. Thermodynamic Scales for Sulfur Atom Transfer and Oxo-for-Sulfido Exchange Reactions. Chem. Rev. 2006, 106, 4747–4783. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, S.; Hidai, M. Hydrosulfido complexes of transition metals. Coord. Chem. Rev. 2001, 213, 211–305. [Google Scholar] [CrossRef]
- Stiefel, E.I. Transition Metal Sulfur Chemistry: Biological and Industrial Significance and Key Trends. In Transition Metal Sulfur Chemistry; ACS Symposium Series; American Chemical Society: Washington, WA, USA, 1996; pp. 2–38. [Google Scholar]
- Liu, H.; Jiang, X. Transfer of Sulfur: From Simple to Diverse. Chem.—Asian J. 2013, 8, 2546–2563. [Google Scholar] [CrossRef]
- Nakano, K.; Tatsumi, G.; Nozaki, K. Synthesis of Sulfur-Rich Polymers: Copolymerization of Episulfide with Carbon Disulfide by Using [PPN]Cl/(salph)Cr(III)Cl System. J. Am. Chem. Soc. 2007, 129, 15116–15117. [Google Scholar] [CrossRef]
- Matsuda, T.; Funae, Y.; Yoshida, M.; Yamamoto, T.; Takaya, T. Optical material of high refractive index resin composed of sulfur-containing aliphatic and alicyclic methacrylates. J. Appl. Polym. Sci. 2000, 76, 45–49. [Google Scholar] [CrossRef]
- Rys, A.Z.; Harpp, D.N. Catalytic sulfuration of dienes with metallocene polysulfides. Tetrahedron Lett. 1998, 39, 9139–9142. [Google Scholar] [CrossRef]
- Farrell, W.S.; Zavalij, P.Y.; Sita, L.R. Catalytic Production of Isothiocyanates via a Mo(II)/Mo(IV) Cycle for the “Soft” Sulfur Oxidation of Isonitriles. Organometallics 2016, 35, 2361–2366. [Google Scholar] [CrossRef]
- Adam, W.; Bargon, R.M.; Schenk, W.A. Direct Episulfidation of Alkenes and Allenes with Elemental Sulfur and Thiiranes as Sulfur Sources, Catalyzed by Molybdenum Oxo Complexes. J. Am. Chem. Soc. 2003, 125, 3871–3876. [Google Scholar] [CrossRef] [PubMed]
- Adam, W.; Bargon, R.M.; Bosio, S.G.; Schenk, W.A.; Stalke, D. Direct Synthesis of Isothiocyanates from Isonitriles by Molybdenum-Catalyzed Sulfur Transfer with Elemental Sulfur. J. Org. Chem. 2002, 67, 7037–7041. [Google Scholar] [CrossRef] [PubMed]
- Ooi, M.L.; Lim, C.E.; Wong, R.C.S.; Ng, S.W. Isolation and characterization of thiolato complexes of [CpCr(SBz)]2S, isomeric trans [CpMo(CO)(SBz)]2 and [CpMo(SBz)2]2 (Cp = (η5-C5H5), Bz = PhCH2) from the reactivity of bibenzyl disulfide towards [CpM(CO)3]2 (M = Cr, Mo). Inorg. Chim. Acta 2017, 467, 364–371. [Google Scholar] [CrossRef]
- Pétillon, F.Y.; Schollhammer, P.; Talarmin, J. Recent advances in the chemistry of tris(thiolato) bridged cyclopentadienyl dimolybdenum complexes. Coord. Chem. Rev. 2017, 331, 73–92. [Google Scholar] [CrossRef]
- Ward, J.P.; Lim, P.J.; Evans, D.J.; White, J.M.; Young, C.G. Tungsten Ligand-Based Sulfur-Atom-Transfer Catalysts: Synthesis, Characterization, Sustained Anaerobic Catalysis, and Mode of Aerial Deactivation. Inorg. Chem. 2020, 59, 16824–16828. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Tajima, R.; Sakurai, T.; Ohi, H.; Miyake, H.; Itoh, S.; Tsukube, H. Reversible Sulfurization–Desulfurization of Tungsten Bis(dithiolene) Complexes. Angew. Chem. Int. Ed. 2006, 45, 3520–3522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eagle, A.A.; Gable, R.W.; Thomas, S.; Sproules, S.A.; Young, C.G. Sulfur atom transfer reactions of tungsten(VI) and tungsten(IV) chalcogenide complexes. Polyhedron 2004, 23, 385–394. [Google Scholar] [CrossRef]
- Ibdah, A.; Jenks, W.S.; Espenson, J.H. Kinetics, Mechanism, and Computational Studies of Rhenium-Catalyzed Desulfurization Reactions of Thiiranes (Thioepoxides). Inorg. Chem. 2006, 45, 5351–5357. [Google Scholar] [CrossRef] [Green Version]
- Arisawa, M.; Ichikawa, T.; Yamaguchi, M. Synthesis of thiiranes by rhodium-catalyzed sulfur addition reaction to reactive alkenes. Chem. Commun. 2015, 51, 8821–8824. [Google Scholar] [CrossRef] [PubMed]
- Arisawa, M.; Tanaka, K.; Yamaguchi, M. Rhodium-catalyzed sulfur atom exchange reaction between organic polysulfides and sulfur. Tetrahedron Lett. 2005, 46, 4797–4800. [Google Scholar] [CrossRef]
- Moon, S.; Kato, M.; Nishii, Y.; Miura, M. Synthesis of Benzo[b]thiophenes through Rhodium-Catalyzed Three-Component Reaction using Elemental Sulfur. Adv. Synth. Catal. 2020, 362, 1669–1673. [Google Scholar] [CrossRef]
- Huang, C.Y.; Doyle, A.G. The chemistry of transition metals with three-membered ring heterocycles. Chem. Rev. 2014, 114, 8153–8198. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ellern, A.; Espenson, J.H. Rhenium(V) Complexes with Thiolato and Dithiolato Ligands: Synthesis, Structures, and Monomerization Reactions. Inorg. Chem. 2005, 44, 3690–3699. [Google Scholar] [CrossRef]
- Koshino, N.; Espenson, J.H. Kinetics and Mechanism of Oxygen Atom Transfer from Methyl Phenyl Sulfoxide to Triarylphosphines Catalyzed by an Oxorhenium(V) Dimer. Inorg. Chem. 2003, 42, 5735–5742. [Google Scholar] [CrossRef]
- Birgisson, B.O.; Monger, L.J.; Damodaran, K.K.; Suman, S.G. Synthesis and characterization of asymmetric [Mo2O2(μ-S)2(S2)(L)] complexes (L = bipy, en, dien) and their heterogeneous reaction with propylene sulfide. Inorg. Chim. Acta 2020, 119272. [Google Scholar] [CrossRef]
- Dave, M.; Rajagopal, A.; Damm-Ruttensperger, M.; Schwarz, B.; Nägele, F.; Daccache, L.; Fantauzzi, D.; Jacob, T.; Streb, C. Understanding homogeneous hydrogen evolution reactivity and deactivation pathways of molecular molybdenum sulfide catalysts. Sustain. Energy Fuels 2018, 2, 1020–1026. [Google Scholar] [CrossRef]
- Coucouvanis, D. Syntheses, Structures, and Reactions of Binary and Tertiary Thiomolybdate Complexes Containing the (O)Mo(Sx) AND (S)Mo(Sx) Functional Groups (x = 1, 2, 4). In Advances in Inorganic Chemistry; Sykes, A.G., Ed.; Academic Press: Cambridge, MA, USA, 1998; Volume 45, pp. 1–73. [Google Scholar]
- McKee, M.L. Fluctional Molecules. Wires Comput. Mol. Sci. 2011, 1, 943–951. [Google Scholar] [CrossRef]
- Faller, J.W. Fluxional and Nonrigid Behavior of Transition Metal Organometallic π-Complexes. In Advances in Organometallic Chemistry; Stone, F.G.A., West, R., Eds.; Academic Press: Cambridge, MA, USA, 1977; Volume 16, pp. 211–239. [Google Scholar]
- Scorzelli, A.G.; Macalush, B.E.; Naik, D.V.; Moehring, G.A. Comparative study of fluxional processes at two different classes of eight-coordinate rhenium(V) polyhydride complexes. Inorg. Chim. Acta 2021, 516, 120120. [Google Scholar] [CrossRef]
- Ólafsson, S.N.; Kvaran, Á.; Jonsdottir, S.; Suman, S.G. Non-rigid coordination behavior of the ambidentate phosphinoyldithioformate ligands, [S2CP(O)R2]-, (R = Ph, CH2Ph) in organometallic Lead(IV) and Mercury(II) compounds. J. Organomet. Chem. 2018, 854, 38–48. [Google Scholar] [CrossRef]
- Ólafsson, S.N.; Bjornsson, R.; Helgason, Ö.; Jonsdottir, S.; Suman, S.G. Coordination geometry determination of stannane compounds with phosphinoyldithioformate ligands using multinuclear NMR, Sn Mössbauer and DFT methods. J. Organomet. Chem. 2016, 825–826, 125–138. [Google Scholar] [CrossRef]
- Suman, S.G.; Gretarsdottir, J.M.; Penwell, P.E.; Gunnarsson, J.P.; Frostason, S.; Jonsdottir, S.; Damodaran, K.K.; Hirschon, A. Reaction Chemistry of the syn-Mo2O2(μ-S)2(S2)(DMF)3 Complex with Cyanide and Catalytic Thiocyanate Formation. Inorg. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Coucouvanis, D.; Toupadakis, A.; Lane, J.D.; Koo, S.M.; Kim, C.G.; Hadjikyriacou, A. Reactivity of the Mo(O)(S) functional group in the [(L)Mo(O)(μ-S)2Mo(O)(S)]n- dimeric thiomolybdate complexes, (L = C5H5—, n = 1; S42-, n = 2) and implications regarding the function of xanthine oxidase. Synthesis and structural characterization of [(DMF)3Mo(O)(μ-S)2Mo(O)(S2)], [Ph4P][(C5H5)Mo(O)(μ-S)2Mo(O)(S2)], [Ph4P]2[(S4)Mo(O)(μ-S)2Mo(O)(S)] and (Et4N)4{[(S4)Mo(O)(μ-S)2Mo(O)(S)]2}. J. Am. Chem. Soc. 1991, 113, 5271–5282. [Google Scholar] [CrossRef]
- Ólafsson, S.N.; Flensburg, C.; Andersen, P. Lead(iv) complexes with phosphinoyldithioformates, R2P(O)CS2~ (R = Ph or PhCH2). Syntheses and characterization. Crystal structures of PPhJ[S2CP(O)Ph2]-0.5H2O, [PbPh2{S2CP(O)Ph2}2] and [PbPh2Cl{S2CP(O)Ph2}]. J. Chem. Soc. Dalton Trans. 2000, 4360–4368. [Google Scholar] [CrossRef]
- Djordjevic, C.; Vuletic, N.; Jacobs, B.A.; Lee-Renslo, M.; Sinn, E. Molybdenum(VI) Peroxo α-Amino Acid Complexes: Synthesis, Spectra, and Properties of MoO(O2)2(α-aa)(H2O) for α-aa = Glycine, Alanine, Proline, Valine, Leucine, Serine, Asparagine, Glutamine, and Glutamic Acid. X-ray Crystal Structures of the Glycine, Alanine, and Proline Compounds. Inorg. Chem. 1997, 36, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Clegg, W.; Mohan, N.; Mueller, A.; Neumann, A.; Rittner, W.; Sheldrick, G.M. Crystal and molecular structure of [N(CH3)4]2[Mo2O2S2(S2)2]: A compound with two S22- ligands. Inorg. Chem. 1980, 19, 2066–2069. [Google Scholar] [CrossRef]
- Singh, V.; Kumar, A.; Prasad, R.; Rajput, G.; Drew, M.G.B.; Singh, N. The interplay of secondary Hg⋯S, Hg⋯N and Hg⋯π bonding interactions in supramolecular structures of phenylmercury(ii) dithiocarbamates. CrystEngComm 2011, 13, 6817–6826. [Google Scholar] [CrossRef]
- Ólafsson, S.N.; Petersen, T.N.; Andersen, P. Crystal structure of diphenylbis(diphenyldithio-phosphinato)lead(IV), PbPh2(S2PPh2)2. Acta Chem. Scand. 1996, 50, 745–748. [Google Scholar] [CrossRef]
- Ogienko, D.S.; Smolentsev, A.I.; Bashirov, D.A.; Afonin, M.Y.; Petrov, P.A.; Konchenko, S.N. Syntheses and structures of complexes {Mo2S2O2}2+ with labile Cl− and DMF ligands. Russ. J. Coord. Chem. 2015, 41, 759–764. [Google Scholar] [CrossRef]
- Denney, D.B.; Boskin, M.J. Concerning the Mechanism of the Reaction of Trisubstituted Phosphines with Episulfides1. J. Am. Chem. Soc. 1960, 82, 4736–4738. [Google Scholar] [CrossRef]
- Yoo, B.W.; Kim, J.Y. Selective and efficient desulfurization of thiiranes with Mo(CO)6. J. Sulfur Chem. 2020, 41, 13–18. [Google Scholar] [CrossRef]
- Caló, V.; Lopez, L.; Nacci, A.; Mele, G. Aminium Salts Induced Desulphurization of Allyl and Diallyl Thiiranes. Synthesis of Dienes and Trienes. Tetrahedron 1995, 51, 8935–8940. [Google Scholar] [CrossRef]
- Arterburn, J.B.; Perry, M.C. Rhenium catalyzed sulfurization of phosphorus(III) compounds with thiiranes: New reagents for phosphorothioate ester synthesis. Tetrahedron Lett. 1997, 38, 7701–7704. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Nakagawa, Y.; Mizuno, N. High Turnover Numbers for the Catalytic Selective Epoxidation of Alkenes with 1 atm of Molecular Oxygen. Angew. Chem. Int. Ed. 2001, 40, 3639–3641. [Google Scholar] [CrossRef]
- Jacob, J.; Espenson, J.H. Stereospecific rhenium catalyzed desulfurization of thiiranes. Chem. Commun. 1999, 1003–1004. [Google Scholar] [CrossRef]
- Nguyen, T.B. Recent Advances in Organic Reactions Involving Elemental Sulfur. Adv. Synth. Catal. 2017, 359, 1066–1130. [Google Scholar] [CrossRef]
- Wang, J.-J.; Kryatova, O.P.; Rybak-Akimova, E.V.; Holm, R.H. Comparative Kinetics and Mechanism of Oxygen and Sulfur Atom Transfer Reactions Mediated by Bis(dithiolene) Complexes of Molybdenum and Tungsten. Inorg. Chem. 2004, 43, 8092–8101. [Google Scholar] [CrossRef]
- Dubois, M.R. Carbon-chalcogen bond cleavage reactions characterized for dinuclear sulfur-bridged cyclopentadienyl molybdenum complexes. Polyhedron 1997, 16, 3089–3098. [Google Scholar] [CrossRef]
- Romano, F.; Linden, A.; Mba, M.; Zonta, C.; Licini, G. Molybdenum(VI) Amino Triphenolate Complexes as Catalysts for Sulfoxidation, Epoxidation and Haloperoxidation. Adv. Synth. Catal. 2010, 352, 2937–2942. [Google Scholar] [CrossRef]
- Gabay, J.; Dietz, S.; Bernatis, P.; DuBois, M.R. Dinuclear cyclopentadienylmolybdenum complexes containing thioether ligands. Ligand substitution and desulfurization reactions. Organometallics 1993, 12, 3630–3635. [Google Scholar] [CrossRef]
- Cabon, N.; Le Goff, A.; Le Roy, C.; Pétillon, F.Y.; Schollhammer, P.; Talarmin, J.; McGrady, J.E.; Muir, K.W. Transformations and Agostic Interactions of Hydrocarbyl Ligands Bonded to the Sulfur-Rich Dimolybdenum Site {Mo2Cp2(μ-SMe)3}: Chemical and Electrochemical Formation of μ-Alkyl and μ-Vinyl Compounds from a μ-Alkylidene Derivative. Organometallics 2005, 24, 6268–6278. [Google Scholar] [CrossRef]
- Cabon, N.; Pétillon, F.Y.; Orain, P.-Y.; Schollhammer, P.; Talarmin, J.; Muir, K.W. Controlled nucleophilic activation of different sites in [Mo2Cp2L2(μ-SMe)2(μ-L′)]+ cations (L = ButNC, xylNC, CO; L′ = SMe or PPh2). J. Organomet. Chem. 2005, 690, 4583–4601. [Google Scholar] [CrossRef]
- Bruker. Apex III; Bruker AXS Inc.: Madison, WI, USA, 2015. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]




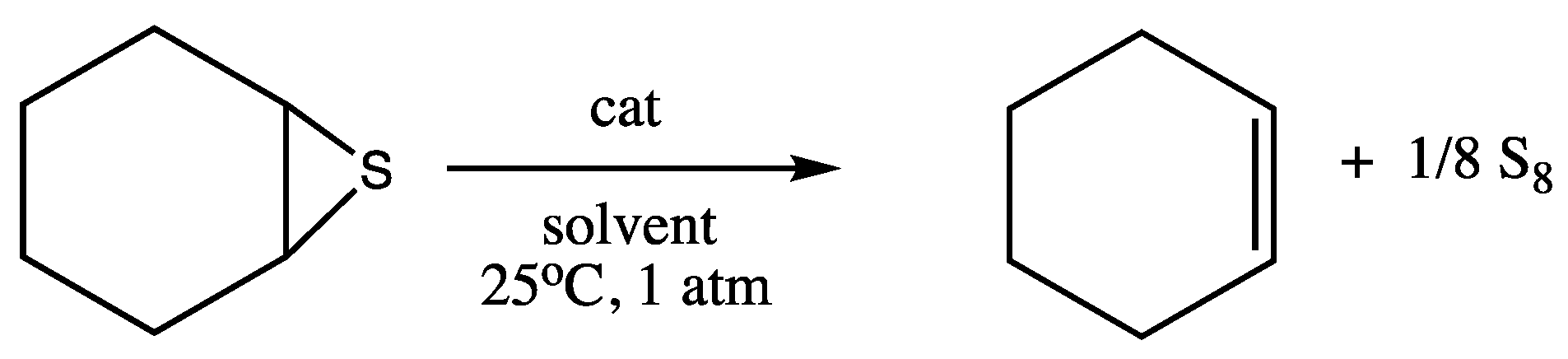




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd/Method | IR, KBr, cm−1 | IR, CH2Cl2 cm−1 | Raman, neat, cm−1 | Scheme 2 |
|---|---|---|---|---|
| 1 | (P=O), (CS2), (Mo=O) | (P=O), (CS2), (Mo=O) | (P=O), (CS2), (Mo=O) | |
| 1077 1071 945 | 1079 1050 947 | 1052 918 | ||
| 1121 915 933 | 1118 937 | II III IV | ||
| 1251 1054 | ||||
| 2 | 1080 1071 932(br.) | 1077 1057 946 | 1073 930 | II, III, IV |
| 1251 1054 | 932 | |||
| [Ph2P(O)CS2]— | 1176 a 1032 a 915 a | 1168 b 1034 b | ||
| [Bn2P(O)CS2]— | 1200 a 1030 a | |||
| 910 a |
| Complex | Mol%, Catalyst | TON | TOF, h−1 | t50%, hr. (a) |
|---|---|---|---|---|
| 1 | 0.1 | 500 | 140 | 3.58 |
| 0.3 | 167 | 102 | 1.63 | |
| 0.5 | 100 | 93 | 1.07 | |
| 1.0 | 50 | 58 | 0.87 | |
| 2 | 0.1 | 500 | 698 | 0.72 |
| 0.3 | 167 | 435 | 0.38 | |
| 0.5 | 100 | 286 | 0.35 | |
| 1.0 | 50 | 194 | 0.26 | |
| (Me4N)2 [Mo2O2(µ-S)2(Cl)4] | 0.1 | 500 | 48 | 10.5 |
| 0.3 | 167 | 25 | 6.6 | |
| 0.5 | 100 | 24 | 4.2 | |
| 1.0 | 50 | 24 | 2.1 |
| Complex | Mol%, Catalyst | TON | TOF, h−1 | t50%, hr. (a) |
|---|---|---|---|---|
| 1 | 1.0 | 50 | 0.50 | 100 |
| 3.0 | 16.7 | 0.36 | 47 | |
| 2 | 0.1 | 500 | 12.91 | 39 |
| 0.5 | 100 | 4.56 | 22 | |
| 1.0 | 50 | 2.46 | 20 | |
| 2.0 | 25 | 5.85 | 4.3 | |
| 3.0 | 16.7 | 4.56 | 3.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Razinkov, D.; Ingvarsdottir, H.I.; Kvaran, Á.; Jonsdottir, S.; Suman, S.G. Coordination Properties of Non-Rigid Phosphinoyldithioformate Complexes of the [Mo2O2(µ-S)2]2+ Cation in Catalytic Sulfur Transfer Reactions with Thiiranes. Catalysts 2021, 11, 593. https://doi.org/10.3390/catal11050593
Razinkov D, Ingvarsdottir HI, Kvaran Á, Jonsdottir S, Suman SG. Coordination Properties of Non-Rigid Phosphinoyldithioformate Complexes of the [Mo2O2(µ-S)2]2+ Cation in Catalytic Sulfur Transfer Reactions with Thiiranes. Catalysts. 2021; 11(5):593. https://doi.org/10.3390/catal11050593
Chicago/Turabian StyleRazinkov, Dmitrii, Hafdís I. Ingvarsdottir, Ágúst Kvaran, Sigridur Jonsdottir, and Sigridur G. Suman. 2021. "Coordination Properties of Non-Rigid Phosphinoyldithioformate Complexes of the [Mo2O2(µ-S)2]2+ Cation in Catalytic Sulfur Transfer Reactions with Thiiranes" Catalysts 11, no. 5: 593. https://doi.org/10.3390/catal11050593