
Multienzymatic Processes Involving Baeyer–Villiger Monooxygenases



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Multi-Step Reactions Including BVMO Activity Catalyzed by Whole Cells
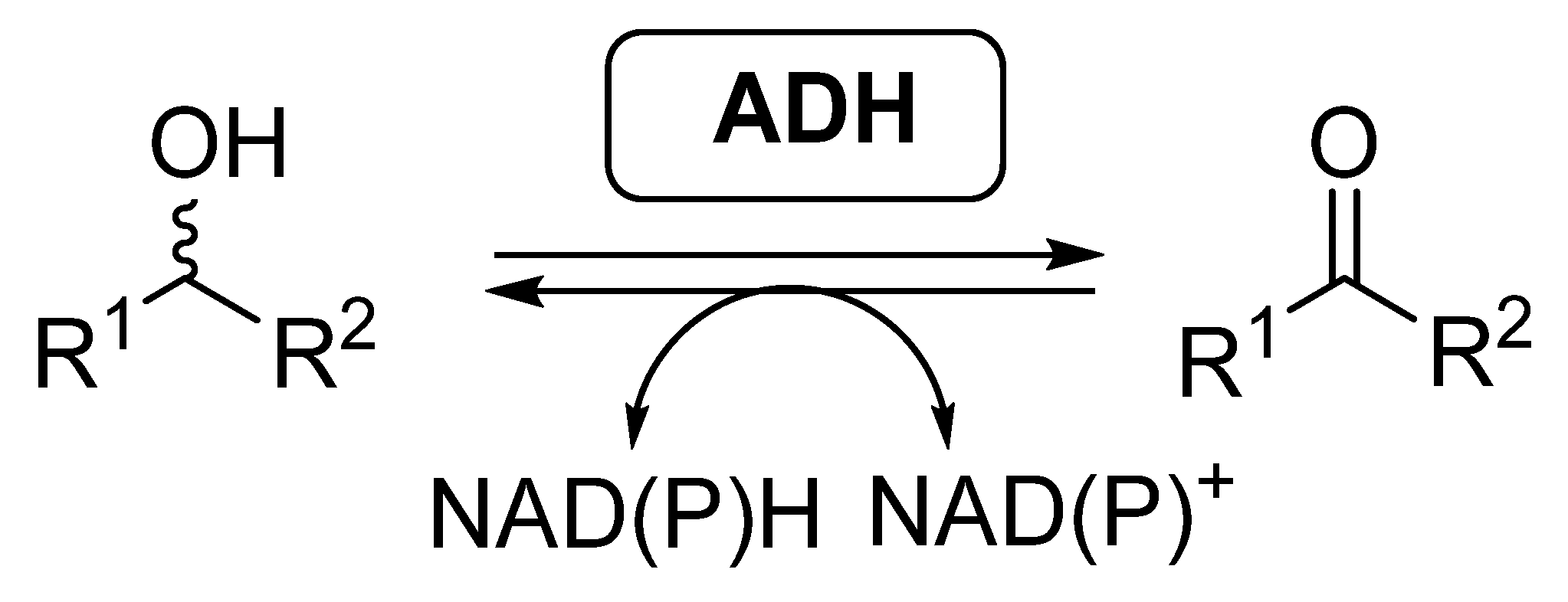
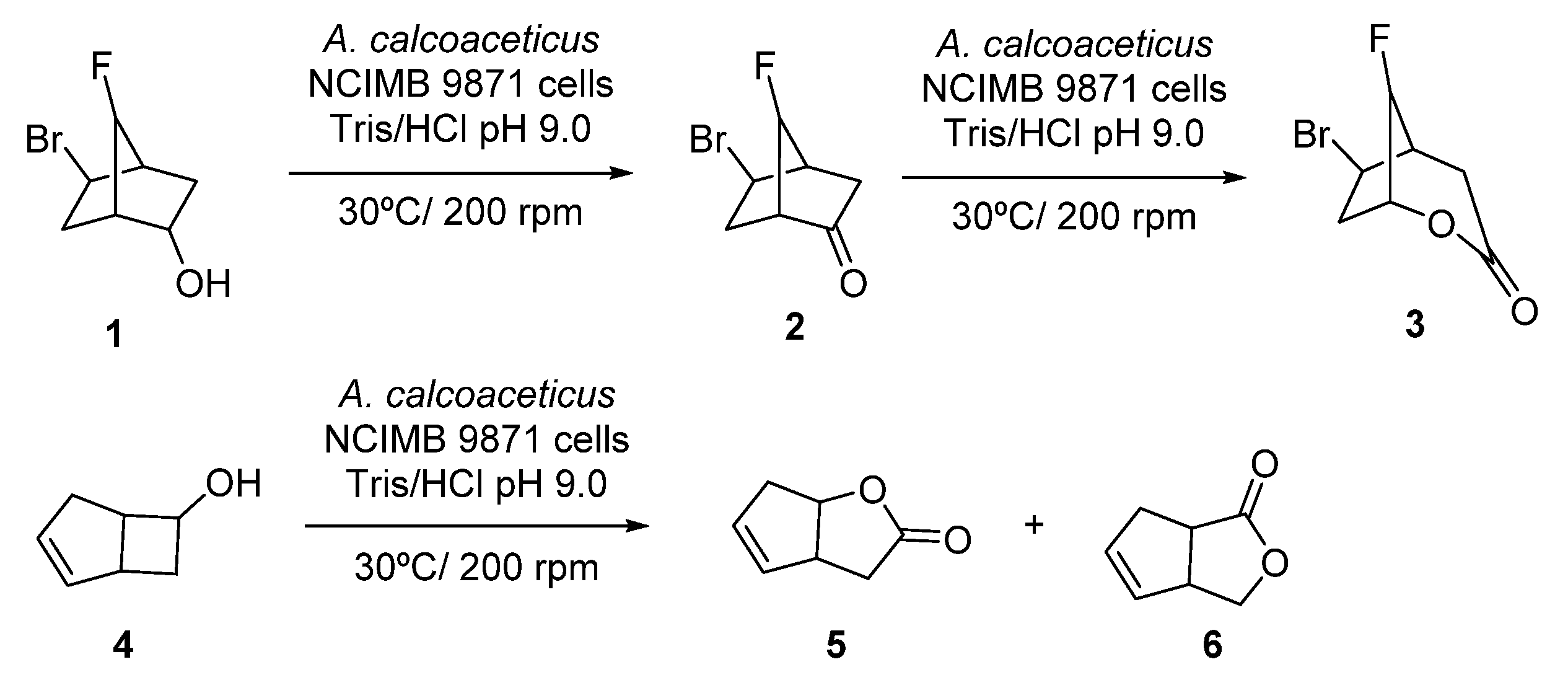
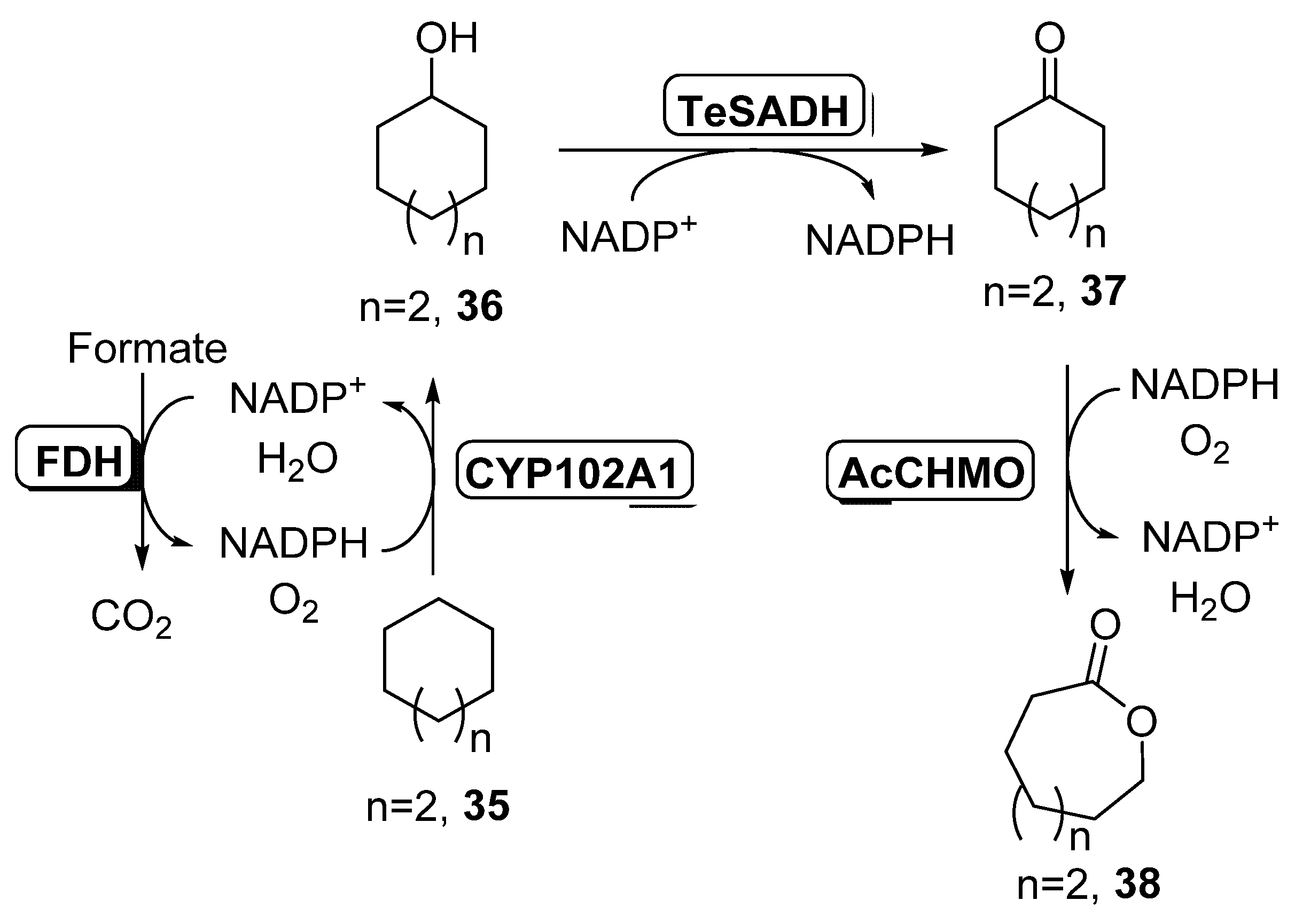
2.1. Multi-Step Reactions Including Alcohol Dehydrogenases/BVMO Activity Catalyzed by Whole Cells
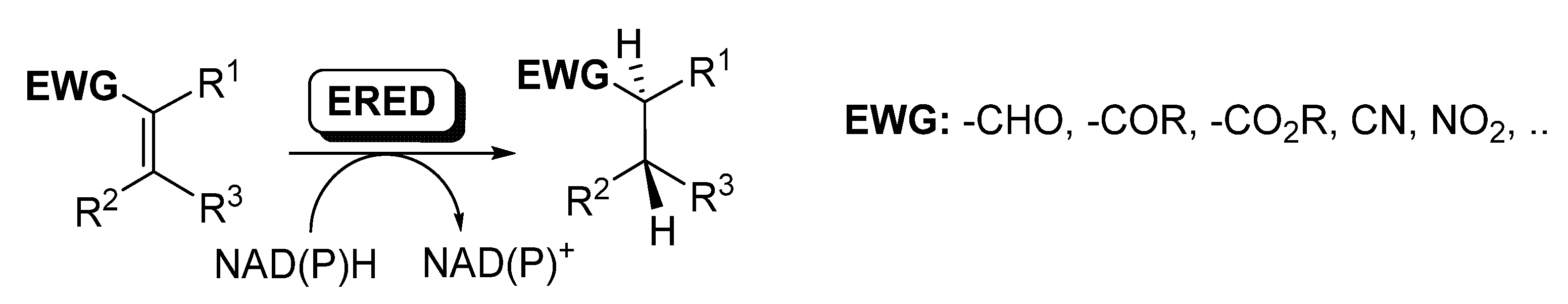
2.2. Multi-Step Reactions Including ERED/BVMO Activity Catalyzed by Whole Cells
3. Multi-Step Reactions Including BVMO Activity Catalyzed by Isolated Enzymes
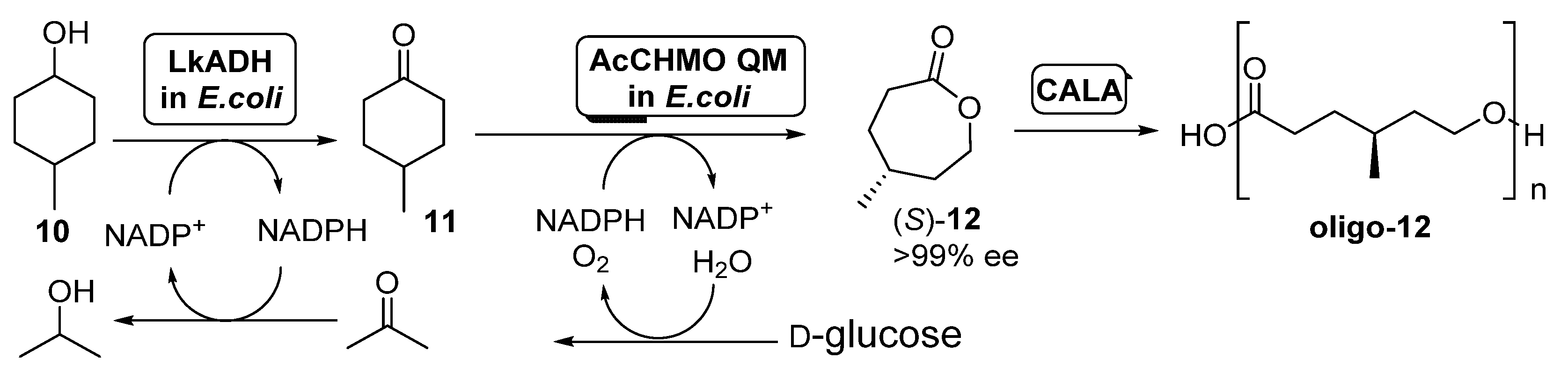
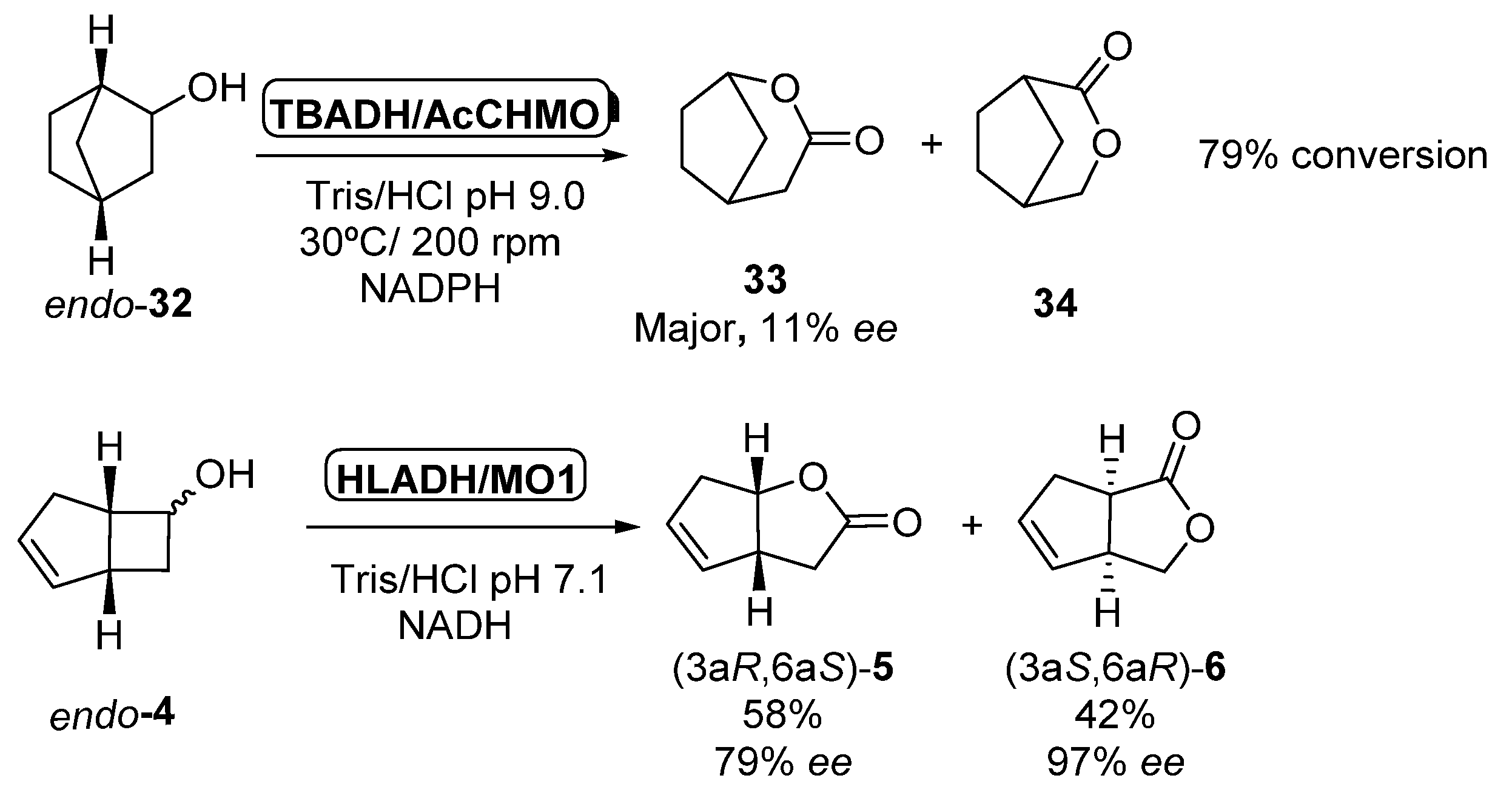
3.1. Linear ADHs/BVMOs Cascades
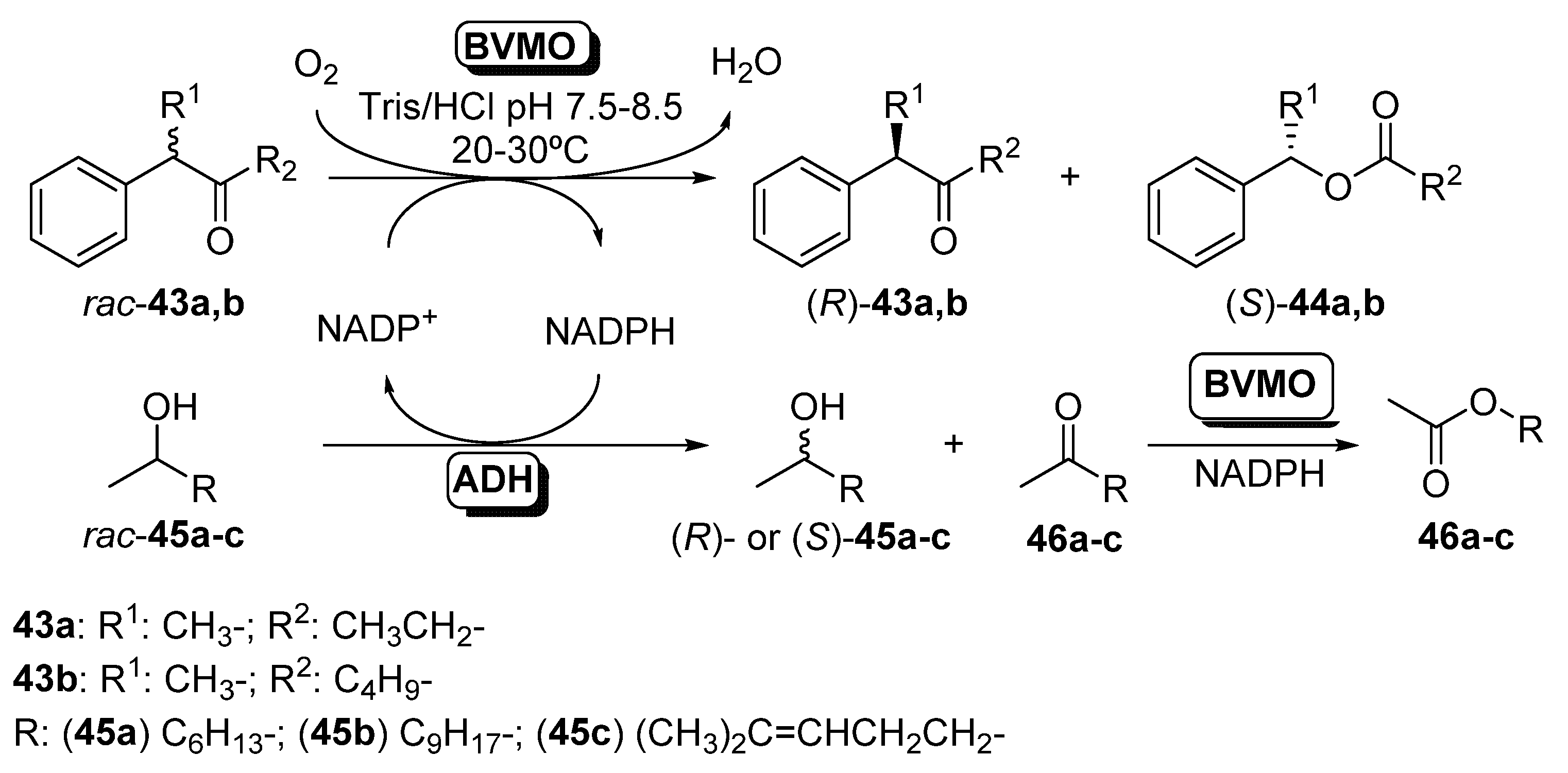
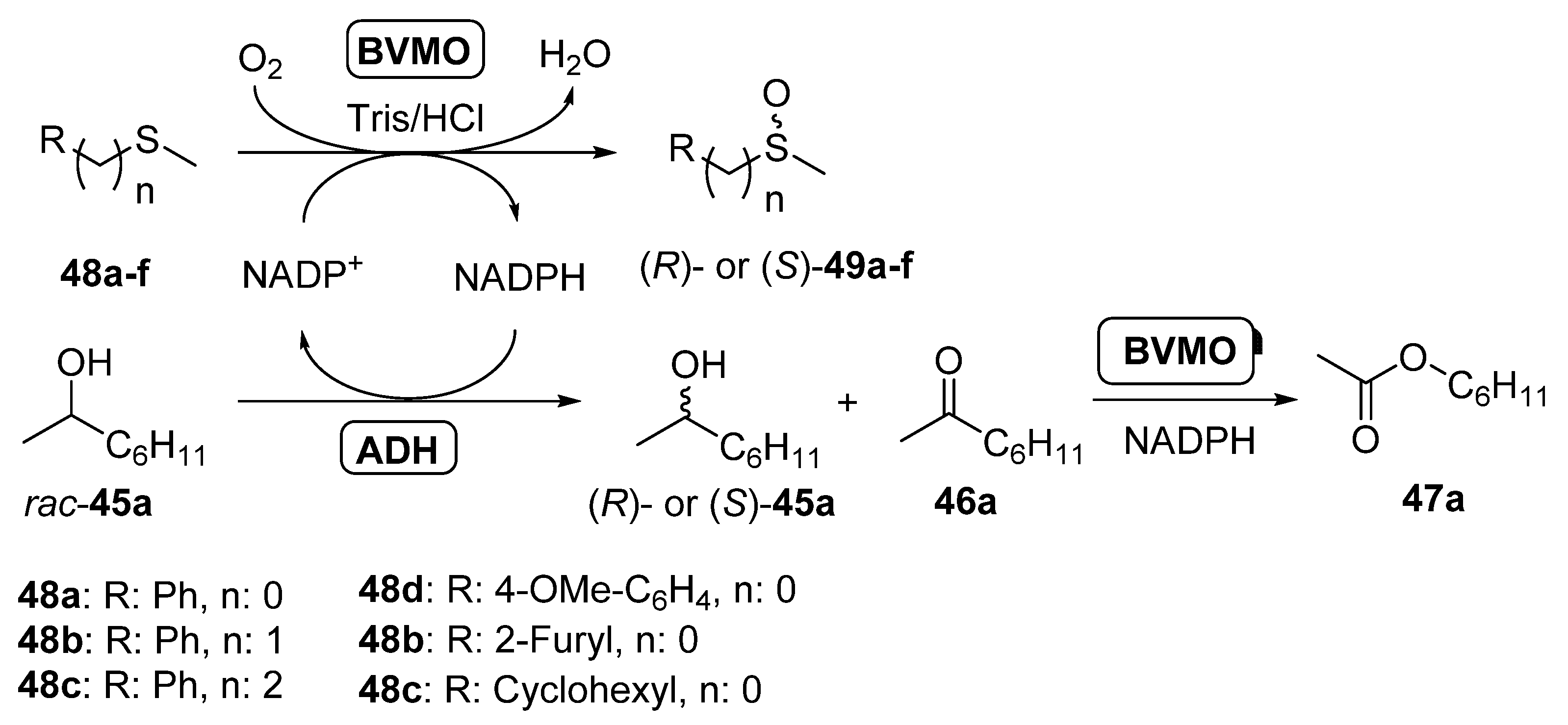
3.2. Parallel ADHs/BVMOs Cascades
3.3. Sequential EREDs/BVMOs Cascades
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kitano, H. Systems biology: A brief overview. Science 2002, 295, 1662–1664. [Google Scholar] [CrossRef] [Green Version]
- García-Junceda, E. Multi-Step Enzyme Catalysis: Biotransformations and Chemoenzymatic Synthesis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar] [CrossRef] [Green Version]
- García-Junceda, E.; Lavandera, I.; Rother, D.; Schrittwieser, J.H. (Chemo)enzymatic cascades-Nature’s synthetic strategy transferred to the laboratory. J. Mol. Catal. B Enzym. 2015, 114, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Oroz-Guinea, I.; Fernández-Lucas, J.; Hormigo, D.; García-Junceda, E. Designed Enzymatic Cascades. In Science of Synthesis: Biocatalysis in Organic Synthesis 3; Faber, K., Fessner, W.D., Turner, N.J., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 2015; Volume 3. [Google Scholar]
- Schrittwieser, J.H.; Velikogne, S.; Hall, M.; Kroutil, W. Artificial Biocatalytic Linear Cascades for Preparation of Organic Molecules. Chem. Rev. 2018, 118, 270–348. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, S.; Castiglione, K.; Kourist, R. Overcoming the Incompatibility Challenge in Chemoenzymatic and Multi-Catalytic Cascade Reactions. Chem. Eur. J. 2018, 24, 1755–1768. [Google Scholar] [CrossRef]
- Schrittwieser, J.H.; Velikogne, S.; Kroutil, W. Artificial Biocatalytic Cascades to Alcohols and Amines. In Modern Biocatalysis: Advances Towards Synthetic Biological Systems; Williams, G., Hall, M., Eds.; Royal Soc Chemistry: Cambridge, UK, 2018; Volume 32, pp. 387–438. [Google Scholar]
- Walsh, C.T.; Moore, B.S. Enzymatic Cascade Reactions in Biosynthesis. Angew. Chem. Int. Ed. 2019, 58, 6846–6879. [Google Scholar] [CrossRef]
- Kara, S.; Rudroff, F. (Eds.) Enzyme Cascade Design and Modelling; Springer International Publishing: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- McIntosh, J.A.; Owens, A.E. Enzyme engineering for biosynthetic cascades. Curr. Opin. Green Sustain. Chem. 2021, 29, 100448. [Google Scholar] [CrossRef]
- Nazor, J.; Liu, J.; Huisman, G. Enzyme evolution for industrial biocatalytic cascades. Curr. Opin. Biotechnol. 2021, 69, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Uppada, V.; Bhaduri, S.; Noronha, S.B. Cofactor regeneration—An important aspect of biocatalysis. Curr. Sci. 2014, 106, 946–957. [Google Scholar]
- Berenguer-Murcia, A.; Fernandez-Lafuente, R. New Trends in the Recycling of NAD(P)H for the Design of Sustainable Asymmetric Reductions Catalyzed by Dehydrogenases. Curr. Org. Chem. 2010, 14, 1000–1021. [Google Scholar] [CrossRef]
- Truppo, M.D. 7.4 Cofactor Recycling for Enzyme Catalyzed Processes. In Comprehensive Chirality; Elsevier Ltd.: Amsterdam, The Netherlands, 2012; Volume 7, pp. 46–70. [Google Scholar]
- Mordhorst, S.; Andexer, J.N. Round, round we go-strategies for enzymatic cofactor regeneration. Nat. Prod. Rep. 2020, 37, 1316–1333. [Google Scholar] [CrossRef] [PubMed]
- Reis, R.A.G.; Li, H.; Johnson, M.; Sobrado, P. New frontiers in flavin-dependent monooxygenases. Arch. Biochem. Biophys. 2021, 699, 14. [Google Scholar] [CrossRef] [PubMed]
- Toplak, M.; Matthews, A.; Teufel, R. The devil is in the details: The chemical basis and mechanistic versatility of flavoprotein monooxygenases. Arch. Biochem. Biophys. 2021, 698, 14. [Google Scholar] [CrossRef]
- Dockrey, S.A.B.; Narayan, A.R.H. Flavin-dependent biocatalysts in synthesis. Tetrahedron 2019, 75, 1115–1121. [Google Scholar] [CrossRef]
- Hall, M. Flavoenzymes for biocatalysis. In Enzymes; Chaiyen, P., Tamanoi, F., Tamanoi, F., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 47, pp. 37–62. [Google Scholar]
- Schmidt, S.; Bornscheuer, U.T. Baeyer-Villiger monooxygenases: From protein engineering to biocatalytic applications. In Enzymes; Chaiyen, P., Tamanoi, F., Tamanoi, F., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 47, pp. 231–281. [Google Scholar]
- de Gonzalo, G.; Mihovilovic, M.D.; Fraaije, M.W. Recent Developments in the Application of Baeyer-Villiger Monooxygenases as Biocatalysts. ChemBioChem 2010, 11, 2208–2231. [Google Scholar] [CrossRef] [Green Version]
- Leisch, H.; Morley, K.; Lau, P.C.K. Baeyer-Villiger Monooxygenases: More Than Just Green Chemistry. Chem. Rev. 2011, 111, 4165–4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balke, K.; Kadow, M.; Mallin, H.; Sass, S.; Bornscheuer, U.T. Discovery, application and protein engineering of Baeyer-Villiger monooxygenases for organic synthesis. Org. Biomol. Chem. 2012, 10, 6249–6265. [Google Scholar] [CrossRef]
- Bucko, M.; Gemeiner, P.; Schenkmayerova, A.; Krajcovic, T.; Rudroff, F.; Mihovilovic, M.D. Baeyer-Villiger oxidations: Biotechnological approach. Appl. Microbiol. Biotechnol. 2016, 100, 6585–6599. [Google Scholar] [CrossRef] [PubMed]
- Knaus, T.; Toogood, H.S.; Scrutton, N.S. Ene-reductases and their Applications. In Green Biocatalysis; Patel, R.N., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 473–488. [Google Scholar] [CrossRef]
- Furst, M.; Gran-Scheuch, A.; Aalbers, F.S.; Fraaije, M.W. Baeyer-Villiger Monooxygenases: Tunable Oxidative Biocatalysts. ACS Catal. 2019, 9, 11207–11241. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Wang, H.; Liu, J.; Li, S.; Guo, J.; Li, H.; Jia, X.; Huo, H.; Zheng, Z.; You, S.; et al. Old yellow enzymes: Structures and structure-guided engineering for stereocomplementary bioreduction. Appl. Microbiol. Biotechnol. 2020, 104, 8155–8170. [Google Scholar] [CrossRef]
- Huijbers, M.M.E.; Montersino, S.; Westphal, A.H.; Tischler, D.; van Berkel, W.J.H. Flavin dependent monooxygenases. Arch. Biochem. Biophys. 2014, 544, 2–17. [Google Scholar] [CrossRef]
- Cummings Ryerson, C.; Ballou, D.P.; Walsh, C. Mechanistic Studies on Cyclohexanone Oxygenase. Biochemistry 1982, 21, 2644–2655. [Google Scholar] [CrossRef]
- van Berkel, W.J.H.; Kamerbeek, N.M.; Fraaije, M.W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006, 124, 670–689. [Google Scholar] [CrossRef] [Green Version]
- Mthethwa, K.S.; Kassier, K.; Engel, J.; Kara, S.; Smit, M.S.; Opperman, D.J. Fungal BVMOs as alternatives to cyclohexanone monooxygenase. Enzym. Microb. Technol. 2017, 106, 11–17. [Google Scholar] [CrossRef] [PubMed]
- van Beek, H.L.; Beyer, N.; Janssen, D.B.; Fraaije, M.W. Lyophilization conditions for the storage of monooxygenases. J. Biotechnol. 2015, 203, 41–44. [Google Scholar] [CrossRef]
- Balke, K.; Beier, A.; Bornscheuer, U.T. Hot spots for the protein engineering of Baeyer-Villiger monooxygenases. Biotechnol. Adv. 2018, 36, 247–263. [Google Scholar] [CrossRef]
- Fraaije, M.W.; Wu, J.; Heuts, D.P.H.M.; Van Hellemond, E.W.; Spelberg, J.H.L.; Janssen, D.B. Discovery of a thermostable Baeyer-Villiger monooxygenase by genome mining. Appl. Microbiol. Biotechnol. 2005, 66, 393–400. [Google Scholar] [CrossRef]
- de Gonzalo, G.; Ottolina, G.; Zambianchi, F.; Fraaije, M.W.; Carrea, G. Biocatalytic properties of Baeyer-Villiger monooxygenases in aqueous-organic media. J. Mol. Catal. B Enzym. 2006, 39, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, C.; de Gonzalo, G.; Fraaije, M.W.; Gotor, V. Ionic liquids for enhancing the enantioselectivity of isolated BVMO-catalysed oxidations. Green Chem. 2010, 12, 2255–2260. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.D. Cyclohexanone monooxygenase: A useful reagent for asymmetric baeyer-villiger reactions. Curr. Org. Chem. 1998, 2, 195–216. [Google Scholar]
- Secundo, F.; Fialà, S.; Fraaije, M.W.; De Gonzalo, G.; Meli, M.; Zambianchi, F.; Ottolina, G. Effects of water miscible organic solvents on the activity and conformation of the Baeyer-Villiger monooxygenases from Thermobifida fusca and Acinetobacter calcoaceticus: A comparative study. Biotechnol. Bioeng. 2011, 108, 491–499. [Google Scholar] [CrossRef]
- Sartori, S.K.; Diaz, M.A.N.; Diaz-Muñoz, G. Lactones: Classification, synthesis, biological activities, and industrial applications. Tetrahedron 2021, 84, 132001. [Google Scholar] [CrossRef]
- Wu, S.K.; Li, Z. Whole-Cell Cascade Biotransformations for One-Pot Multistep Organic Synthesis. ChemCatChem 2018, 10, 2164–2178. [Google Scholar] [CrossRef]
- Fessner, W.D. Systems Biocatalysis: Development and engineering of cell-free “artificial metabolism” for preparative multi-enzymatic synthesis. New Biotech. 2015, 32, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Carballeira, J.D.; Quezada, M.A.; Hoyos, P.; Simeo, Y.; Hernaiz, M.J.; Alcantara, A.R.; Sinisterra, J.V. Microbial cells as catalysts for stereoselective red-ox reactions. Biotechnol. Adv. 2009, 27, 686–714. [Google Scholar] [CrossRef]
- Kadisch, M.; Willrodt, C.; Hillen, M.; Buhler, B.; Schmid, A. Maximizing the stability of metabolic engineering-derived whole-cell biocatalysts. Biotechnol. J. 2017, 12, 29. [Google Scholar] [CrossRef]
- Polakovic, M.; Svitel, J.; Bucko, M.; Filip, J.; Nedela, V.; Ansorge-Schumacher, M.B.; Gemeiner, P. Progress in biocatalysis with immobilized viable whole cells: Systems development, reaction engineering and applications. Biotechnol. Lett. 2017, 39, 667–683. [Google Scholar] [CrossRef]
- Garzon-Posse, F.; Becerra-Figueroa, L.; Hernandez-Arias, J.; Gamba-Sanchez, D. Whole Cells as Biocatalysts in Organic Transformations. Molecules 2018, 23, 37. [Google Scholar] [CrossRef] [Green Version]
- Musa, M.M.; Phillips, R.S. Recent advances in alcohol dehydrogenase-catalyzed asymmetric production of hydrophobic alcohols. Catal. Sci. Technolog. 2011, 1, 1311–1323. [Google Scholar] [CrossRef]
- Kratzer, R.; Woodley, J.M.; Nidetzky, B. Rules for biocatalyst and reaction engineering to implement effective, NAD(P)H-dependent, whole cell bioreductions. Biotechnol. Adv. 2015, 33, 1641–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.G.; Yin, H.H.; Yu, D.F.; Chen, X.; Tang, X.L.; Zhang, X.J.; Xue, Y.P.; Wang, Y.J.; Liu, Z.Q. Recent advances in biotechnological applications of alcohol dehydrogenases. Appl. Microbiol. Biotechnol. 2017, 101, 987–1001. [Google Scholar] [CrossRef]
- Gonzalo, G.; Lavandera, I. Recent advances in selective biocatalytic (Hydrogen Transfer) reductions. In Homogeneous Hydrogenation with Non-Precious Catalysts; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2019; pp. 227–259. [Google Scholar] [CrossRef]
- Willetts, A.J.; Knowles, C.J.; Levitt, M.S.; Roberts, S.M.; Sandey, H.; Shipston, N.F. Biotransformation of endo-bicyclo[2.2.1]heptan-2-ols and endo-bicyclo[3.2.0]-hept-2-en-6-ol into the corresponding lactones. J. Chem. Soc. Perkin Trans. 1 1991, 1608–1610. [Google Scholar] [CrossRef]
- Lipik, V.T.; Kong, J.F.; Chattopadhyay, S.; Widjaja, L.K.; Liow, S.S.; Venkatraman, S.S.; Abadie, M.J.M. Thermoplastic biodegradable elastomers based on ε-caprolactone and L-lactide block co-polymers: A new synthetic approach. Acta Biomater. 2010, 6, 4261–4270. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Aihara, K.; Yamanishi, H.; Fukuoka, H.; Tanaka, R.; Cai, Z.; Shiono, T. Synthesis of biodegradable thermoplastic elastomers from ε-caprolactone and lactide. J. Polym. Sci. Part A 2015, 53, 489–495. [Google Scholar] [CrossRef]
- Schmidt, S.; Scherkus, C.; Muschiol, J.; Menyes, U.; Winkler, T.; Hummel, W.; Groger, H.; Liese, A.; Herz, H.G.; Bornscheuer, U.T. An Enzyme Cascade Synthesis of epsilon-Caprolactone and its Oligomers. Angew. Chem. Int. Ed. 2015, 54, 2784–2787. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Buchsenschutz, H.C.; Scherkus, C.; Liese, A.; Groger, H.; Bornscheuer, U.T. Biocatalytic Access to Chiral Polyesters by an Artificial Enzyme Cascade Synthesis. ChemCatChem 2015, 7, 3951–3955. [Google Scholar] [CrossRef]
- Scherkus, C.; Schmidt, S.; Bornscheuer, U.T.; Groeger, H.; Kara, S.; Liese, A. Kinetic insights into e-caprolactone synthesis: Improvement of an enzymatic cascade reaction. Biotechnol. Bioeng. 2017, 114, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Kohl, A.; Srinivasamurthy, V.; Boettcher, D.; Kabisch, J.; Bornscheuer, U.T. Co-expression of an alcohol dehydrogenase and a cyclohexanone monooxygenase for cascade reactions facilitates the regeneration of the NADPH cofactor. Enz. Microb. Technol. 2018, 108, 53–58. [Google Scholar] [CrossRef]
- Srinivasamurthy, V.S.T.; Bottcher, D.; Engel, J.; Kara, S.; Bornscheuer, U.T. A whole-cell process for the production of epsilon-caprolactone in aqueous media. Process Biochem. 2020, 88, 22–30. [Google Scholar] [CrossRef]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, E.Y.; Baek, A.H.; Bornscheuer, U.T.; Park, J.B. Enzyme fusion for whole-cell biotransformation of long-chain sec-alcohols into esters. Appl. Microbiol. Biotechnol. 2015, 99, 6267–6275. [Google Scholar] [CrossRef]
- Toogood, H.S.; Scrutton, N.S. New developments in ‘ene’-reductase catalysed biological hydrogenations. Curr. Opin. Chem. Biol. 2014, 19, 107–115. [Google Scholar] [CrossRef]
- Toogood, H.S.; Scrutton, N.S. Discovery, Characterization, Engineering, and Applications of Ene-Reductases for Industrial Biocatalysis. ACS Catal. 2018, 8, 3532–3549. [Google Scholar] [CrossRef]
- Nett, N.; Duewel, S.; Schmermund, L.; Benary, G.E.; Ranaghan, K.; Mulholland, A.; Opperman, D.J.; Hoebenreich, S. A robust and stereocomplementary panel of ene-reductase variants for gram-scale asymmetric hydrogenation. Mol. Cat. 2021, 502, 111404. [Google Scholar] [CrossRef]
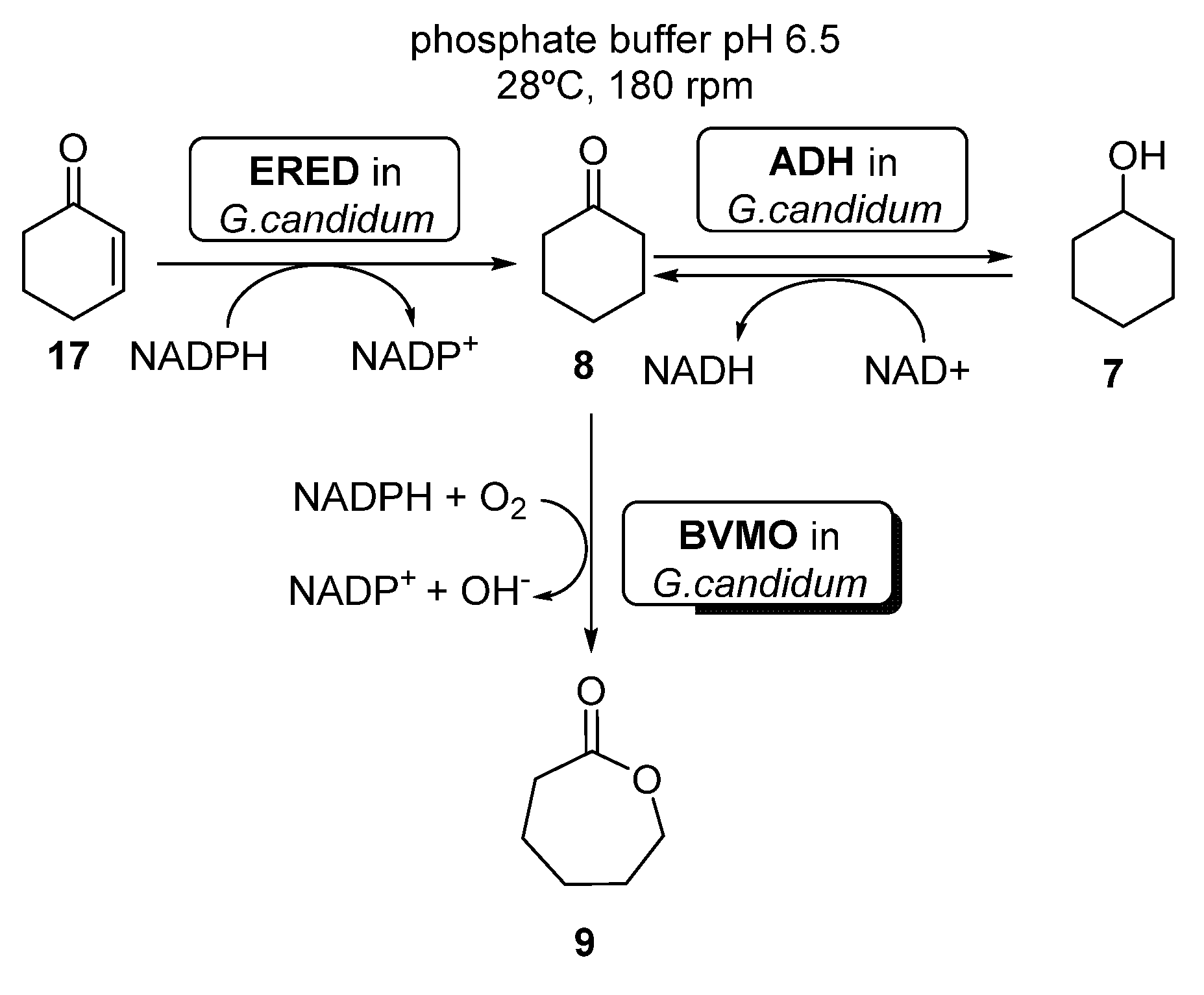
- Silva, A.L.P.; Batista, P.K.; Filho, A.D.; do Nascimento Junior, C.S.; Reboucas, J.S.; Vale, J.A. Rapid conversion of cyclohexenone, cyclohexanone and cyclohexanol to epsilon-caprolactone by whole cells of Geotrichum candidum CCT 1205. Biocatal. Biotransfor. 2017, 35, 185–190. [Google Scholar] [CrossRef]
- Mihovilovic, M.D.; Snajdrova, R.; Grotzl, B. Microbial Baeyer-Villiger oxidation of 4,4-disubstituted cyclohexan—And cyclohexenones by recombinant whole-cells expressing monooxygenases of bacterial origin. J. Mol. Catal. B Enzym. 2006, 39, 135–140. [Google Scholar] [CrossRef]
- Silva, A.L.P.; Caridade, T.N.D.; Magalhaes, R.R.; de Sousa, K.T.; de Sousa, C.C.; Vale, J.A. Biocatalytic production of e-caprolactone using Geotrichum candidum cells immobilized on functionalized silica. Appl. Microbiol. Biotechnol. 2020, 104, 8887–8895. [Google Scholar] [CrossRef]
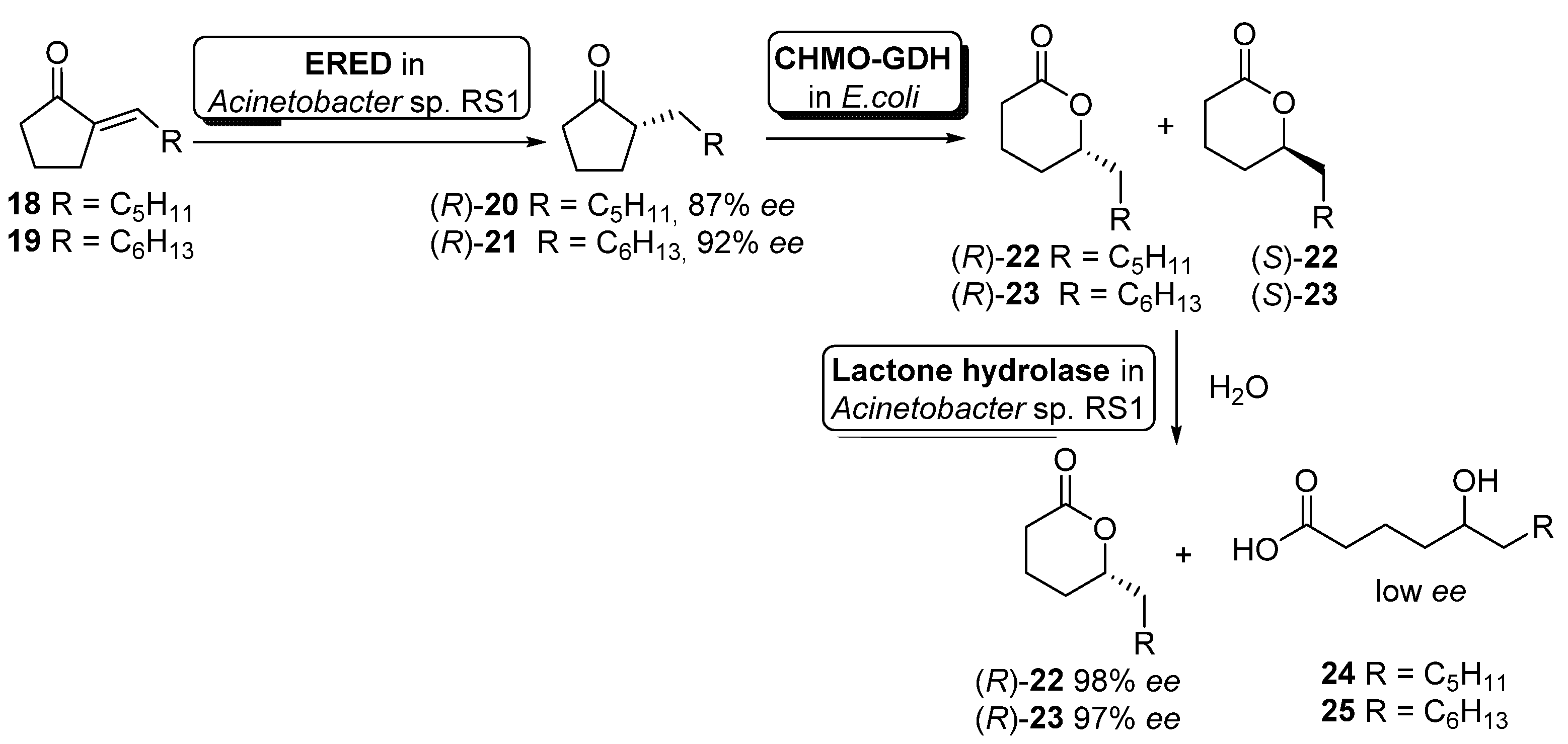
- Liu, J.; Li, Z. Cascade Biotransformations via Enantioselective Reduction, Oxidation, and Hydrolysis: Preparation of (R)-delta-Lactones from 2-Alkylidenecyclopentanones. ACS Catal. 2013, 3, 908–911. [Google Scholar] [CrossRef]
- Shin, J.; Martello, M.T.; Shrestha, M.; Wissinger, J.E.; Tolman, W.B.; Hillmyer, M.A. Pressure-sensitive adhesives from renewable triblock copolymers. Macromolecules 2011, 44, 87–94. [Google Scholar] [CrossRef]
- Wanamaker, C.L.; O’Leary, L.E.; Lynd, N.A.; Hillmeyer, M.A.; Tolman, W.B. Renewable-resource thermoplastic elastomers based on polylactide and polymenthide. Biomacromolecules 2007, 8, 3634–3640. [Google Scholar] [CrossRef]
- Gurusamy-Thangavelu, S.A.; Emond, S.J.; Kulshrestha, A.; Hillmyer, M.A.; MacOsko, C.W.; Tolman, W.B.; Hoye, T.R. Polyurethanes based on renewable polyols from bioderived lactones. Polym. Chem. 2012, 3, 2941–2948. [Google Scholar] [CrossRef]
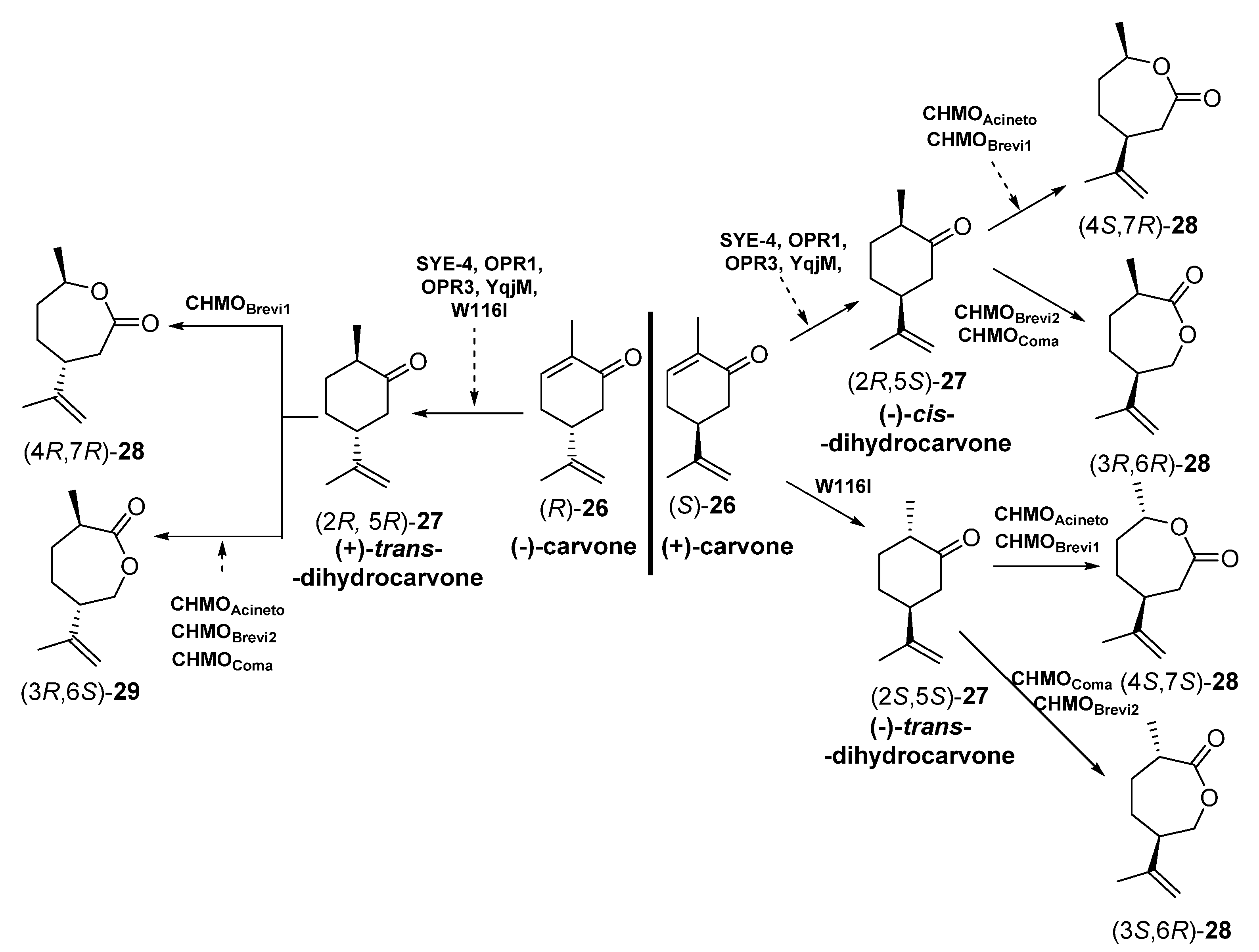
- Iqbal, N.; Stewart, J.D.; Macheroux, P.; Rudroff, F.; Mihovilovic, M.D. Novel concurrent redox cascades of (R)- and (S)-carvones enables access to carvo-lactones with distinct regio- and enantioselectivity. Tetrahedron 2018, 74, 7389–7394. [Google Scholar] [CrossRef]
- Brigé, A.; Van Den Hemel, D.; Carpentier, W.; De Smet, L.; Van Beeumen, J.J. Comparative characterization and expression analysis of the four Old Yellow Enzyme homologues from Shewanella oneidensis indicate differences in physiological function. Biochem. J. 2006, 394, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.; Stueckler, C.; Kroutil, W.; Macheroux, P.; Faber, K. Asymmetric bioreduction of activated alkenes using cloned 12-oxophytodienoate reductase isoenzymes OPR-1 and OPR-3 from Lycopersicon esculentum (tomato): A striking change of stereoselectivity. Angew. Chem. Int. Ed. 2007, 46, 3934–3937. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Stueckler, C.; Ehammer, H.; Pointner, E.; Oberdorfer, G.; Gruber, K.; Hauer, B.; Stuermer, R.; Kroutil, W.; Macheroux, P.; et al. Asymmetric bioreduction of C=C bonds using enoate reductases OPR1, OPR3 and YqjM: Enzyme-based stereocontrol. Adv. Synth. Catal. 2008, 350, 411–418. [Google Scholar] [CrossRef]
- Padhi, S.K.; Bougioukou, D.J.; Stewart, J.D. Site-saturation mutagenesis of tryptophan 116 of Saccharomyces pastorianus old yellow enzyme uncovers stereocomplementary variants. J. Am. Chem. Soc. 2009, 131, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, N.A.; Norris, D.B.; Trudgill, P.W. The Purification and Properties of Cyclohexanone Oxygenase from Nocardia globerula CL1 and Acinetobacter NCIB 9871. Eur. J. Biochem. 1976, 63, 175–192. [Google Scholar] [CrossRef]
- Brzostowicz, P.C.; Gibson, K.L.; Thomas, S.M.; Blasko, M.S.; Rouvière, P.E. Simultaneous identification of two cyclohexanone oxidation genes from an environmental Brevibacterium isolate using MRNA differential display. J. Bacteriol. 2000, 182, 4241–4248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, M.; Trudgill, P.W. Purification and Properties of Cyclopentanone Oxygenase of Pseudomonas NCIB 9872. Eur. J. Biochem. 1976, 63, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Krow, G.R. The Baeyer–Villiger Oxidation of Ketones and Aldehydes. In Organic Reactions; Paquette, L.A., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1993; Volume 43, pp. 251–353. [Google Scholar]
- Oberleitner, N.; Ressmann, A.K.; Bica, K.; Gartner, P.; Fraaije, M.W.; Bornscheuer, U.T.; Rudroff, F.; Mihovilovic, M.D. From waste to value—Direct utilization of limonene from orange peel in a biocatalytic cascade reaction towards chiral carvolactone. Green Chem. 2017, 19, 367–371. [Google Scholar] [CrossRef]
- Oberleitner, N.; Peters, C.; Muschiol, J.; Kadow, M.; Sass, S.; Bayer, T.; Schaaf, P.; Iqbal, N.; Rudroff, F.; Mihovilovic, M.D.; et al. An Enzymatic Toolbox for Cascade Reactions: A Showcase for an In Vivo Redox Sequence in Asymmetric Synthesis. ChemCatChem 2013, 5, 3524–3528. [Google Scholar] [CrossRef]
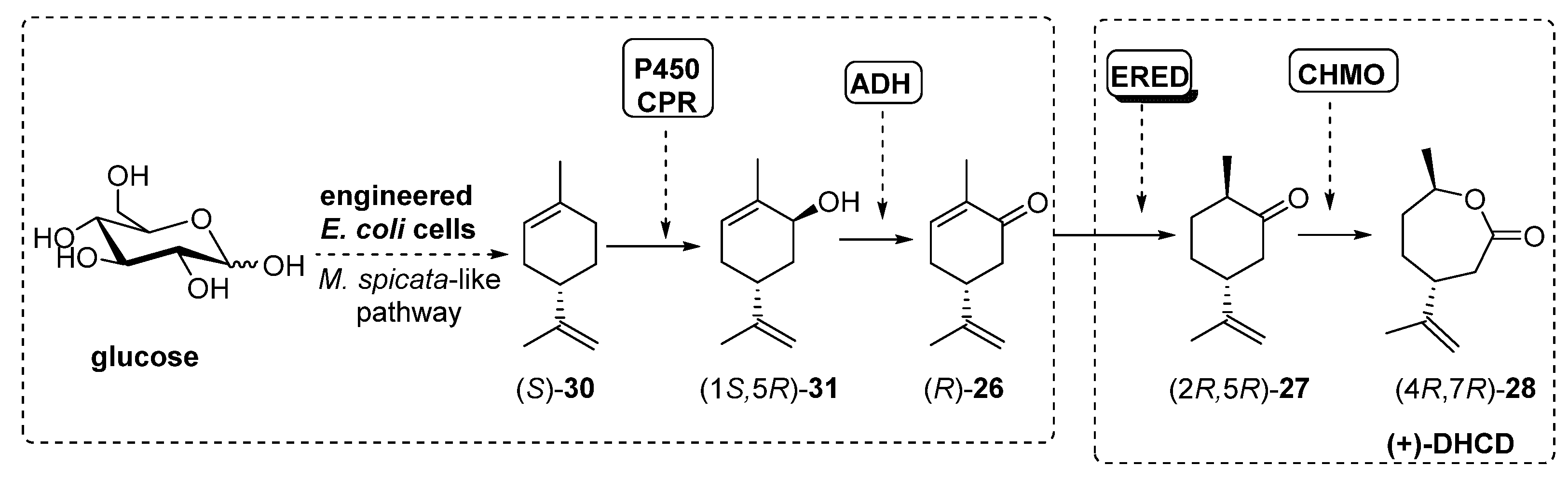
- Avalos, G.A.A.; Toogood, H.S.; Tait, S.; Messiha, H.L.; Scrutton, N.S. From Bugs to Bioplastics: Total (+)-Dihydrocarvide Biosynthesis by Engineered Escherichia coli. ChemBioChem 2019, 20, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fryszkowska, A.; Toogood, H.; Sakuma, M.; Gardiner, J.M.; Stephens, G.M.; Scrutton, N.S. Asymmetrie reduction of activated alkenes by pentaerythritol tetranitrate reductase: Specificity and control of stereochemical outcome by reaction optimisation. Adv. Synth. Catal. 2009, 351, 2976–2990. [Google Scholar] [CrossRef] [PubMed]
- French, C.E.; Nicklin, S.; Bruce, N.C. Sequence and properties of pentaerythritol tetranitrate reductase from Enterobacter cloacae PB2. J. Bacteriol. 1996, 178, 6623–6627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messiha, H.L.; Ahmed, S.T.; Karuppiah, V.; Suardíaz, R.; Ascue Avalos, G.A.; Fey, N.; Yeates, S.; Toogood, H.S.; Mulholland, A.J.; Scrutton, N.S. Biocatalytic Routes to Lactone Monomers for Polymer Production. Biochemistry 2018, 57, 1997–2008. [Google Scholar] [CrossRef]
- Gagnon, R.; Grogan, G.; Levitt, M.S.; Roberts, S.M.; Wan, P.W.H.; Willetts, A.J. Biological Baeyer-Villiger oxidation of some monocyclic and bicyclic ketones using monooxygenases from Acinetobacter calcoaceticus NCIMB 9871 and Pseudomonas putida NCIMB 10007. J. Chem. Soc. Perkin Trans 1 1994, 2537–2543. [Google Scholar] [CrossRef]
- Mallin, H.; Wulf, H.; Bornscheuer, U.T. A self-sufficient Baeyer-Villiger biocatalysis system for the synthesis of ε-caprolactone from cyclohexanol. Enzym. Microb. Technol. 2013, 53, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Staudt, S.; Bornscheuer, U.T.; Menyes, U.; Hummel, W.; Gröger, H. Direct biocatalytic one-pot-transformation of cyclohexanol with molecular oxygen into ε-caprolactone. Enzym. Microb. Technol. 2013, 53, 288–292. [Google Scholar] [CrossRef]
- Aalbers, F.S.; Fraaije, M.W. Coupled reactions by coupled enzymes: Alcohol to lactone cascade with alcohol dehydrogenase–cyclohexanone monooxygenase fusions. Appl. Microbiol. Biotechnol. 2017, 101, 7557–7565. [Google Scholar] [CrossRef] [PubMed]
- de Gonzalo, G.; Franconetti, A. Enantioselective sulfoxidations employing the thermostable cyclohexanone monooxygenase from Thermocrispum municipale. Enzym. Microb. Technol. 2018, 113, 24–28. [Google Scholar] [CrossRef]
- Reimer, A.; Wedde, S.; Staudt, S.; Schmidt, S.; Höffer, D.; Hummel, W.; Kragl, U.; Bornscheuer, U.T.; Gröger, H. Process Development through Solvent Engineering in the Biocatalytic Synthesis of the Heterocyclic Bulk Chemical ε-Caprolactone. J. Heterocycl. Chem. 2017, 54, 391–396. [Google Scholar] [CrossRef]
- Pennec, A.; Hollmann, F.; Smit, M.S.; Opperman, D.J. One-pot conversion of cycloalkanes to lactones. ChemCatChem 2015, 7, 236–239. [Google Scholar] [CrossRef]
- Fasan, R. Tuning P450 Enzymes as Oxidation Catalysts. ACS Catal. 2012, 2, 647–666. [Google Scholar] [CrossRef]
- Urlacher, V.B.; Girhard, M. Cytochrome P450 Monooxygenases in Biotechnology and Synthetic Biology. Trends Biotechnol. 2019, 37, 882–897. [Google Scholar] [CrossRef]
- Sattler, J.H.; Fuchs, M.; Mutti, F.G.; Grischek, B.; Engel, P.; Pfeffer, J.; Woodley, J.M.; Kroutil, W. Introducing an In Situ Capping Strategy in Systems Biocatalysis To Access 6-Aminohexanoic acid. Angew. Chem. Int. Ed. 2014, 53, 14153–14157. [Google Scholar] [CrossRef] [PubMed]
- Rioz-Martinez, A.; Bisogno, F.R.; Rodriguez, C.; de Gonzalo, G.; Lavandera, I.; Pazmino, D.E.T.; Fraaije, M.W.; Gotor, V. Biocatalysed concurrent production of enantioenriched compounds through parallel interconnected kinetic asymmetric transformations. Org. Biomol. Chem. 2010, 8, 1431–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gonzalo, G.; Torres Pazmiño, D.E.; Ottolina, G.; Fraaije, M.W.; Carrea, G. Oxidations catalyzed by phenylacetone monooxygenase from Thermobifida fusca. Tetrahedron Asymmetry 2005, 16, 3077–3083. [Google Scholar] [CrossRef]
- De Gonzalo, G.; Torres Pazmiño, D.E.; Ottolina, G.; Fraaije, M.W.; Carrea, G. 4-Hydroxyacetophenone monooxygenase from Pseudomonas fluorescens ACB as an oxidative biocatalyst in the synthesis of optically active sulfoxides. Tetrahedron Asymmetry 2006, 17, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, C.; Gonzalo, G.D.; Torres Pazmiño, D.E.; Fraaije, M.W.; Gotor, V. Baeyer-Villiger monooxygenase-catalyzed kinetic resolution of racemic α-alkyl benzyl ketones: Enzymatic synthesis of α-alkyl benzylketones and α-alkyl benzylesters. Tetrahedron Asymmetry 2009, 20, 1168–1173. [Google Scholar] [CrossRef] [Green Version]
- Bisogno, F.R.; Rioz-Martinez, A.; Rodriguez, C.; Lavandera, I.; de Gonzalo, G.; Pazmino, D.E.T.; Fraaije, M.W.; Gotor, V. Oxidoreductases Working Together: Concurrent Obtaining of Valuable Derivatives by Employing the PIKAT Method. ChemCatChem 2010, 2, 946–949. [Google Scholar] [CrossRef] [Green Version]
- Torres Pazmiño, D.E.; Snajdrova, R.; Rial, D.V.; Mihovilovic, M.D.; Fraaije, M.W. Altering the substrate specificity and enantioselectivity of phenylacetone monooxygenase by structure-inspired enzyme redesign. Adv. Synth. Catal. 2007, 349, 1361–1368. [Google Scholar] [CrossRef]
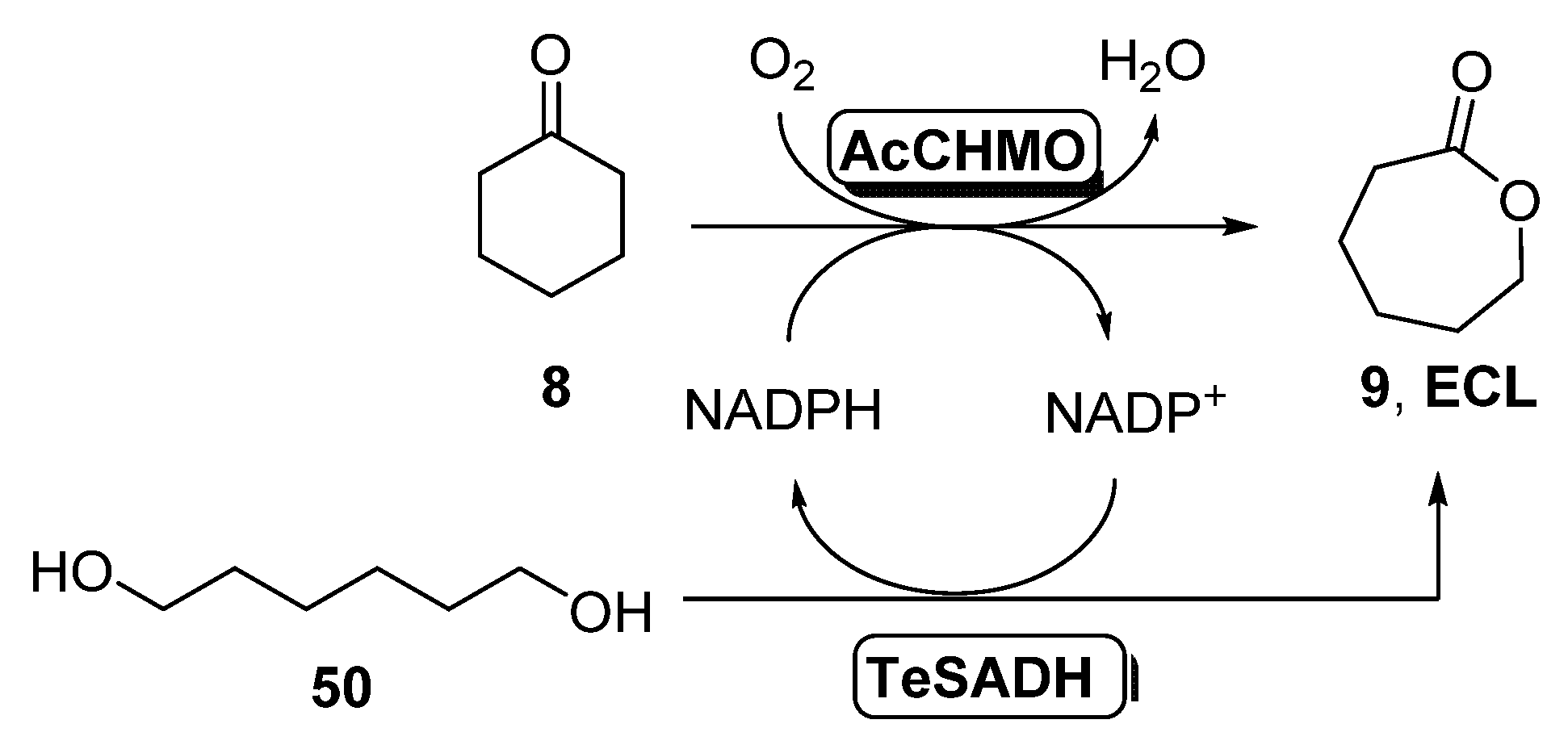
- Bornadel, A.; Hatti-Kaul, R.; Hollmann, F.; Kara, S. A Bi-enzymatic Convergent Cascade for epsilon-Caprolactone Synthesis Employing 1,6-Hexanediol as a “Double-Smart Cosubstrate”. ChemCatChem 2015, 7, 2442–2445. [Google Scholar] [CrossRef]
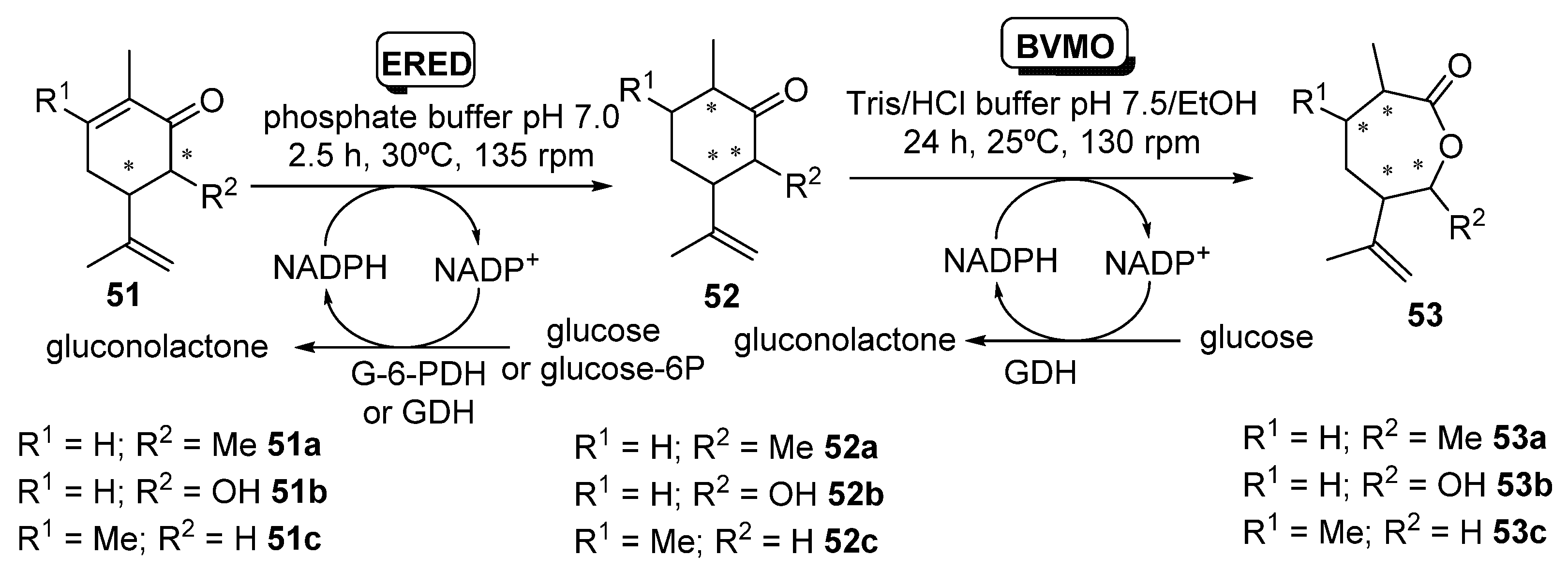
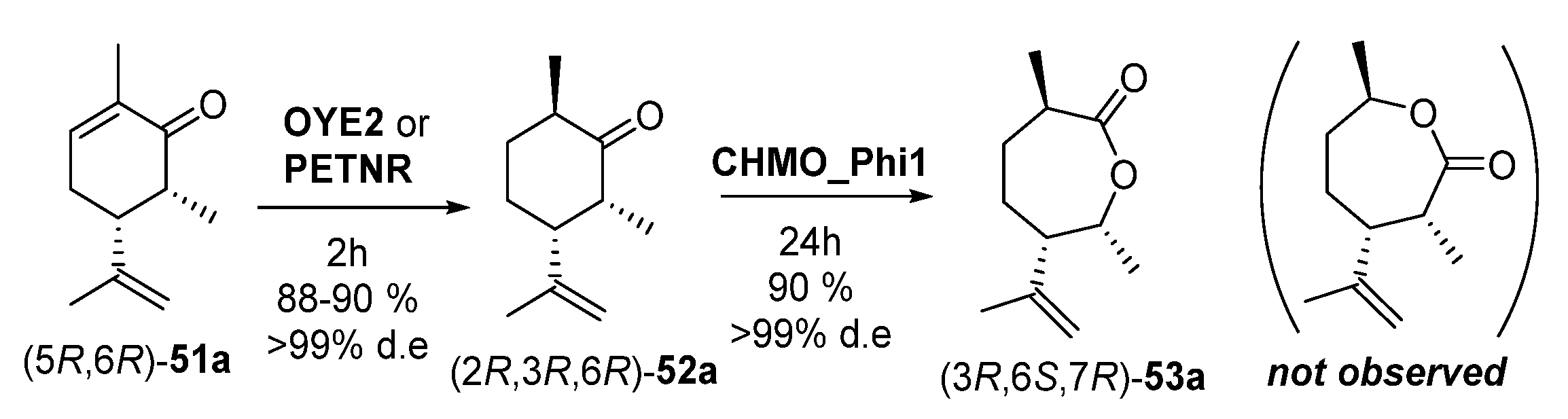
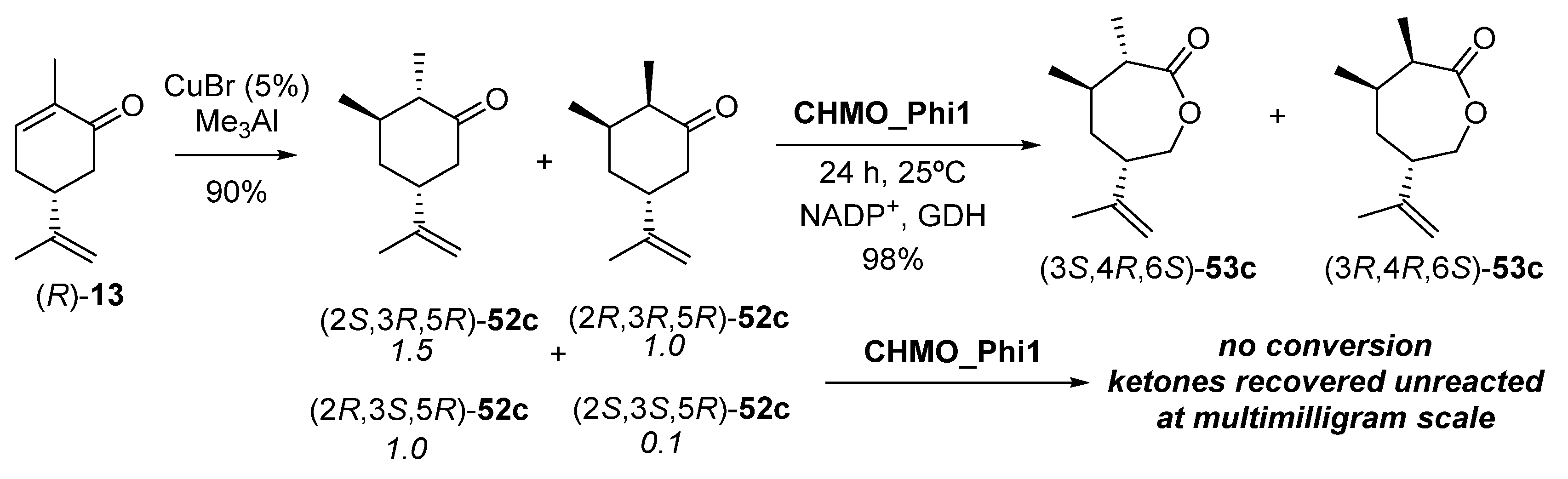
- Issa, I.S.; Toogood, H.S.; Johannissen, L.O.; Raftery, J.; Scrutton, N.S.; Gardiner, J.M. C3 and C6 Modification-Specific OYE Biotransformations of Synthetic Carvones and Sequential BVMO Chemoenzymatic Synthesis of Chiral Caprolactones. Chem. Eur. J. 2019, 25, 2983–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niino, Y.S.; Chakraborty, S.; Brown, B.J.; Massey, V. A new old yellow enzyme of Saccharomyces cerevisiae. J. Biol. Chem. 1995, 270, 1983–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Gonzalo, G.; Alcántara, A.R. Multienzymatic Processes Involving Baeyer–Villiger Monooxygenases. Catalysts 2021, 11, 605. https://doi.org/10.3390/catal11050605
de Gonzalo G, Alcántara AR. Multienzymatic Processes Involving Baeyer–Villiger Monooxygenases. Catalysts. 2021; 11(5):605. https://doi.org/10.3390/catal11050605
Chicago/Turabian Stylede Gonzalo, Gonzalo, and Andrés R. Alcántara. 2021. "Multienzymatic Processes Involving Baeyer–Villiger Monooxygenases" Catalysts 11, no. 5: 605. https://doi.org/10.3390/catal11050605