
CO2 Self-Poisoning and Its Mitigation in CuO Catalyzed CO Oxidation: Determining and Speeding up the Rate-Determining Step


Abstract
:1. Introduction
2. Results and Discussion
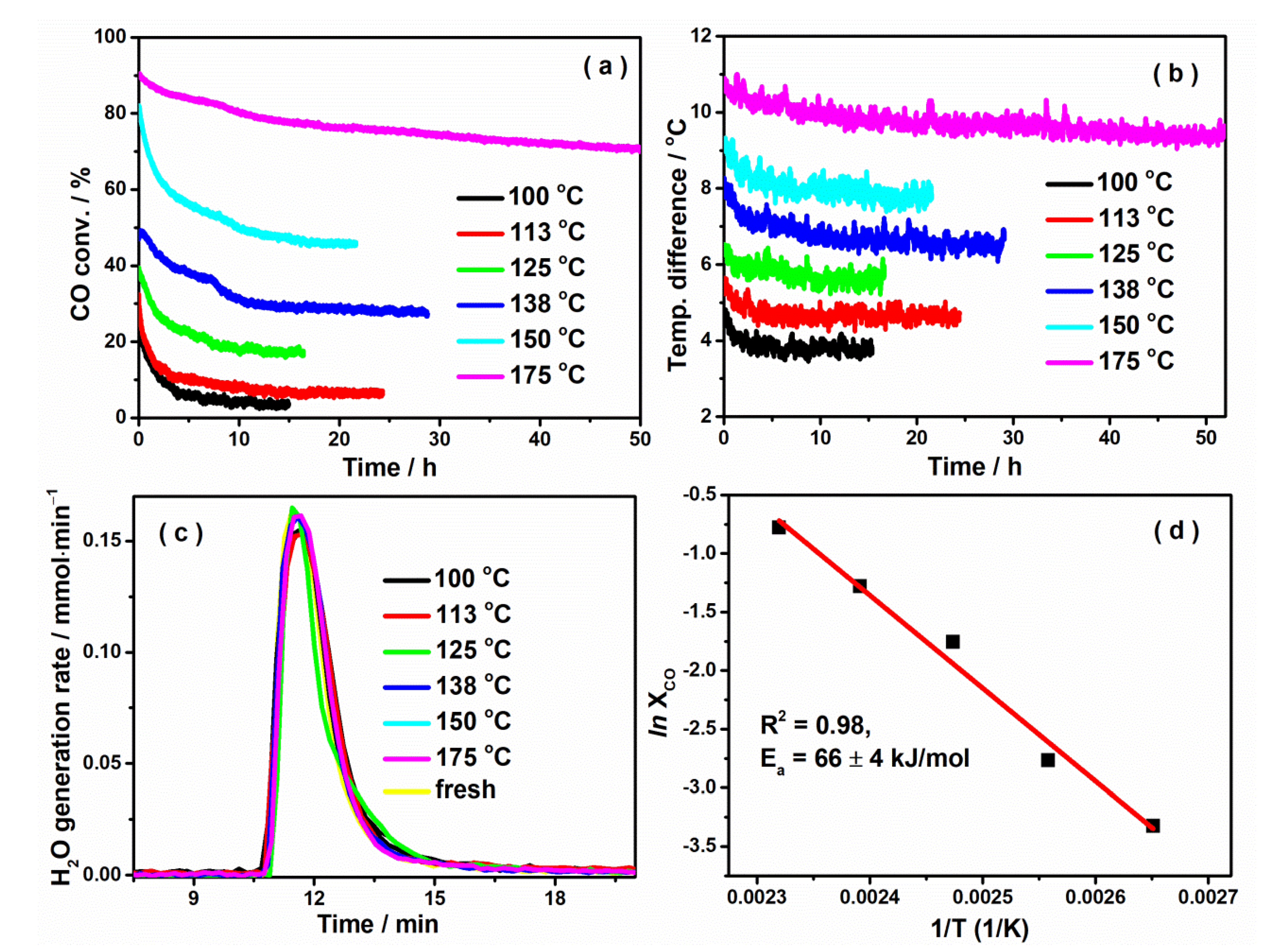
2.1. Catalyst Deactivation Occurs on a CuO/Al2O3 Catalyst in CO Oxidation
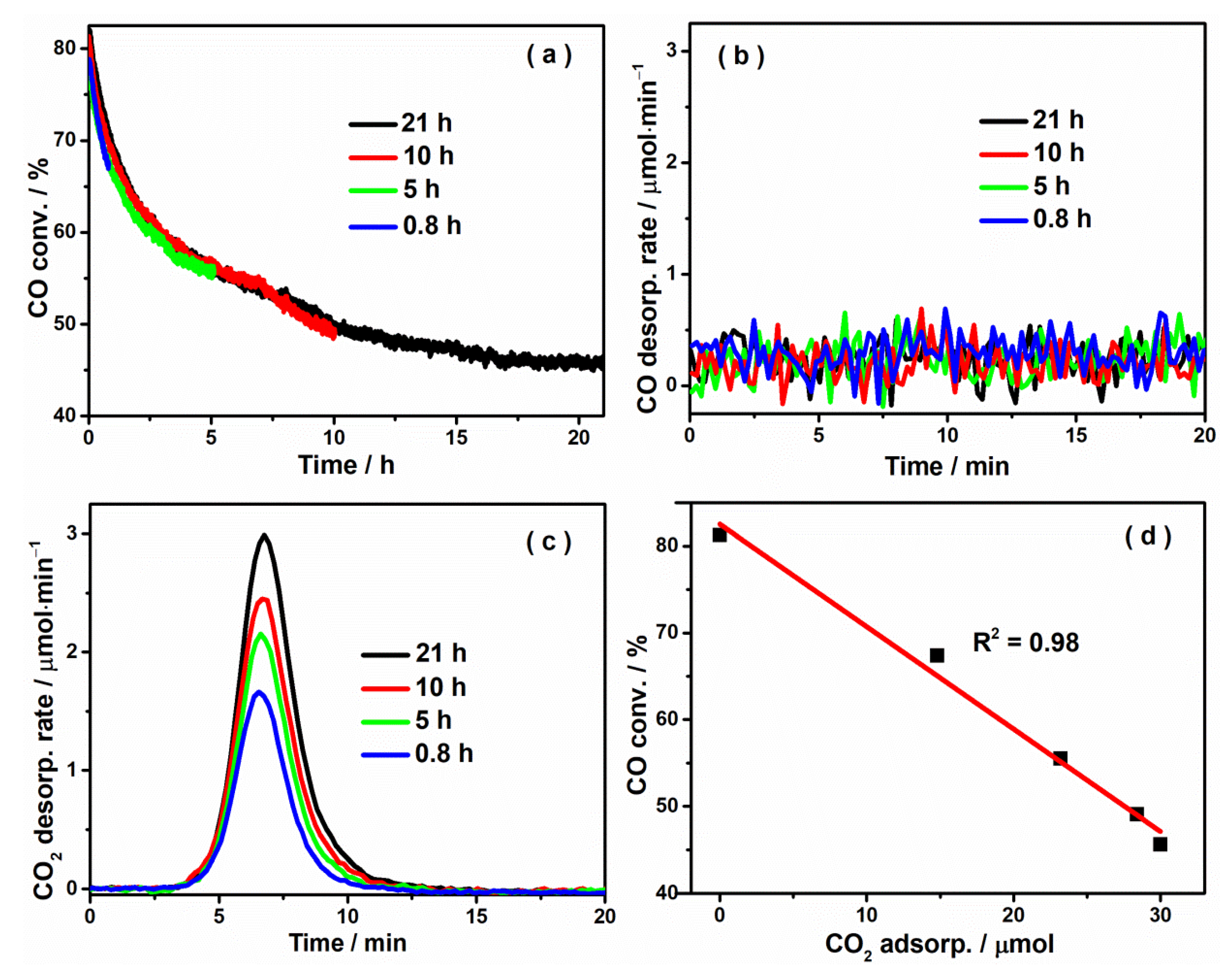
2.2. CO2 Adsorption Is a Major Cause for the Observed Catalyst Deactivation
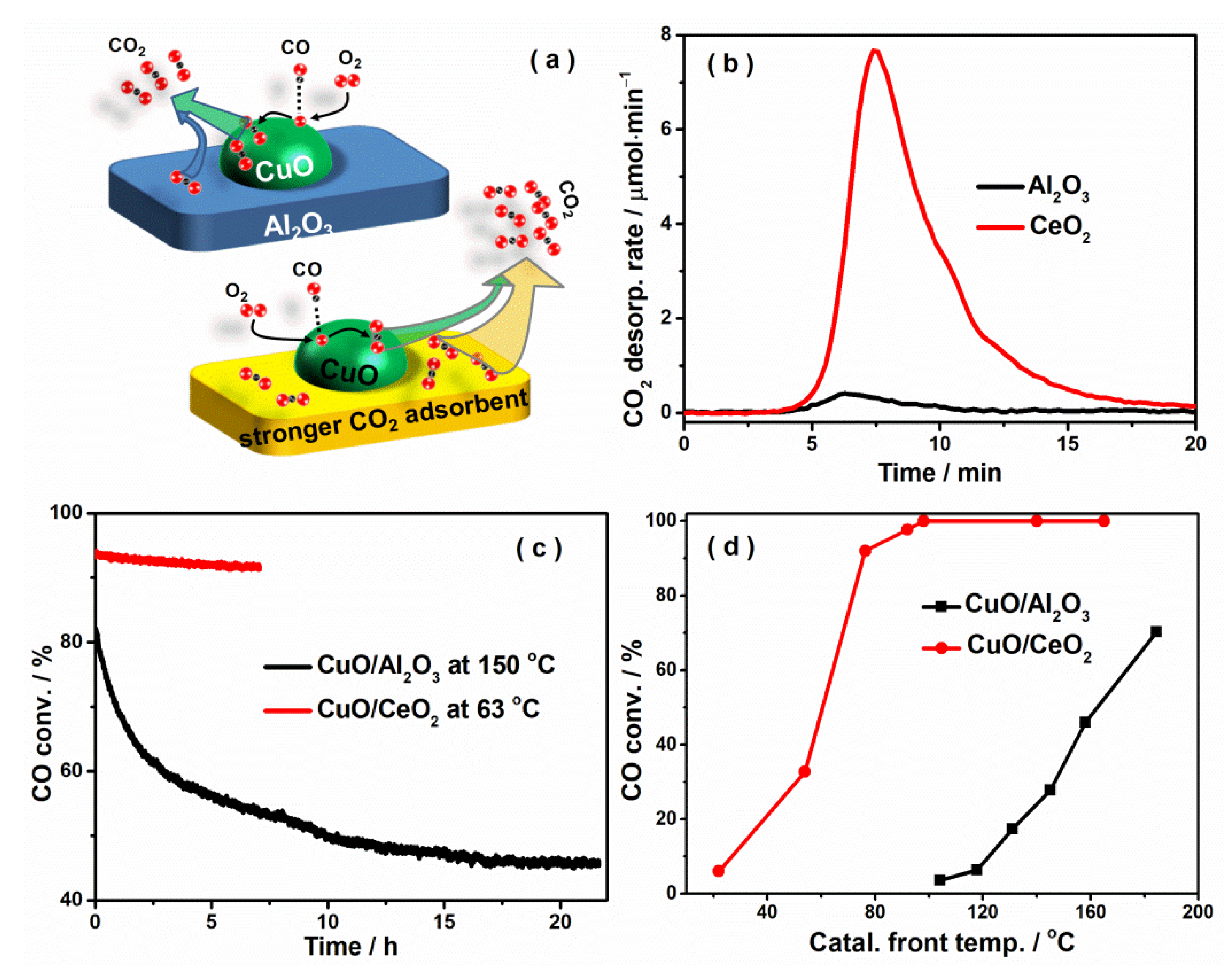
2.3. CO2 Adsorption Inhibits O2 Adsorption
2.4. CO2 Self-Poisoning Is Mitigated by Introducing a Strong CO2 Adsorbent as the Support
3. Experimental Section
3.1. Catalyst Synthesis
3.2. Catalytic Performance Evaluation
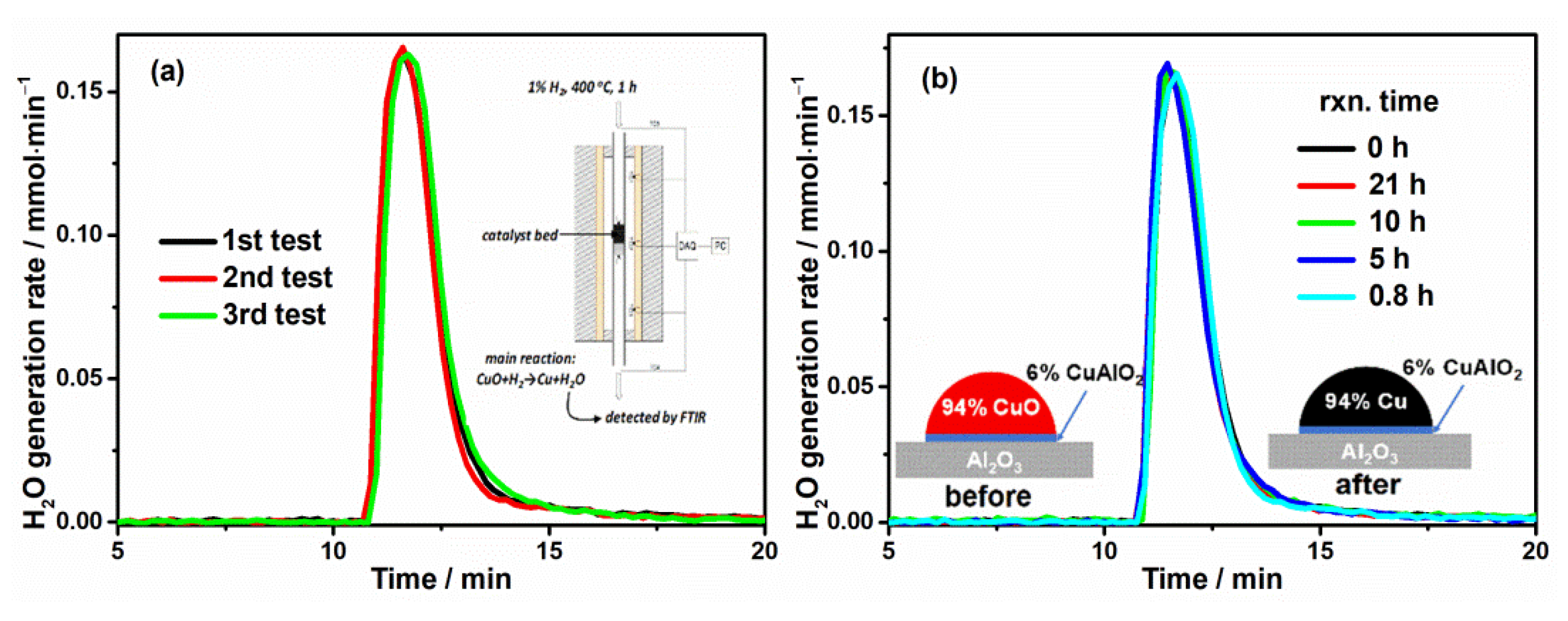
3.3. Catalyst Activation Process
3.4. In-Situ Quantification of Adsorption
3.5. In-Situ Measurement of Average Cu Chemical State
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Freund, H.J.; Meijer, G.; Scheffler, M.; Schlögl, R.; Wolf, M. CO oxidation as a prototypical reaction for heterogeneous processes. Angew. Chem. Int. Ed. 2011, 50, 10064–10094. [Google Scholar] [CrossRef] [PubMed]
- Van Spronsen, M.A.; Frenken, J.W.; Groot, I.M. Surface science under reaction conditions: CO oxidation on Pt and Pd model catalysts. Chem. Soc. Rev. 2017, 46, 4347–4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, L.; Mei, D.; Xiong, H.; Peng, B.; Ren, Z.; Hernandez, X.I.P.; DeLaRiva, A.; Wang, M.; Engelhard, M.H.; Kovarik, L.; et al. Activation of surface lattice oxygen in single-atom Pt/CeO2 for low-temperature CO oxidation. Science 2017, 358, 1419–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wang, Y.-Z.; Wang, X.-X.; Song, K.-K.; Jian, X.-D.; Qian, P.; Bai, Y.; Su, Y.-J. Size and Stoichiometry Effect of FePt Bimetal Nanoparticle Catalyst for CO Oxidation: A DFT Study. J. Phys. Chem. C 2020, 124, 8706–8715. [Google Scholar] [CrossRef]
- Ning, C.; Zhang, Y.; Meng, Z.; Xiao, C.; Lu, R. Computational Screening of Single Non-Noble Transition-Metal Atoms Confined Inside Boron Nitride Nanotubes for CO Oxidation. J. Phys. Chem. C 2020, 124, 2030–2038. [Google Scholar] [CrossRef]
- Yang, F.; Graciani, J.; Evans, J.; Liu, P.; Hrbek, J.; Sanz, J.F.; Rodriguez, J.A. CO oxidation on inverse CeOx/Cu(111) catalysts: High catalytic activity and ceria-promoted dissociation of O2. J. Am. Chem. Soc. 2011, 133, 3444–3451. [Google Scholar] [CrossRef]
- Abdel-Mageed, A.M.; Rungtaweevoranit, B.; Parlinska-Wojtan, M.; Pei, X.; Yaghi, O.M.; Behm, R.J. Highly Active and Stable Single-Atom Cu Catalysts Supported by a Metal-Organic Framework. J. Am. Chem. Soc. 2019, 141, 5201–5210. [Google Scholar] [CrossRef]
- Xie, X.; Li, Y.; Liu, Z.Q.; Haruta, M.; Shen, W. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 2009, 458, 746–749. [Google Scholar] [CrossRef]
- Hu, X.; Chen, J.; Li, S.; Chen, Y.; Qu, W.; Ma, Z.; Tang, X. The Promotional Effect of Copper in Catalytic Oxidation by Cu-Doped α-MnO2 Nanorods. J. Phys. Chem. C 2020, 124, 701–708. [Google Scholar] [CrossRef]
- Maynes, A.J.; Driscoll, D.M.; DeSario, P.A.; Pietron, J.J.; Pennington, A.M.; Rolison, D.R.; Morris, J.R. Electronic Metal-Support Interactions in the Activation of CO Oxidation over a Cu/TiO2 Aerogel Catalyst. J. Phys. Chem. C 2020, 124, 21491–21501. [Google Scholar] [CrossRef]
- Royer, S.; Duprez, D. Catalytic Oxidation of Carbon Monoxide over Transition Metal Oxides. ChemCatChem 2011, 3, 24–65. [Google Scholar] [CrossRef]
- Heck, R.M.; Farrauto, R.J.; Gulati, S.T. Catalytic Air Pollution Control: Commercial Technology, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Gawande, M.B.; Goswami, A.; Felpin, F.-X.; Asefa, T.; Huang, X.; Silva, R.; Zou, X.; Zboril, R.; Varma, R.S. Cu and Cu-based nanoparticles: Synthesis and applications in catalysis. Chem. Rev. 2016, 116, 3722–3811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, X.P.; Wang, L.N.; Li, X.N.; Liu, Q.Y.; Zhao, Y.X.; Ma, T.M.; He, S.G. Noble-Metal-Free Single-Atom Catalysts CuAl4O7–9− for CO Oxidation by O2. Angew. Chem. Int. Ed. 2018, 130, 11155–11159. [Google Scholar] [CrossRef]
- Sun, B.-Z.; Chen, W.-K.; Xu, Y.-J. Reaction mechanism of CO oxidation on Cu2O (111): A density functional study. J. Chem. Phys. 2010, 133, 154502. [Google Scholar] [CrossRef] [PubMed]
- El Kasmi, A.; Tian, Z.-Y.; Vieker, H.; Beyer, A.; Chafik, T. Innovative CVD synthesis of Cu2O catalysts for CO oxidation. Appl. Catal. B 2016, 186, 10–18. [Google Scholar] [CrossRef]
- Pierron, E.; Rashkin, J.; Roth, J. Copper oxide on alumina: I. XRD studies of catalyst composition during air oxidation of carbon monoxide. J. Catal. 1967, 9, 38–44. [Google Scholar] [CrossRef]
- Eren, B.; Heine, C.; Bluhm, H.; Somorjai, G.A.; Salmeron, M. Catalyst Chemical State during CO Oxidation Reaction on Cu(111) Studied with Ambient-Pressure X-ray Photoelectron Spectroscopy and Near Edge X-ray Adsorption Fine Structure Spectroscopy. J. Am. Chem. Soc. 2015, 137, 11186–11190. [Google Scholar] [CrossRef] [PubMed]
- Tsakoumis, N.E.; York, A.P.; Chen, D.; Rønning, M. Catalyst characterisation techniques and reaction cells operating at realistic conditions; towards acquisition of kinetically relevant information. Catal. Sci. Technol. 2015, 5, 4859–4883. [Google Scholar] [CrossRef]
- Svintsitskiy, D.A.; Kardash, T.Y.; Stonkus, O.A.; Slavinskaya, E.M.; Stadnichenko, A.I.; Koscheev, S.V.; Chupakhin, A.P.; Boronin, A.I. In situ XRD, XPS, TEM, and TPR study of highly active in CO oxidation CuO nanopowders. J. Phys. Chem. C 2013, 117, 14588–14599. [Google Scholar] [CrossRef]
- Chorkendorff, I.; Niemantsverdriet, J.W. Concepts of Modern Catalysis and Kinetics; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Constales, D.; Yablonsky, G.S.; D’hooge, D.R.; Thybaut, J.W.; Marin, G.B. Advanced Data Analysis and Modelling in Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Raróg, W.; Kowalczyk, Z.; Sentek, J.; Składanowski, D.; Szmigiel, D.; Zieliñski, J. Decomposition of ammonia over potassium promoted ruthenium catalyst supported on carbon. Appl. Catal. A Gen. 2011, 208, 213–216. [Google Scholar] [CrossRef]
- Gulicovski, J.; Rašković-Lovre, Ž.; Kurko, S.; Vujasin, R.; Jovanović, Z.; Matović, L.; Grbović Novaković, J. Influence of vacant CeO2 nanostructured ceramics on MgH2 hydrogen desorption properties. Ceram. Int. 2012, 38, 1181–1186. [Google Scholar] [CrossRef]
- Guo, Y.; Li, C.; Lu, S.; Zhao, C. Low temperature CO catalytic oxidation and kinetic performances of KOH–Hopcalite in the presence of CO2. RSC Adv. 2016, 6, 7181–7188. [Google Scholar] [CrossRef]
- Zhang, X.-m.; Tian, P.; Tu, W.; Zhang, Z.; Xu, J.; Han, Y.-F. Tuning the dynamic interfacial structure of copper–ceria catalysts by indium oxide during CO oxidation. ACS Catal. 2018, 8, 5261–5275. [Google Scholar] [CrossRef]
- Wang, Y.; Widmann, D.; Lehnert, F.; Gu, D.; Schuth, F.; Behm, R.J. Avoiding Self-Poisoning: A Key Feature for the High Activity of Au/Mg(OH)2 Catalysts in Continuous Low-Temperature CO Oxidation. Angew. Chem. Int. Ed. 2017, 56, 9597–9602. [Google Scholar] [CrossRef] [PubMed]
- Del Río, E.; Collins, S.E.; Aguirre, A.; Chen, X.; Delgado, J.J.; Calvino, J.J.; Bernal, S. Reversible deactivation of a Au/Ce0.62Zr0.38O2 catalyst in CO oxidation: A systematic study of CO2-triggered carbonate inhibition. J. Catal. 2014, 316, 210–218. [Google Scholar] [CrossRef]
- Denkwitz, Y.; Schumacher, B.; Kučerová, G.; Behm, R.J. Activity, stability, and deactivation behavior of supported Au/TiO2 catalysts in the CO oxidation and preferential CO oxidation reaction at elevated temperatures. J. Catal. 2009, 267, 78–88. [Google Scholar] [CrossRef]
- Hao, Y.; Mihaylov, M.; Ivanova, E.; Hadjiivanov, K.; Knozinger, H.; Gates, B. CO oxidation catalyzed by gold supported on MgO: Spectroscopic identification of carbonate-like species bonded to gold during catalyst deactivation. J. Catal. 2009, 261, 137–149. [Google Scholar] [CrossRef]
- Ren, J.; Cheng, K.; Li, M.; Zhao, S.; Li, H.; Chen, Y. Bridging the gaps between experimental and mechanistic catalysis research: A case study with CO oxidation over a Pd/Al2O3 catalyst. ChemCatChem 2019, 11, 4427–4433. [Google Scholar] [CrossRef]
- Schmidt-Whitley, R.; Martinez-Clemente, M.; Revcolevschi, A. Growth and microstructural control of single crystal cuprous oxide Cu2O. J. Cryst. Growth 1974, 23, 113–120. [Google Scholar] [CrossRef]
- Il’Ichev, A.; Matyshak, V.; Korchak, V. Oxidation of CO with catalyst oxygen and with oxygen from the gas phase over CuO/CeO2 according to TPD and IR spectroscopic data. Kinet. Catal. 2015, 56, 115–124. [Google Scholar] [CrossRef]
- Zammit, M.; DiMaggio, C.L.; Kim, C.H.; Lambert, C.; Muntean, G.G.; Peden, C.H.; Parks, J.E.; Howden, K. Future Automotive Aftertreatment Solutions: The 150 °C Challenge Workshop Report; U.S. Drive Report; U.S. Drive: Southfield, MI, USA, 2013.
- Beale, A.M.; Gao, F.; Lezcano-Gonzalez, I.; Peden, C.H.; Szanyi, J. Recent advances in automotive catalysis for NOx emission control by small-pore microporous materials. Chem. Soc. Rev. 2015, 44, 7371–7405. [Google Scholar] [CrossRef]
- Wang, F.; Wang, X.; Liu, D.; Zhen, J.; Li, J.; Wang, Y.; Zhang, H. High-Performance ZnCo2O4@CeO2 Core@shell Microspheres for Catalytic CO Oxidation. ACS Appl. Mater. Interfaces 2014, 6, 22216–22223. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Mu, X.; Zhang, Y.; Cui, Y.; Xia, G.; Chen, Z. Rare earth metal modified CuO/γ-Al2O3 catalysts in the CO oxidation. Catal. Commun. 2011, 12, 349–352. [Google Scholar] [CrossRef]
- Tang, C.; Li, J.; Yao, X.; Sun, J.; Cao, Y.; Zhang, L.; Gao, F.; Deng, Y.; Dong, L. Mesoporous NiO-CeO2 catalysts for CO oxidation: Nickel content effect and mechanism aspect. Appl. Catal. A 2015, 494, 77–86. [Google Scholar] [CrossRef]
- Ren, J.; Chen, Y. Pitfalls in identifying active catalyst species. Nat. Commun. 2020, 11, 4564. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Lee, A.C.; Cheng, K.; Li, M.; Chen, Y. Measuring the Unmeasurable by IR Spectroscopy: Carbon Deposition Kinetics in Dry Reforming of Methane. ChemPhysChem 2018, 19, 1814–1819. [Google Scholar] [CrossRef]
- Ren, J.; Li, M.; Lee, A.C.; Cheng, K.; Chen, Y. IR spectroscopic measurement of hydrogen production kinetics in methane dry reforming. Int. J. Hydrog. Energy 2019, 44, 9866–9872. [Google Scholar] [CrossRef]
- Fernández-Garcıa, M.; Rodrıguez-Ramos, I.; Ferreira-Aparicio, P.; Guerrero-Ruiz, A. Tracking down the reduction behavior of copper-on-alumina catalysts. J. Catal. 1998, 178, 253–263. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Furnace Temp./°C | Initial CO Conv. | Steady-State CO Conv. | Relative Conv. Loss * | Initial Cat. Temp. (Ti)/°C | Steady-State Cat. Temp. (Ts)/°C | Relative Conv. Change Due to Temp. ** | CO2 Adsorp./μmol |
|---|---|---|---|---|---|---|---|
| 100 | 30.2% | 3.6% | 89.4% | 105.1 | 104.1 | 5.6% | 10.5 |
| 113 | 32.4% | 6.3% | 80.2% | 118.9 | 117.8 | 5.6% | 15.38 |
| 125 | 48.5% | 17.3% | 63.9% | 131.6 | 131.1 | 2.3% | 25.1 |
| 138 | 39.3% | 27.8% | 30.5% | 146.4 | 145.1 | 5.8% | 39.8 |
| 150 | 82.1% | 45.9% | 43.8% | 159.5 | 158.0 | 6.2% | 30.0 |
| 175 | 90.1% | 70.2% | 22.2% | 186.1 | 184.5 | 5.9% | 66.7 |
| Sample | Initial Conv. | Final Conv. | CO2 Phys. Adsorp./μmol | CO2 Chem. Adsorp./μmol | Total Adsorp./μmol |
|---|---|---|---|---|---|
| 0.8 h | 78.8% | 67.4% | 10.4 | 4.4 | 14.8 |
| 5 h | 77.5% | 55.5% | 17.5 | 5.7 | 23.2 |
| 10 h | 81.3% | 49.1% | 21.8 | 6.6 | 28.4 |
| 21 h | 82.1% | 45.6% | 22.0 | 8.0 | 30.0 |
| CO2 pre-adsorp. | 67.9% | 46.7% | 10.9 | 4.0 | 14.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, K.; Zhao, S.; Ren, J.; Li, H.; Chen, Y. CO2 Self-Poisoning and Its Mitigation in CuO Catalyzed CO Oxidation: Determining and Speeding up the Rate-Determining Step. Catalysts 2021, 11, 654. https://doi.org/10.3390/catal11060654
Cheng K, Zhao S, Ren J, Li H, Chen Y. CO2 Self-Poisoning and Its Mitigation in CuO Catalyzed CO Oxidation: Determining and Speeding up the Rate-Determining Step. Catalysts. 2021; 11(6):654. https://doi.org/10.3390/catal11060654
Chicago/Turabian StyleCheng, Kai, Songjian Zhao, Jiazheng Ren, Haoran Li, and Yongsheng Chen. 2021. "CO2 Self-Poisoning and Its Mitigation in CuO Catalyzed CO Oxidation: Determining and Speeding up the Rate-Determining Step" Catalysts 11, no. 6: 654. https://doi.org/10.3390/catal11060654
APA StyleCheng, K., Zhao, S., Ren, J., Li, H., & Chen, Y. (2021). CO2 Self-Poisoning and Its Mitigation in CuO Catalyzed CO Oxidation: Determining and Speeding up the Rate-Determining Step. Catalysts, 11(6), 654. https://doi.org/10.3390/catal11060654