
Efficient Molybdenum Hydrazonato Epoxidation Catalysts Operating under Green Chemistry Conditions: Water vs. Decane Competition




Abstract
:
1. Introduction
2. Results and Discussion
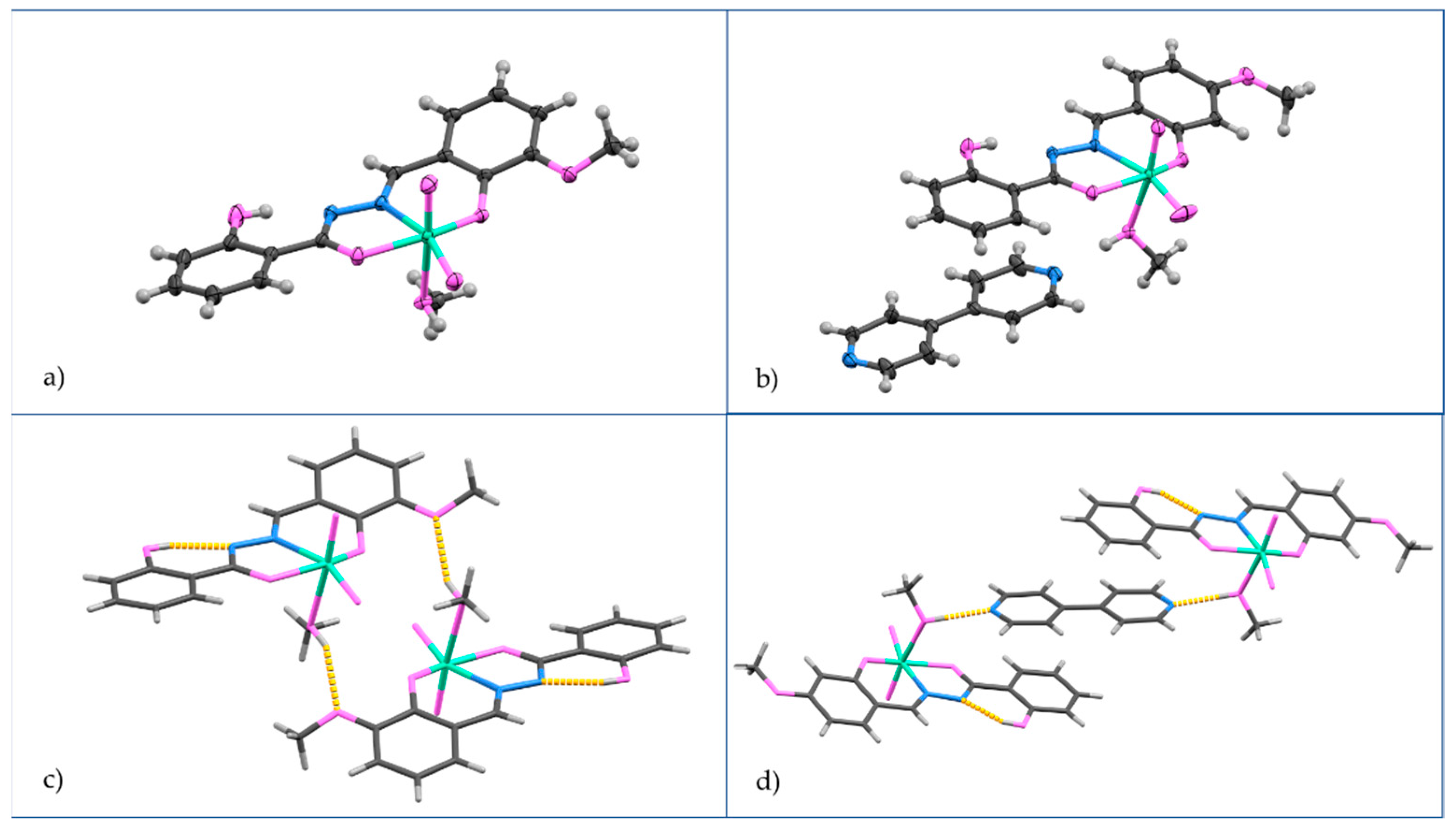
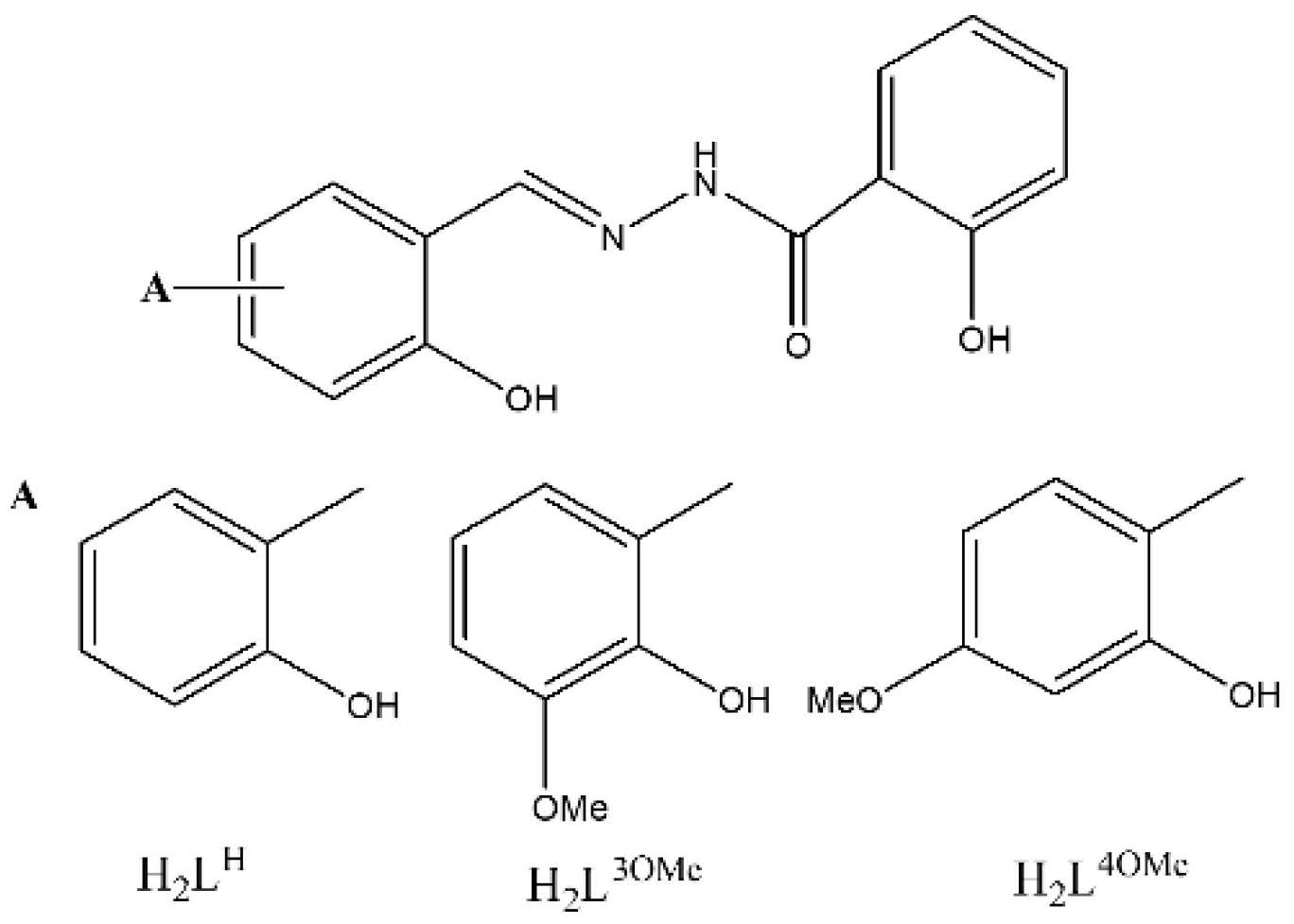
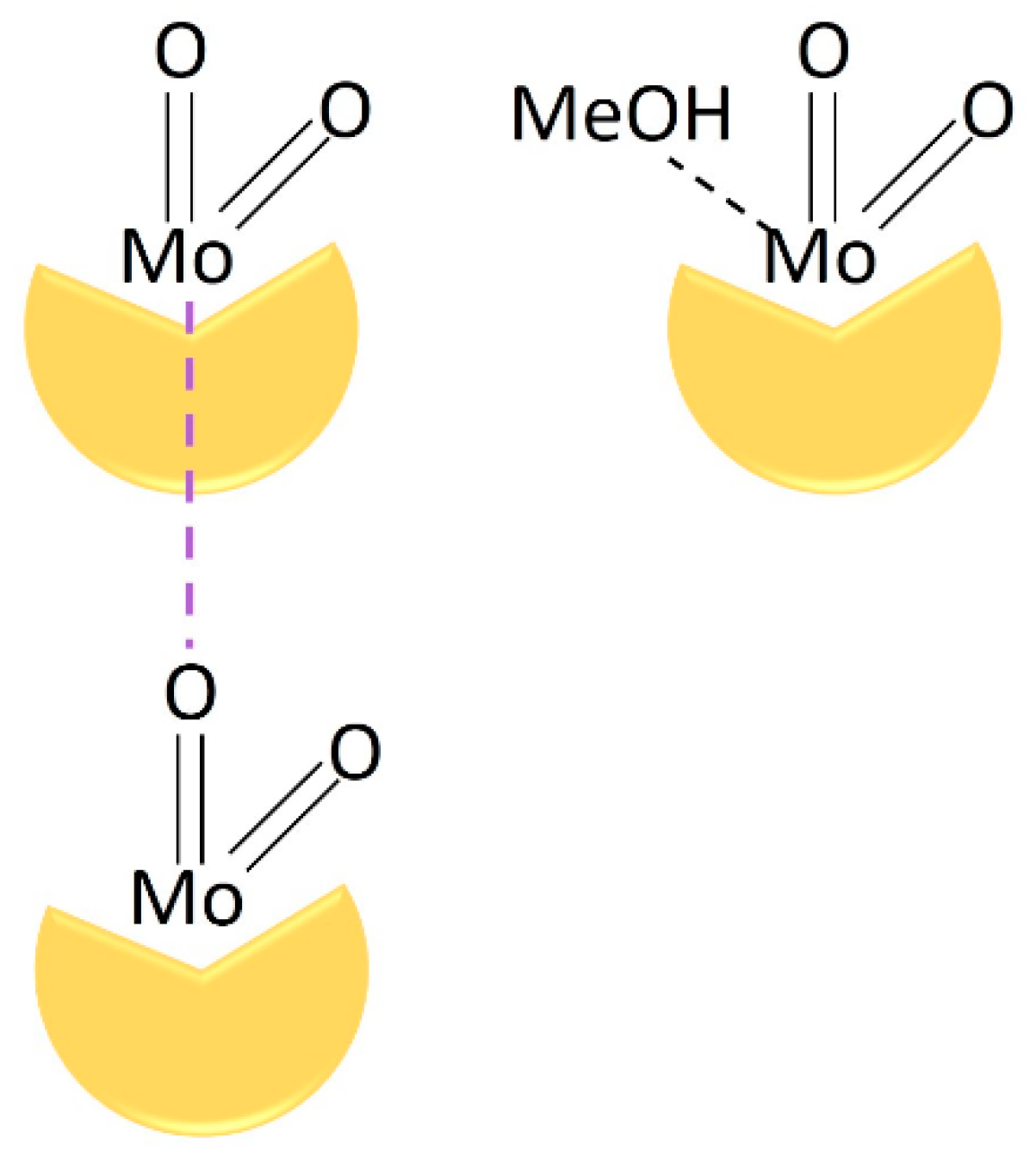
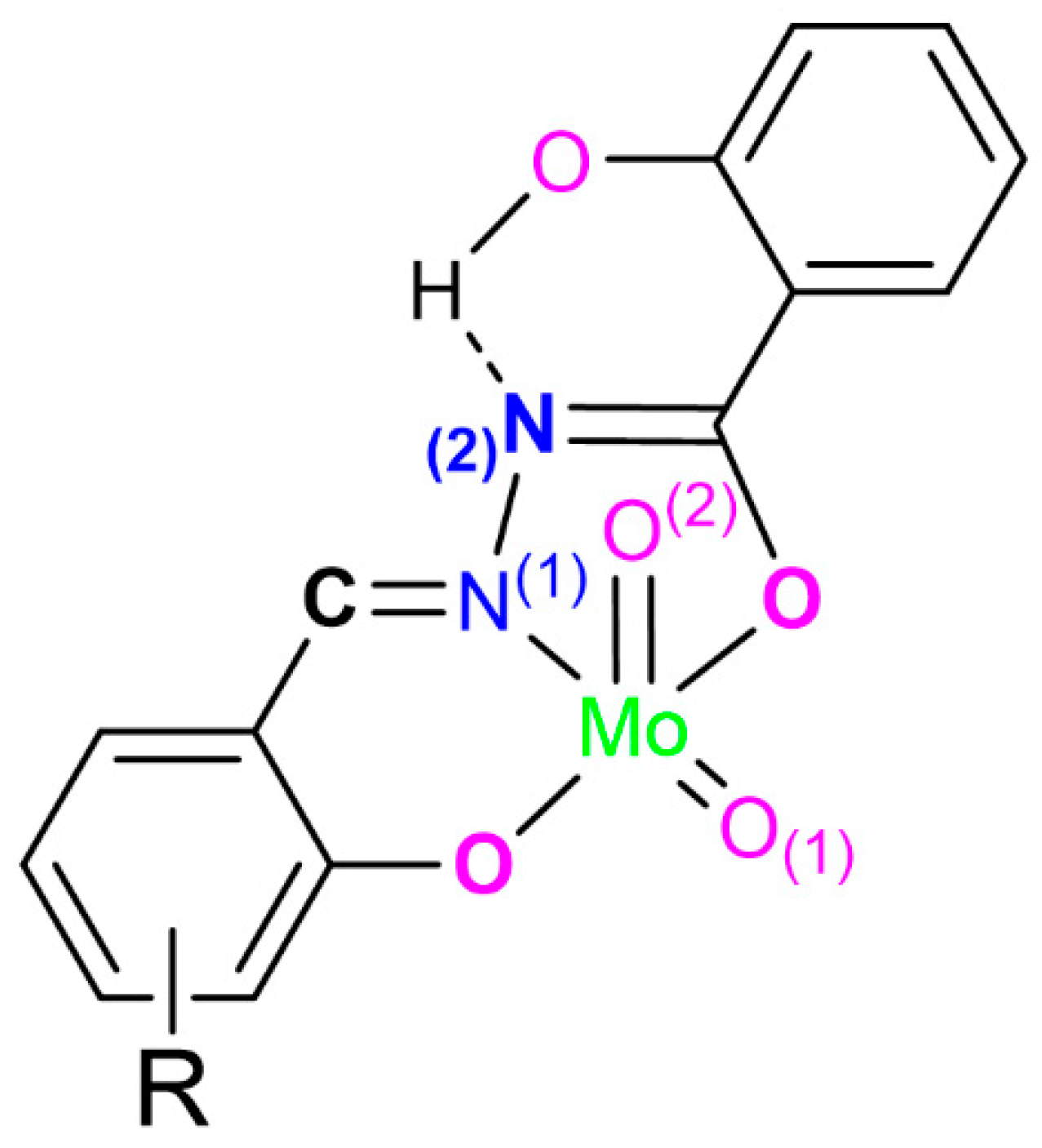
2.1. Catalysts Preparation and Solid-State Characterization
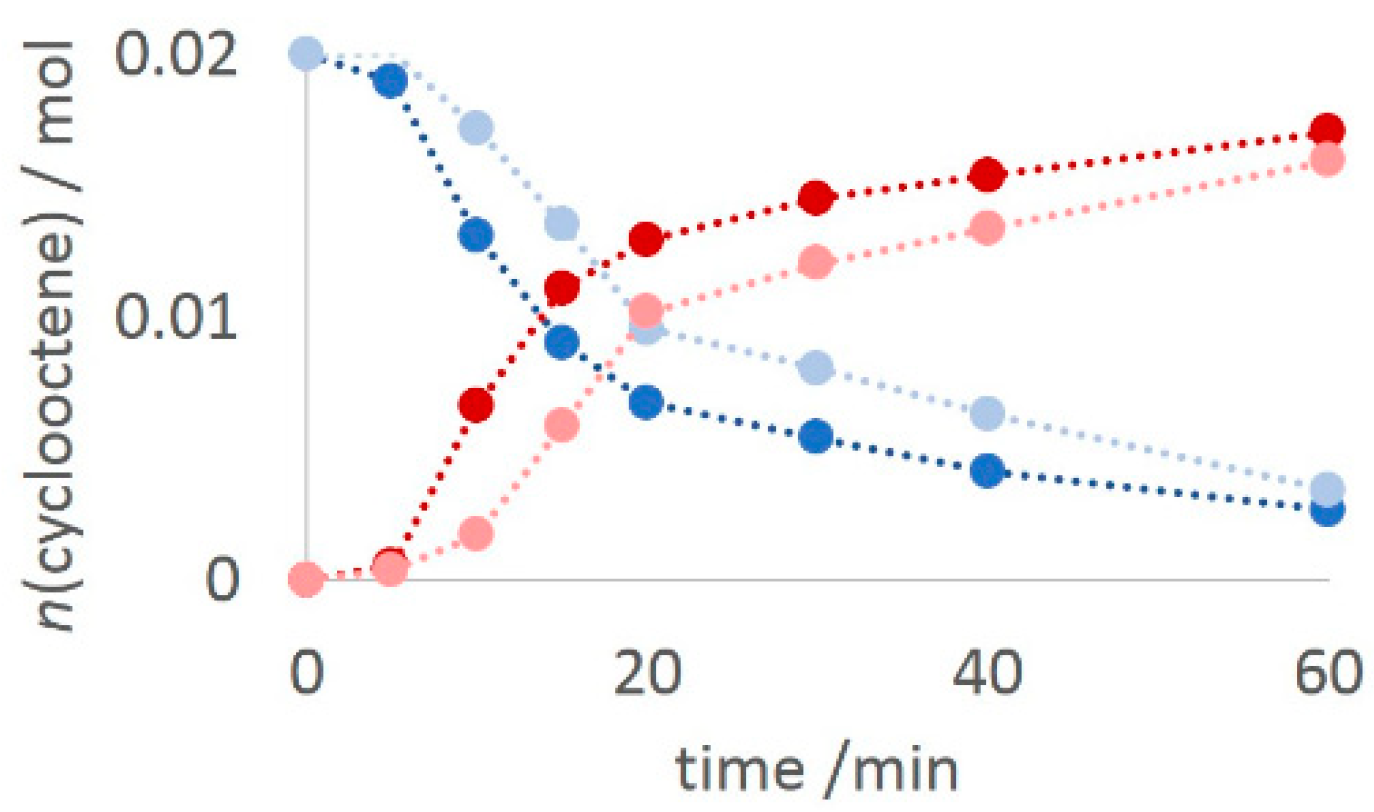
2.2. Catalytic Results
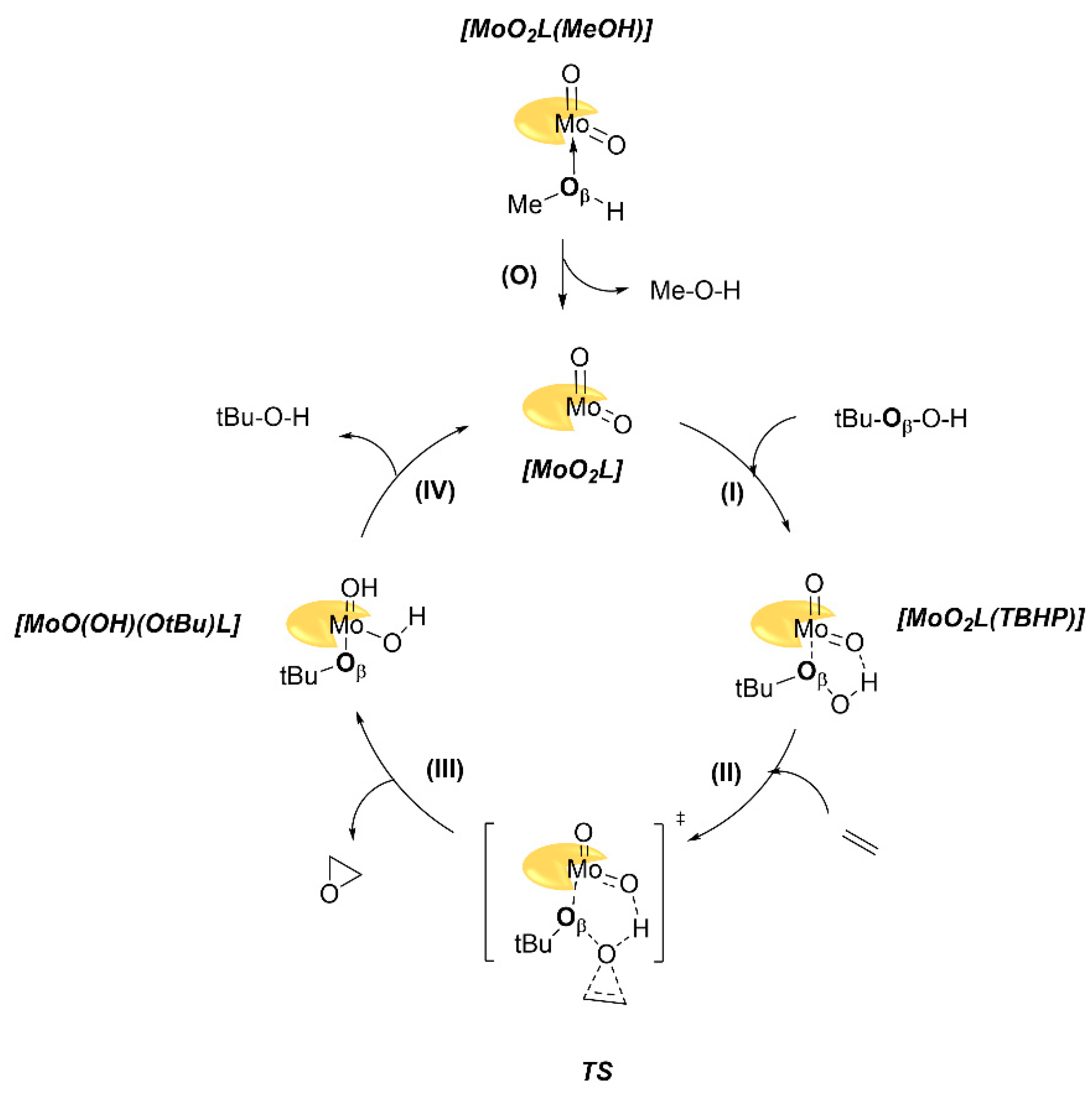
2.3. Theoretical Considerations
3. Materials and Methods
3.1. Synthesis of Ligands
3.2. Synthesis of Molybdenum(VI) Complexes
3.2.1. Oligomeric Complexes (Obtained from DCM)
3.2.2. Monomeric Complexes (Obtained from MeOH)
3.3. General Procedure for the Epoxidation Of Cyclooctene by TBHP
3.4. Theoretical Calculations
3.5. Physical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Note
- Swern, D. (Ed.) Organic Peroxide; Wiley-Interscience: New York, NY, USA, 1971; Volume 2, p. 265. [Google Scholar]
- Sheng, M.N.; Cherry Hill, N.J.; Zajaczek, G.J. Molybdenum-containing catalyst and method for the preparation thereof Atlantic Richfield Co. Patent US 3434975A, 25 May 1969. [Google Scholar]
- Bregeault, J.M. Transition-metal complexes for liquid-phase catalytic oxidation: Some aspects of industrial reactions and of emerging technologies. Dalton Trans. 2003, 17, 3289. [Google Scholar] [CrossRef]
- Sheldon, R.A. Molybdenum-catalysed epoxidation of olefins with alkyl hydroperoxides I. Kinetic and product studies. Recl. Trav. Chim. Pays-Bas 1973, 92, 253–266. [Google Scholar] [CrossRef]
- Chong, A.O.; Sharpless, K.B. Mechanism of the molybdenum and vanadium catalyzed epoxidation of olefins by alkyl hydroperoxides. J. Org. Chem. 1977, 42, 1587–1590. [Google Scholar] [CrossRef]
- Bhattacharya, P.K. Homogeneous catalysis of epoxidation of olefins by mono- and binuclear Schiff base complexes. J. Chem. Sci. 2008, 102, 247–250. [Google Scholar]
- Judmaier, M.E.; Holzer, C.; Volpe, M.; Mösch-Zanetti, N.C. Molybdenum(VI) Dioxo and Oxo-Imido Complexes of Fluorinated β-Ketiminato Ligands and Their Use in OAT Reactions. Inorg. Chem. 2012, 51, 9956–9966. [Google Scholar] [CrossRef]
- Liang, Q.; Newman, P.A.; Daniel, J.S.; Reimann, S.; Hall, B.D.; Dutton, G.; Kuijpers, L.J.M. Constraining the carbon tetrachloride (CCl4) budget using its global trend and inter-hemispheric gradient. Geophys. Res. Lett. 2014, 41, 5307–5315. [Google Scholar] [CrossRef] [Green Version]
- Hossaini, R.; Chipperfield, M.P.; Montzka, S.A.; Rap, A.; Dhomse, S.; Feng, W. Efficiency of short-lived halogens at influencing climate through depletion of stratospheric ozone. Nat. Geosci. 2015, 8, 186–190. [Google Scholar] [CrossRef] [Green Version]
- Byrne, F.P.; Jin, S.; Paggiola, G.; Petchey, T.H.M.; Clark, J.H.; Farmer, T.J.; Hunt, A.J.; McElroy, C.R.; Sherwood, J. Tools and techniques for solvent selection: Green solvent selection guides. Sustain. Chem. Process. 2016, 4, 7. [Google Scholar] [CrossRef] [Green Version]
- Vrdoljak, V.; Pisk, J.; Agustin, D.; Novak, P.; Parlov Vuković, J.; Matković-Čalogović, D. Dioxomolybdenum(VI) and dioxotungsten(VI) complexes chelated with the ONO tridentate hydrazone ligand: Synthesis, structure and catalytic epoxidation activity. New J. Chem. 2014, 38, 6176–6185. [Google Scholar] [CrossRef]
- Cvijanović, D.; Pisk, J.; Pavlović, G.; Šišak-Jung, D.; Matković-Čalogović, D.; Cindrić, M.; Agustin, D.; Vrdoljak, V. Discrete monomeric and dinuclear compounds containing a MoO22+ core and 4-aminobenzhydrazone ligands: Synthesis, structure and organic-solvent-free epoxidation activity. New J. Chem. 2019, 43, 1791–1802. [Google Scholar] [CrossRef]
- Pisk, J.; Rubčić, M.; Kuzman, D.; Cindrić, M.; Agustin, D.; Vrdoljak, V. Molybdenum(VI) complexes of hemilabile aroylhydrazone ligands as efficient catalysts for greener cyclooctene epoxidation: An experimental and theoretical approach. New J. Chem. 2019, 43, 5531–5542. [Google Scholar] [CrossRef]
- Cindrić, M.; Vrdoljak, V.; Strukan, N.; Kamenar, B. Synthesis and characterization of some mono- and dinuclear molybdenum(VI) thiosemicarbazonato complexes. Polyhedron 2005, 24, 369–376. [Google Scholar] [CrossRef]
- Vrdoljak, V.; Cindrić, M.; Milić, D.; Matković-Čalogović, D.; Novak, P.; Kamenar, B. Synthesis of five new molybdenum(VI) thiosemicarbazonato complexes. Crystal structures of salicylaldehyde and 3-methoxy-salicylaldehyde 4-methylthiosemicarbazones and their molybdenum(VI) complexes. Polyhedron 2005, 24, 1717–1726. [Google Scholar] [CrossRef]
- Rao, S.N.; Munshi, K.N.; Rao, N.N.; Bhadbhadak, M.M.; Suresh, E. Synthesis, spectral and X-ray structural characterization of [cis-MoO2(L)(solv)] (L=salicylidene salicyloyl hydrazine) and its use as catalytic oxidant. Polyhedron 1999, 18, 2491–2497. [Google Scholar] [CrossRef]
- Jin, N.-Y. Syntheses, crystal structures, and catalytic properties of dioxomolybdenum(VI) complexes with hydrazone ligands. J. Coord. Chem. 2012, 65, 4013–4022. [Google Scholar] [CrossRef]
- Jia, X.-C.; Li, J.; Liu, Y.-Y.; Wang, N.; Zhang, H.Z. Kristallogr. New Cryst. Struct. 2013, 228, 179. [Google Scholar]
- Wang, W.; Daran, J.-C.; Poli, R.; Agustin, D. OH-substituted tridentate ONO Schiff base ligands and related molybdenum(VI) complexes for solvent-free (ep)oxidation catalysis with TBHP as oxidant. J. Mol. Catal. A Chem 2016, 416, 117–126. [Google Scholar] [CrossRef]
- Wang, W.; Guerrero, T.; Merecias, S.R.; García-Ortega, H.; Santillan, R.; Daran, J.-C.; Farfán, N.; Agustin, D.; Poli, R. Substituent effects on solvent-free epoxidation catalyzed by dioxomolybdenum(VI) complexes supported by ONO Schiff base ligands. Inorganica Chim. Acta 2015, 431, 176–183. [Google Scholar] [CrossRef]
- Cindrić, M.; Pavlović, G.; Katava, R.; Agustin, D. Towards a global greener process: From solvent-less synthesis of molybdenum(VI) ONO Schiff base complexes to catalyzed olefin epoxidation under organic-solvent-free conditions. New J. Chem. 2017, 41, 594–602. [Google Scholar] [CrossRef]
- Chen, G.J.-J.; McDonald, J.W.; Newton, W.E. Synthesis of molybdenum(IV) and molybdenum(V) complexes using oxo abstraction by phosphines. Mechanistic implications. Inorg. Chem. 1976, 15, 2612–2615. [Google Scholar] [CrossRef]
- Yu, S.-B.; Holm, R.H. Aspects of the oxygen atom transfer chemistry of tungsten. Inorg. Chem. 1989, 28, 4385–4391. [Google Scholar] [CrossRef]
- Lu, J.-F. 4-Hydr oxy-N′-(2-hydroxy-3-methoxybenzylidene)benzohydrazide monohydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2008, 64, o2032. [Google Scholar] [CrossRef] [Green Version]
- Rassem, H.-H.; Salhin, A.; Bin Salleh, B.; Rosli, M.M.; Fun, H.-K. (E)-4-Hydroxy-N′-(2-hydroxy-4-methoxybenzylidene) benzohydrazide. Acta Crystallogr. Sect. E Struct. Rep. Online 2012, 68, o2279. [Google Scholar] [CrossRef] [Green Version]
- Salhin, A.; Tameem, A.A.; Saad, B.; Ng, S.-L.; Fun, H.-K. 4-Hydroxy-N′-[(E)-(2-hydroxyphenyl)methylidene]benzohydrazide Acta Crystallogr. Sect. E Struct. Rep. Online 2007, 63, o2880. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J.A.; Peralta, J.E.; Ogliaro, F.; Bearpark, M.; Heyd, J.J.; Brothers, E.; Kudin, K.N.; Staroverov, V.N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J.C.; Iyengar, S.S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J.M.; Klene, M.; Knox, J.E.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Martin, R.L.; Morokuma, K.; Zakrzewski, V.G.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Dapprich, S.; Daniels, A.D.; Farkas, O.; Foresman, J.B.; Ortiz, J.V.; Cioslowski, J.; Fox, D.J. Gaussian, Inc, Wallingford CT (2013).
- Becke, D. Density-functional thermochemistry. III. The role of exact exchange. J.Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.; Ditchfield, R.; Pople, J. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. Accuracy of AH equilibrium geometries by single determinant molecular-orbital theory. Mol. Phys. 1974, 27, 209–214. [Google Scholar] [CrossRef]
- Stevens, W.J.; Basc, H.; Krauss, M. Compact effective potentials and efficient shared-exponent basis sets for the first- and second-row atoms. J. Chem. Phys. 1984, 81, 6026–6033. [Google Scholar] [CrossRef]
- Stevens, W.J.; Krauss, M.; Basch, H.; Jasien, P.G. Relativistic compact effective potentials and efficient, shared-exponent basis sets for the third-, fourth-, and fifth-row atoms. Can. J. Chem. 1992, 70, 612–630. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlisPro. Version 1.171.39.46e; Rigaku Oxford Diffraction: The Woodlands, TX, USA, 2018. [Google Scholar]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H.J. OLEX2: A complete structure solution, refinement and analysis program. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Cryst. 2016, B72, 171. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ONO Plane × Mo Distance/Å | Aryl Interplanar Angle/° |
|---|---|---|
| [MoO2(LH)(MeOH)] | 0.334 | 6.59 |
| [MoO2(L3OMe)(H2O)] | 0.277 | 3.80 |
| [MoO2(L3OMe)(MeOH)] | 0.282 | 20.92 |
| [MoO2(L4OMe)(MeOH)]·MeOH | 0.270 | 2.98 |
| [MoO2(L4OMe)(MeOH)]·4,4-bpy | 0.328 | 9.31 |
| Catalyst | Conversion a /% | Selectivity b /% | TOF20min c | TON d | ||||
|---|---|---|---|---|---|---|---|---|
| TBHPaq | TBHPdec | TBHPaq | TBHPdec | TBHPaq | TBHPdec | TBHPaq | TBHPdec | |
| [MoO2(LH)]n | 90 | 99 | 88 | 91 | 290 | 290 | 361 | 397 |
| [MoO2(LH)(MeOH)] | 92 | 98 | 90 | 90 | 274 | 290 | 368 | 397 |
| [MoO2(L3OMe)]n | 96 | 99 | 93 | 90 | 391 | 625 | 386 | 399 |
| [MoO2(L3OMe)[MeOH)] | 96 | 99 | 95 | 93 | 496 | 796 | 400 | 400 |
| [MoO2(L4OMe)]n | 94 | 99 | 92 | 93 | 319 | 298 | 373 | 399 |
| [MoO2(L4OMe)(MeOH)] | 99 | 99 | 85 | 92 | 326 | 214 | 351 | 400 |
| Absolute Values | Relative to MoO2(L)(MeOH) | ||||||
|---|---|---|---|---|---|---|---|
| Steps | MoO2(LH) | MoO2(L3OMe) | MoO2(L4Ome) | MoO2(LH) | MoO2(L3OMe) | MoO2(L4OMe) | |
| (0) | MeOH decoordination | +9.7 | +9.5 | +9.4 | +9.7 | +9.5 | +9.4 |
| (I) | TBHP coordination | −8.7 | −8.9 | −8.4 | +1.0 | +0.6 | +0.9 |
| (II) | TS barrier | +23.0 | +23.2 | +23.5 | +24.0 | +23.8 | +24.4 |
| (III) | Release of epoxide | −51.4 | −51.7 | −51.7 | −27.3 | −27.9 | −27.3 |
| (IV) | Regeneration of catalyst | −4.3 | −4.1 | −4.7 | −31.7 | −31.9 | −32.0 |
| [MoO2L(MeOH)] | [MoO2(L)] | [MoO2(L)(TBHP)] | TS | ||
|---|---|---|---|---|---|
| Interatomic distances | |||||
| [Mo–Oβ] | LH | 2.498 | - | 2.970 | 2.385 |
| L3OMe | 2.498 | - | 2.975 | 2.388 | |
| L4OMe | 2.507 | - | 3.000 | 2.398 | |
| N2…H | LH | 1.763 | 1.781 | 1.776 | 1.755 |
| L3OMe | 1.761 | 1.779 | 1.775 | 1.753 | |
| L4OMe | 1.764 | 1.781 | 1.777 | 1.756 | |
| N1–Mo | LH | 2.295 | 2.272 | 2.268 | 2.278 |
| L3OMe | 2.297 | 2.275 | 2.271 | 2.281 | |
| L4OMe | 2.283 | 2.255 | 2.255 | 2.266 | |
| Mo=-O1 (±plane) | LH | 1.702 | 1.694 | 1.714 | 1.732 |
| L3OMe | 1.702 | 1.696 | 1.714 | 1.733 | |
| L4OMe | 1.703 | 1.697 | 1.715 | 1.733 | |
| Mo–O2(⊥plane) | LH | 1.694 | 1.695 | 1.690 | 1.692 |
| L3OMe | 1.695 | 1.695 | 1.691 | 1.693 | |
| L4OMe | 1.695 | 1.695 | 1.691 | 1.692 | |
| Interatomic angle | |||||
| OCNO dihedral angle | LH | −9.26 | −6.03 | −11.56 | −16.78 |
| L3OMe | −9.58 | −6.36 | −11.01 | −16.19 | |
| L4OMe | −9.15 | −5.73 | −11.34 | −16.70 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mrkonja, S.; Topić, E.; Mandarić, M.; Agustin, D.; Pisk, J. Efficient Molybdenum Hydrazonato Epoxidation Catalysts Operating under Green Chemistry Conditions: Water vs. Decane Competition. Catalysts 2021, 11, 756. https://doi.org/10.3390/catal11070756
Mrkonja S, Topić E, Mandarić M, Agustin D, Pisk J. Efficient Molybdenum Hydrazonato Epoxidation Catalysts Operating under Green Chemistry Conditions: Water vs. Decane Competition. Catalysts. 2021; 11(7):756. https://doi.org/10.3390/catal11070756
Chicago/Turabian StyleMrkonja, Silvija, Edi Topić, Mirna Mandarić, Dominique Agustin, and Jana Pisk. 2021. "Efficient Molybdenum Hydrazonato Epoxidation Catalysts Operating under Green Chemistry Conditions: Water vs. Decane Competition" Catalysts 11, no. 7: 756. https://doi.org/10.3390/catal11070756
APA StyleMrkonja, S., Topić, E., Mandarić, M., Agustin, D., & Pisk, J. (2021). Efficient Molybdenum Hydrazonato Epoxidation Catalysts Operating under Green Chemistry Conditions: Water vs. Decane Competition. Catalysts, 11(7), 756. https://doi.org/10.3390/catal11070756