
Deactivation and Regeneration of Zeolite Catalysts Used in Pyrolysis of Plastic Wastes—A Process and Analytical Review



Abstract
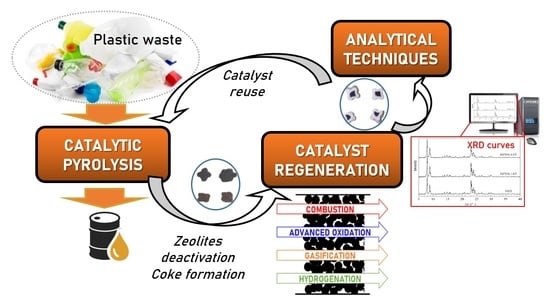
:
1. Introduction
2. Pyrolysis Processes for Plastic Recycling
2.1. Plastic Waste and Recycling Methods
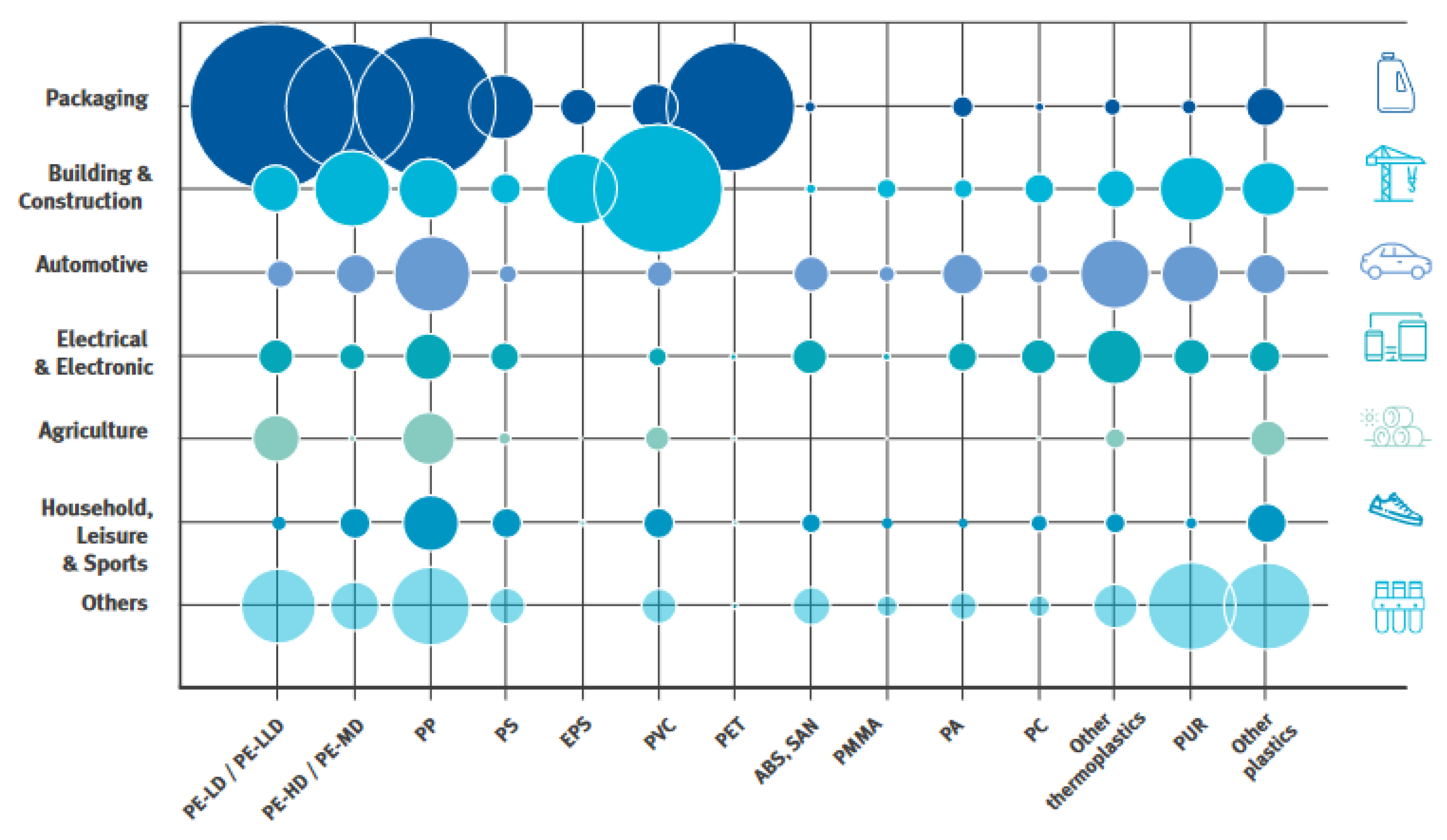
2.1.1. Plastic Production and Generated Waste
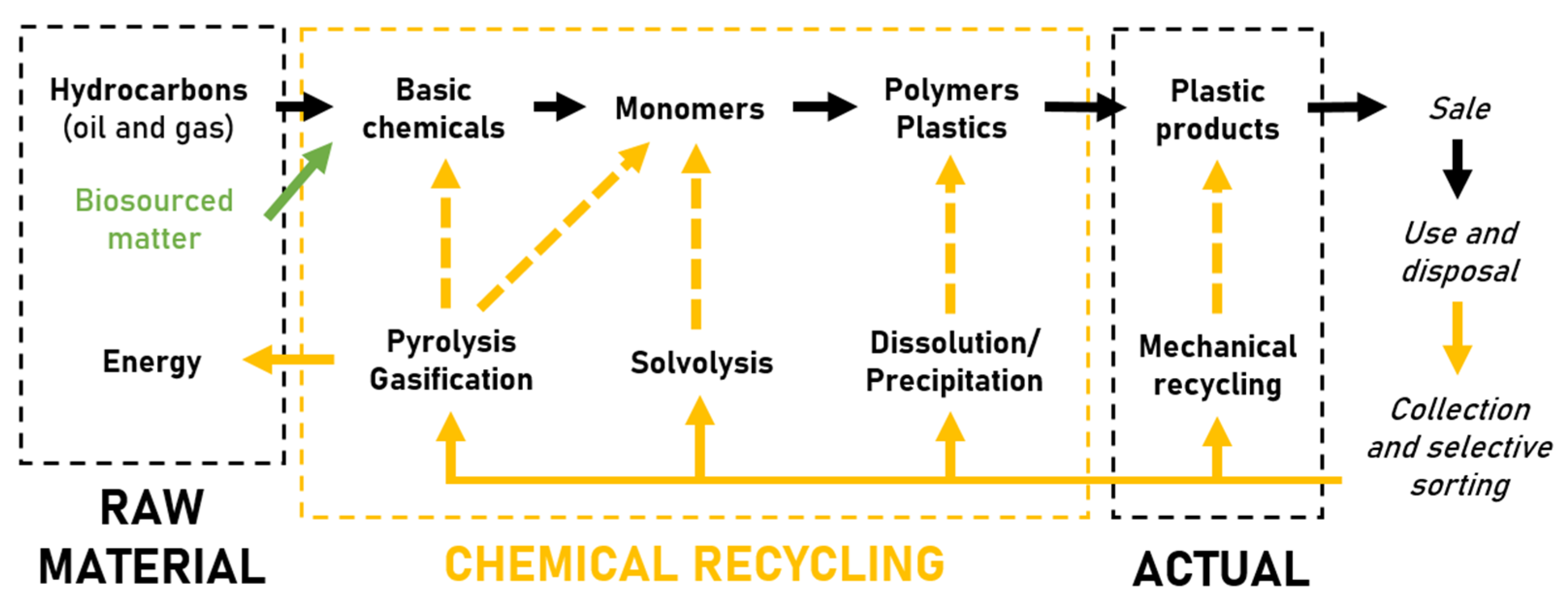
2.1.2. Recycling Methods
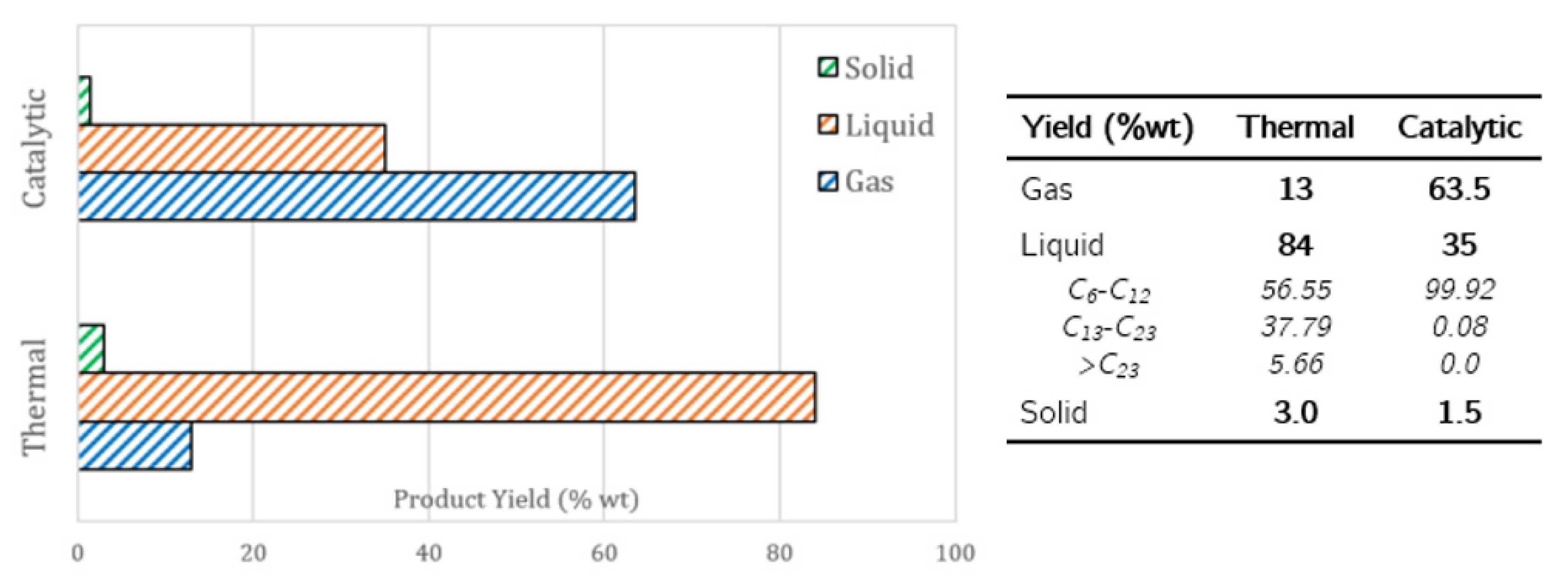
2.2. Catalytic Pyrolysis of Plastic Waste
2.2.1. Zeolite Materials
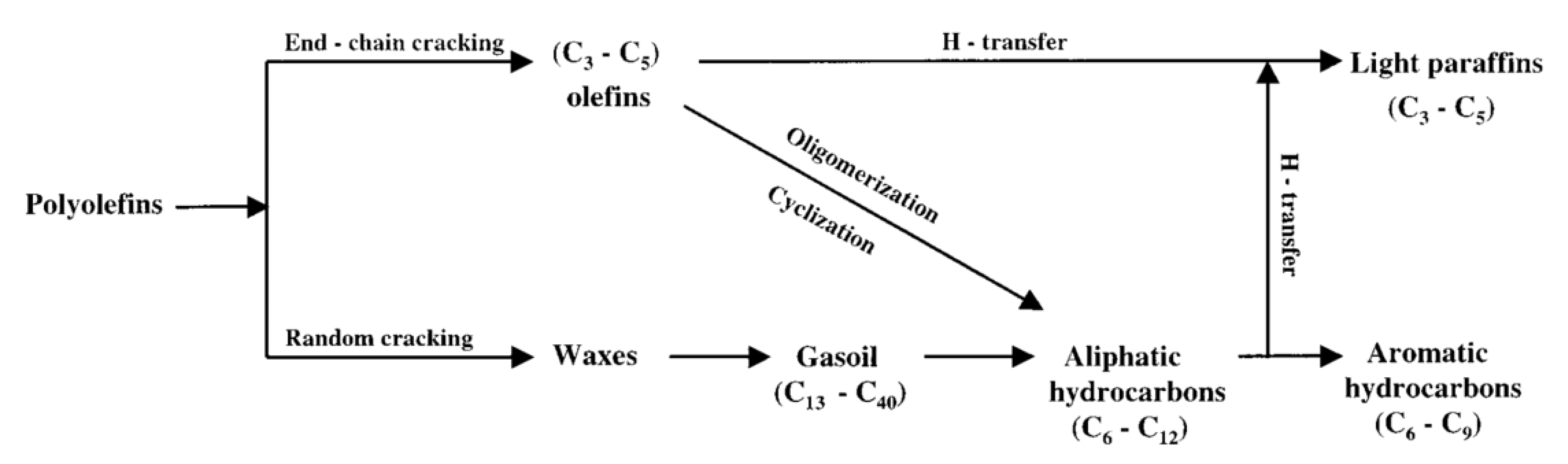
2.2.2. Pyrolysis Reaction Mechanisms
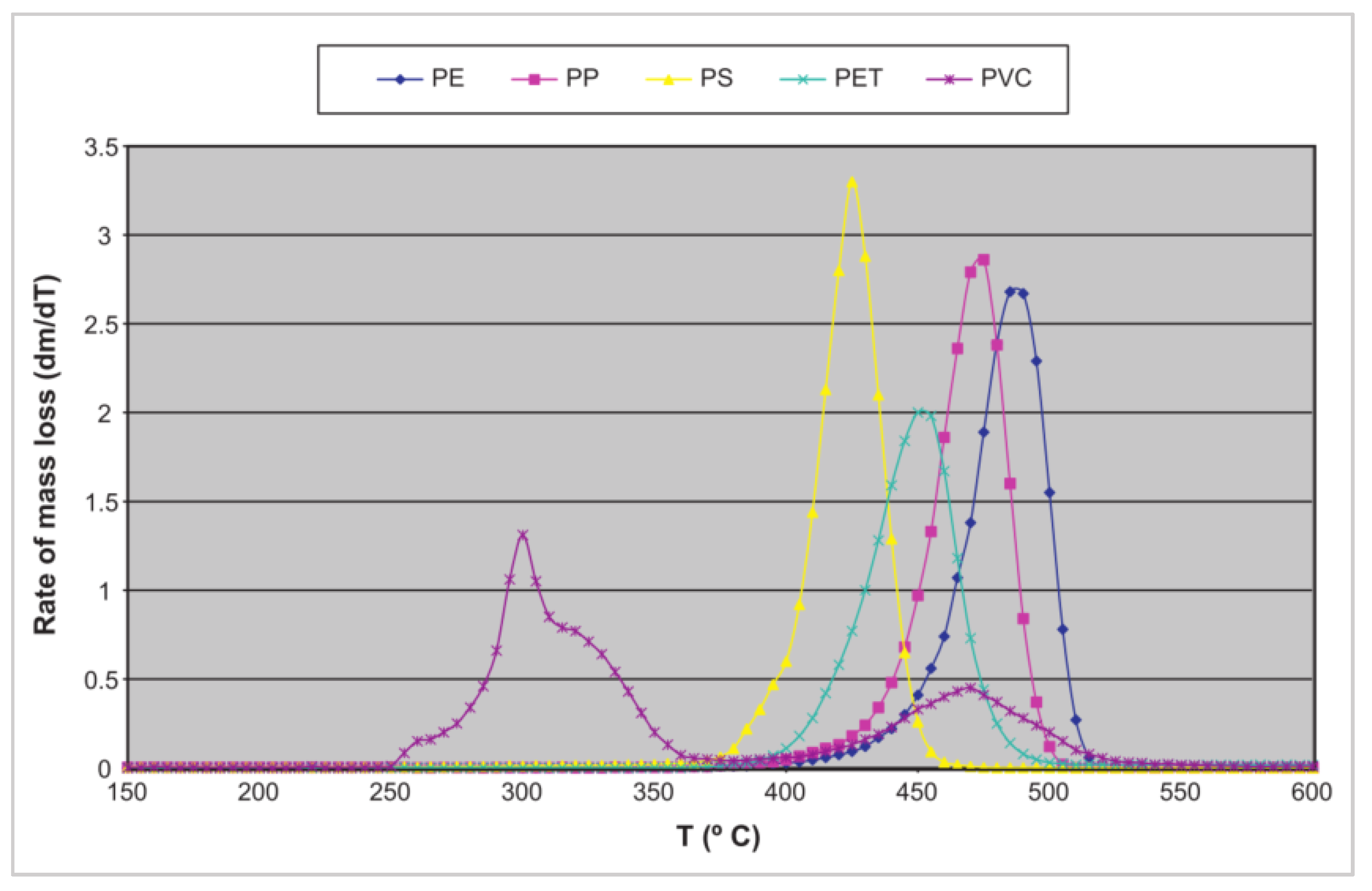
2.2.3. Influencing Factors
- Operating parameters
- Reactor type
2.3. Deactivation of Zeolite Catalysts
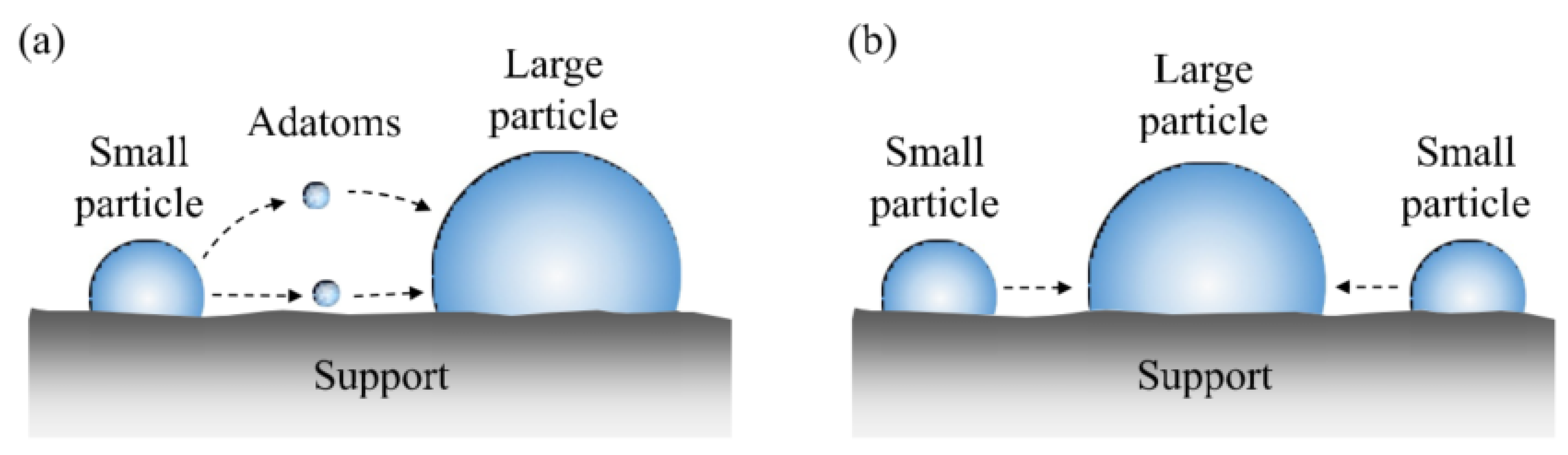
2.3.1. Generalities
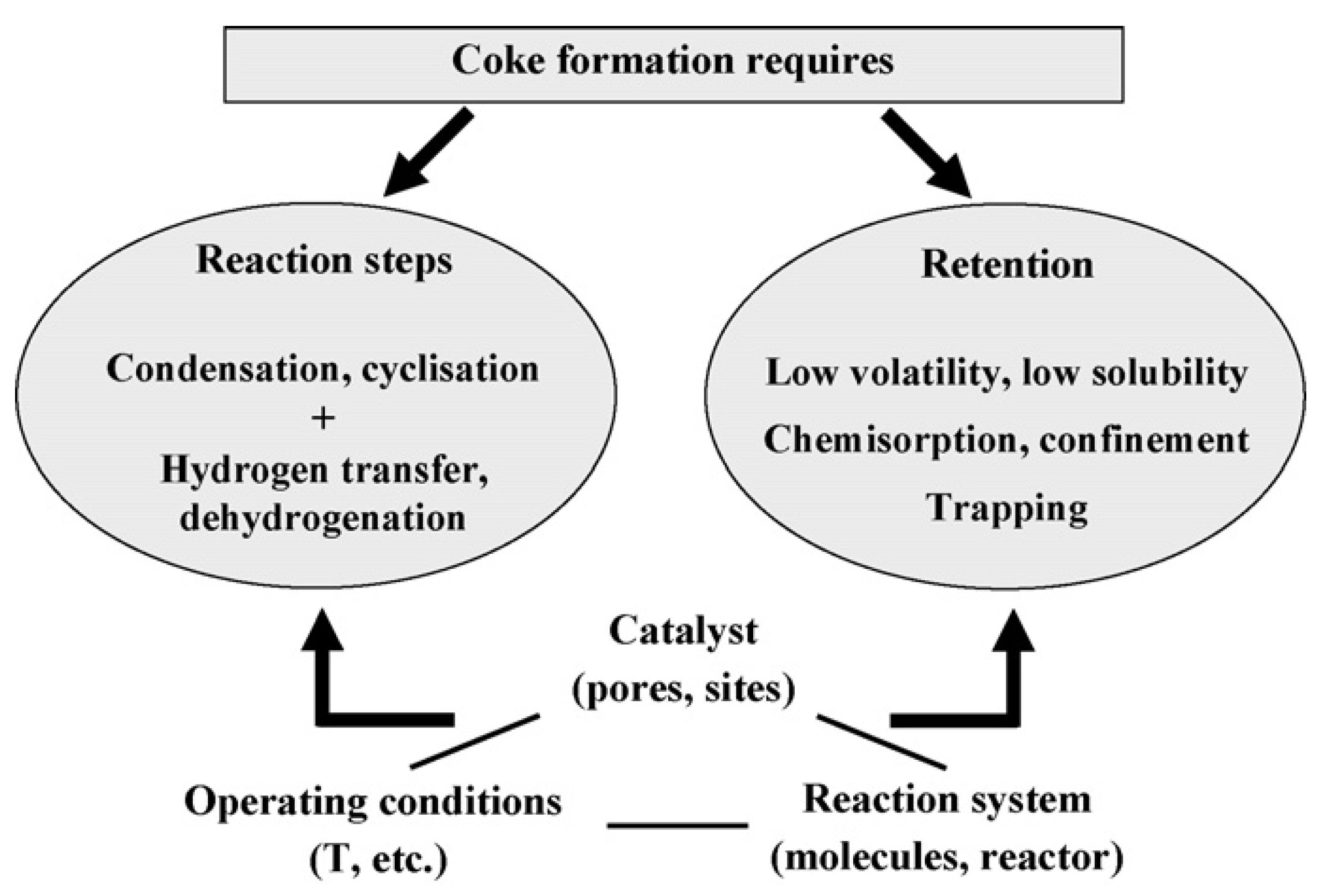
2.3.2. Catalyst Deactivation by Coking
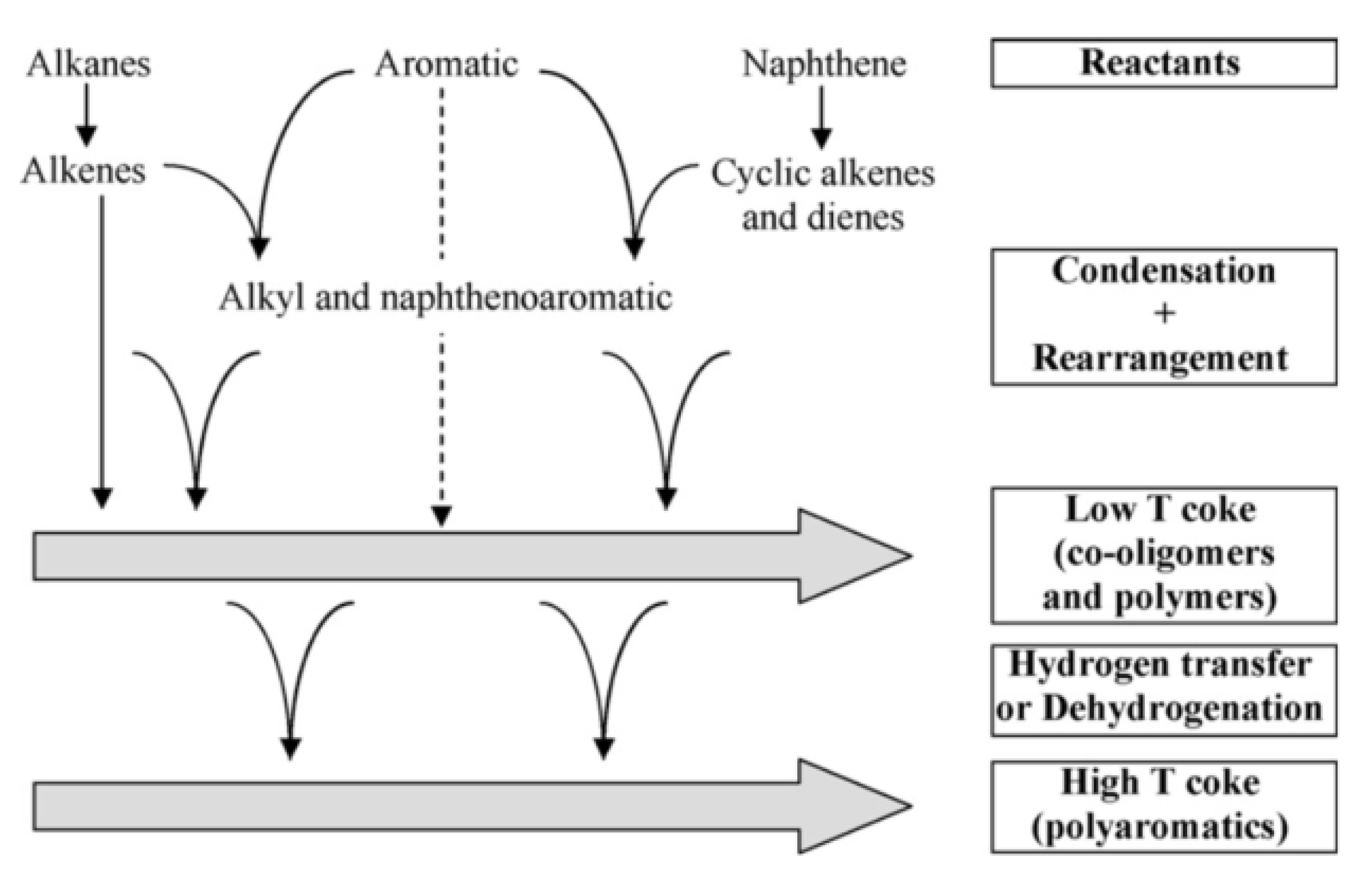
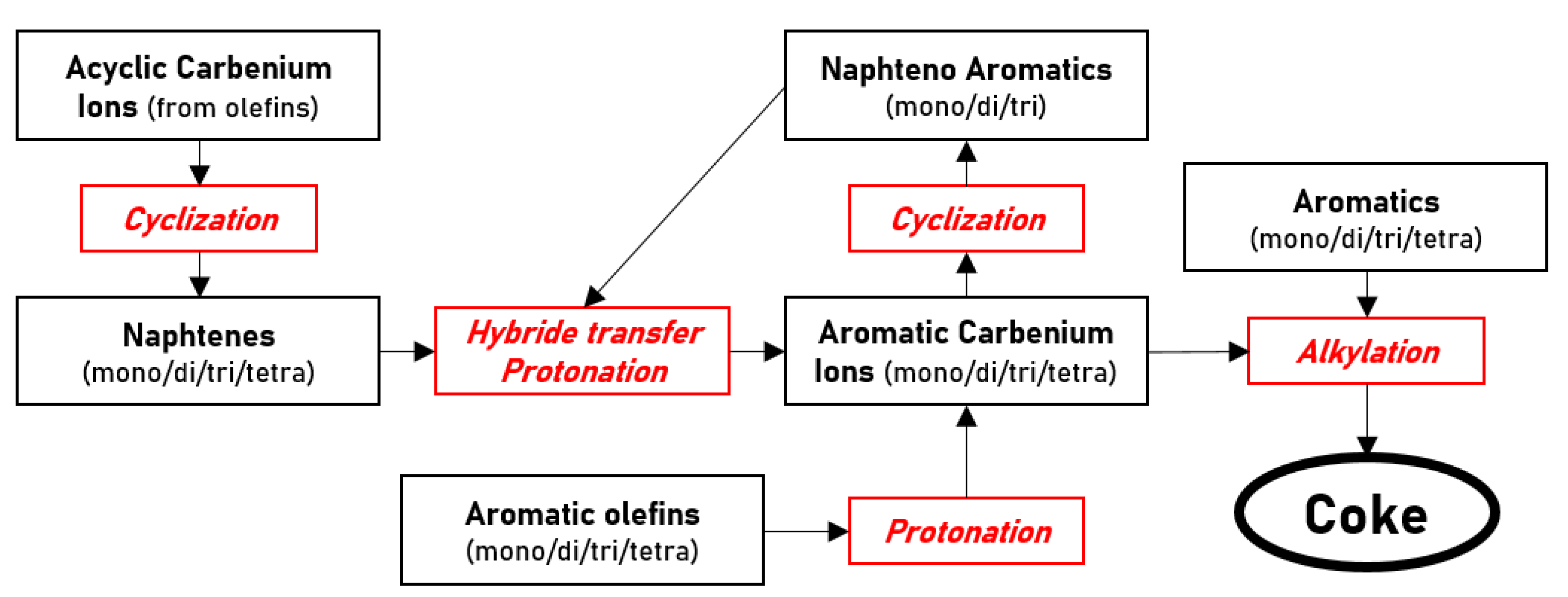
- Coke formation mechanisms
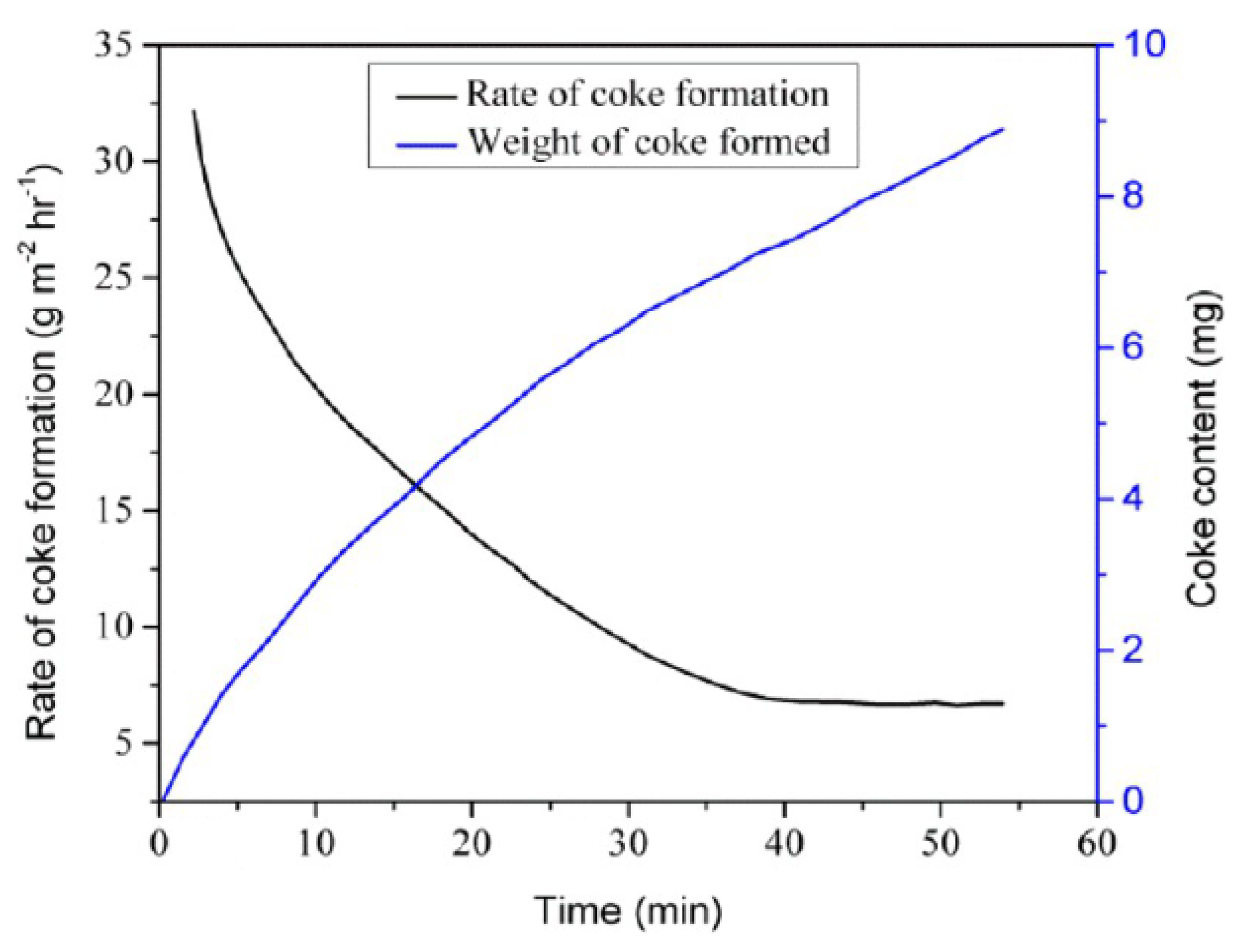
- Kinetics of coking and deactivation
2.3.3. Parameters Influencing Coke Formation
- Reaction system
- Operating conditions
- Catalyst structure and surface
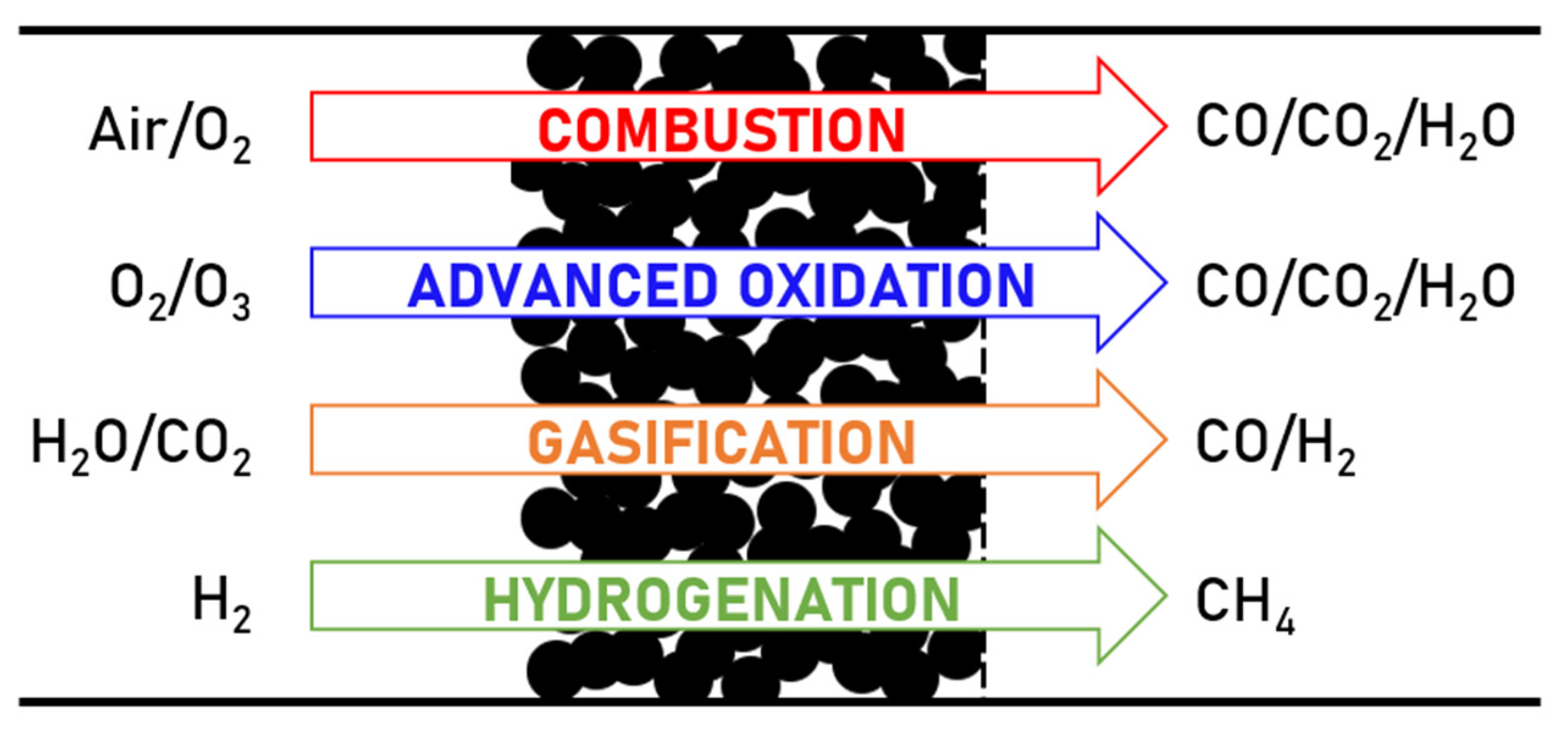
3. Regeneration of Zeolite Catalysts Deactivated by Coke
3.1. Coke Combustion with Air/Oxygen
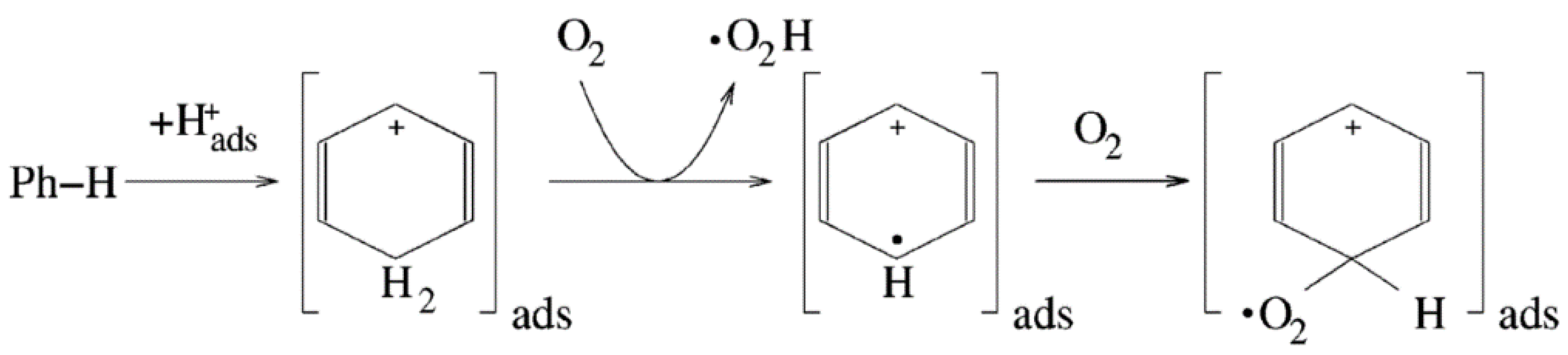
3.1.1. Oxidation Mechanisms
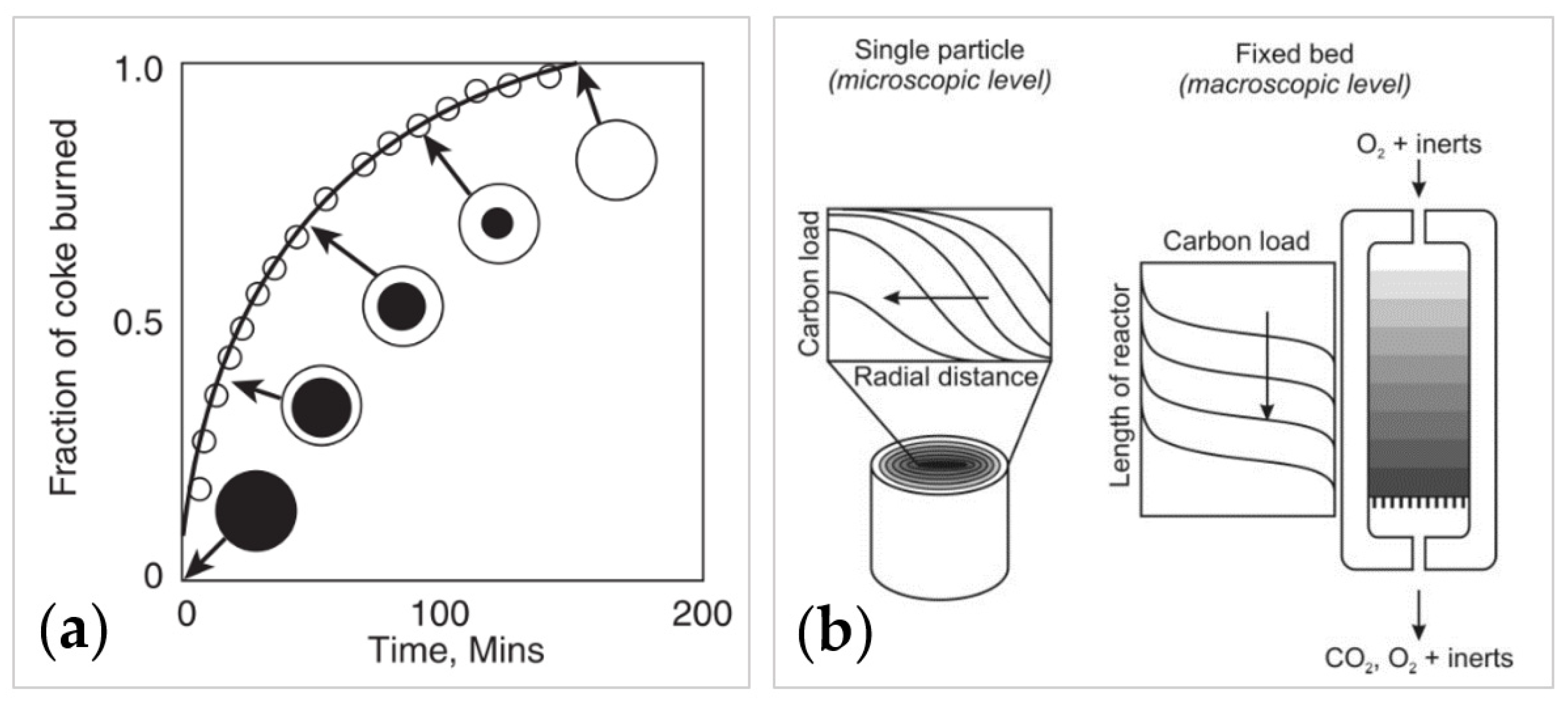
3.1.2. Reaction Model: Kinetics and Mass Transfer
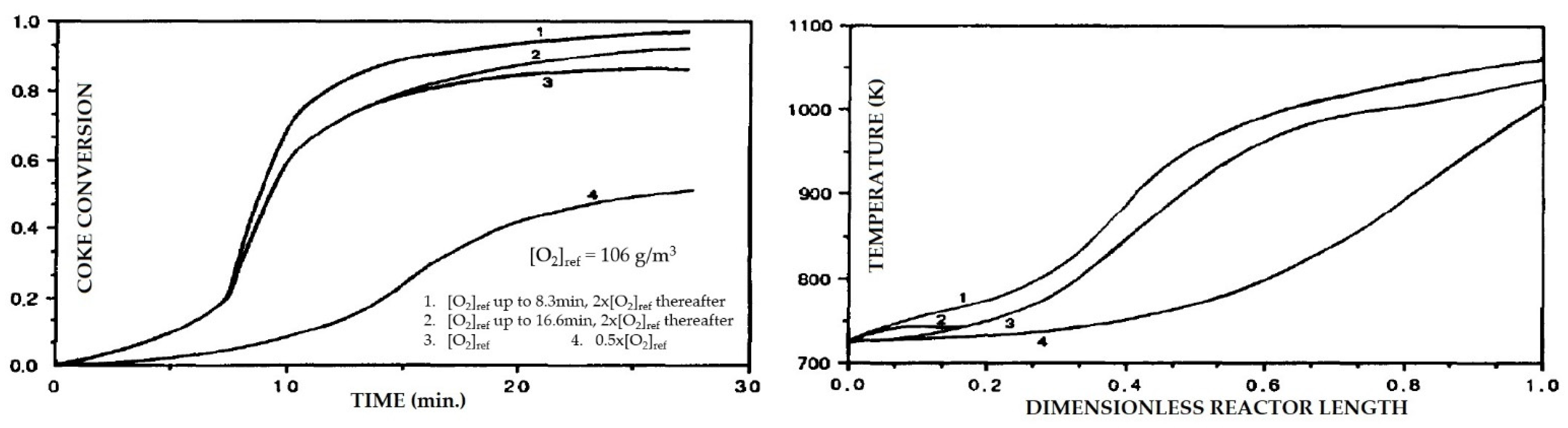
- Kinetics
- Diffusion and mass transfer
3.1.3. Parameters Influencing Coke Formation
- Catalyst structure and composition
- Operating parameters
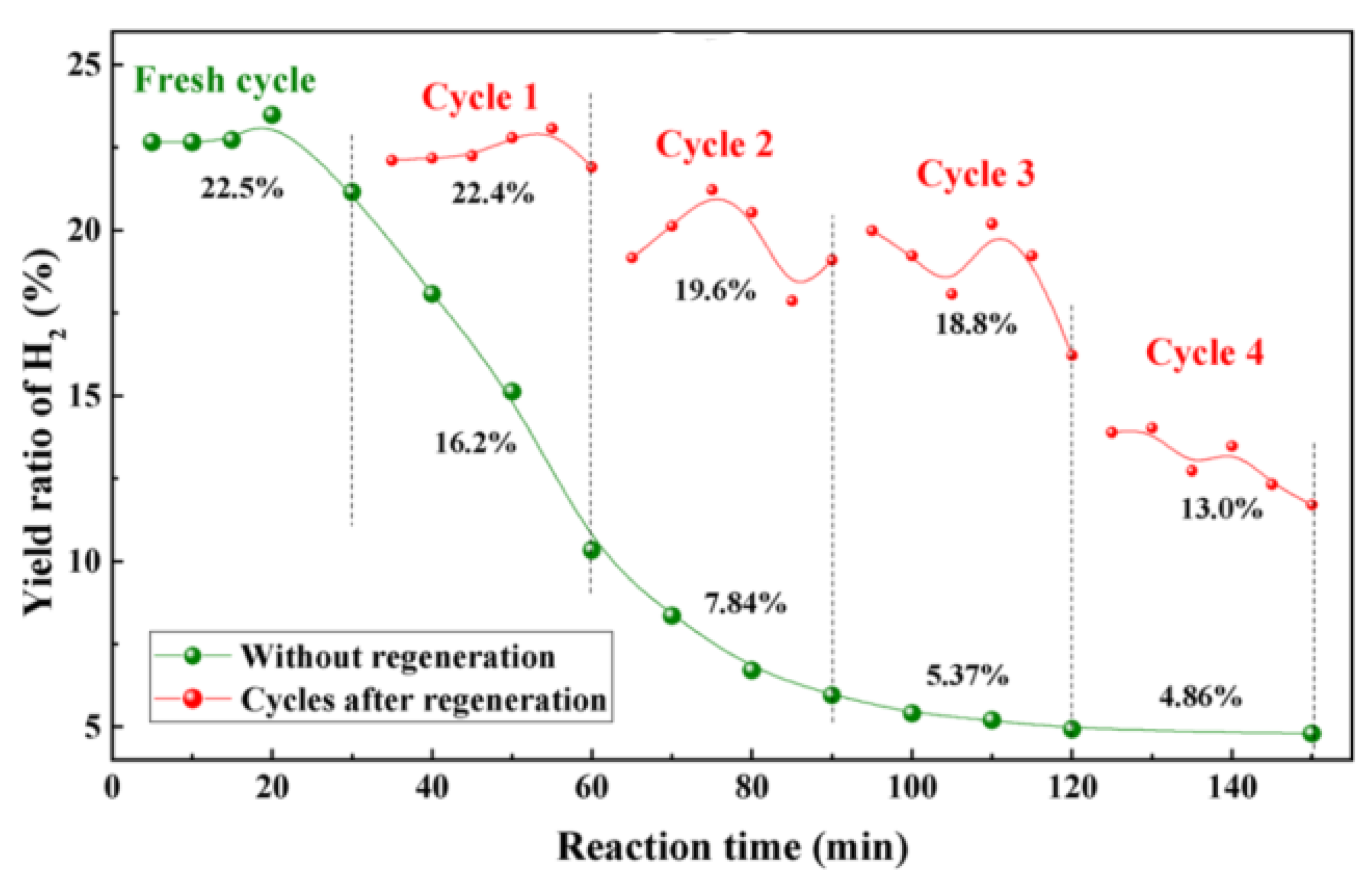
3.1.4. Limitations of Oxygen Oxidation
3.2. Advanced Oxidation Processes (AOP)
3.2.1. Ozonation
3.2.2. Other Methods for Advanced Oxidation
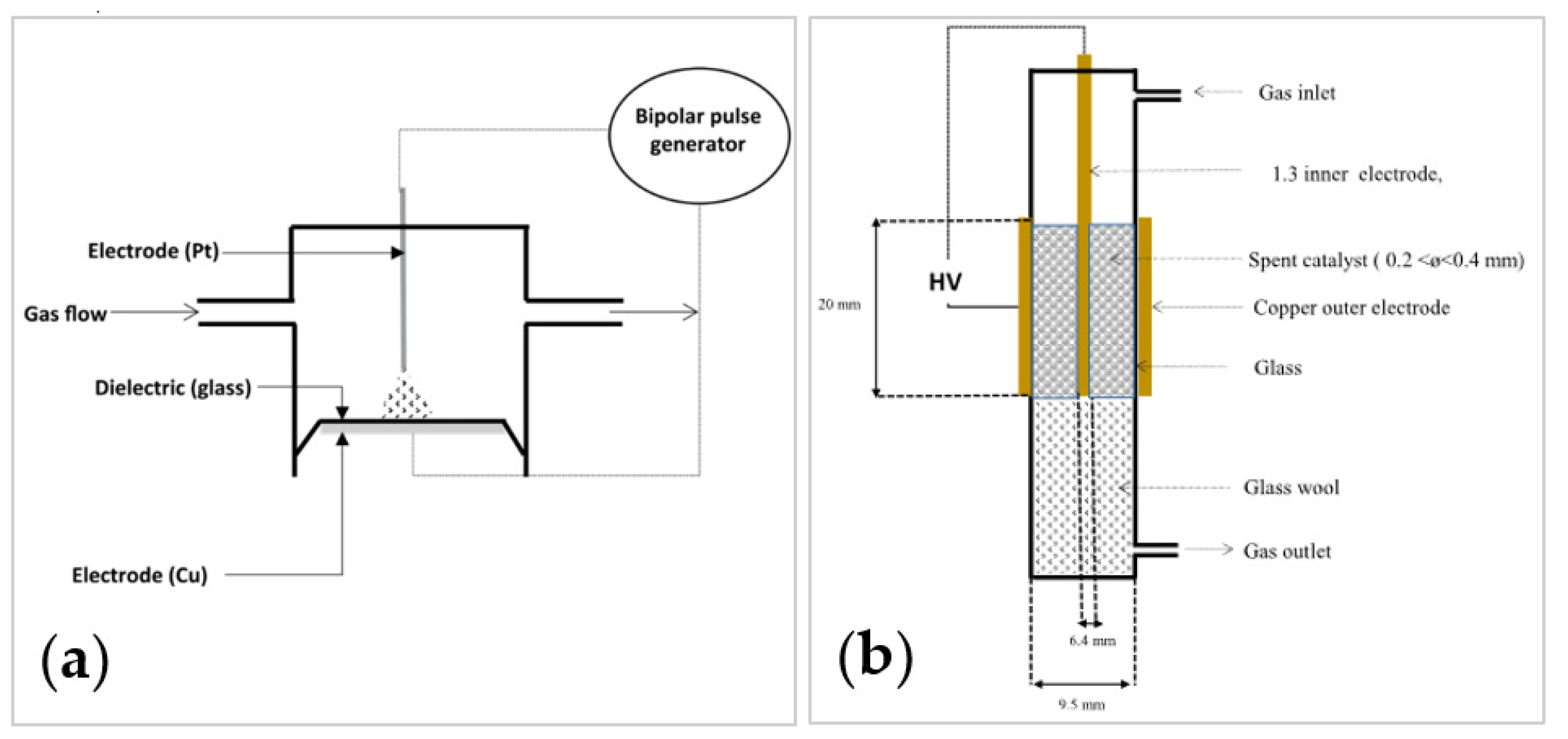
- Non-Thermal Plasma (NTP)
- Hydrogen peroxide and OH-derived Fenton radicals
3.3. Other Regenerating Reactions
3.3.1. Gasification
3.3.2. Hydrogenation
4. Analytical Techniques
4.1. Catalyst Characterization
4.1.1. Structural and Physical Properties
4.1.2. Chemical Properties and Reactivity
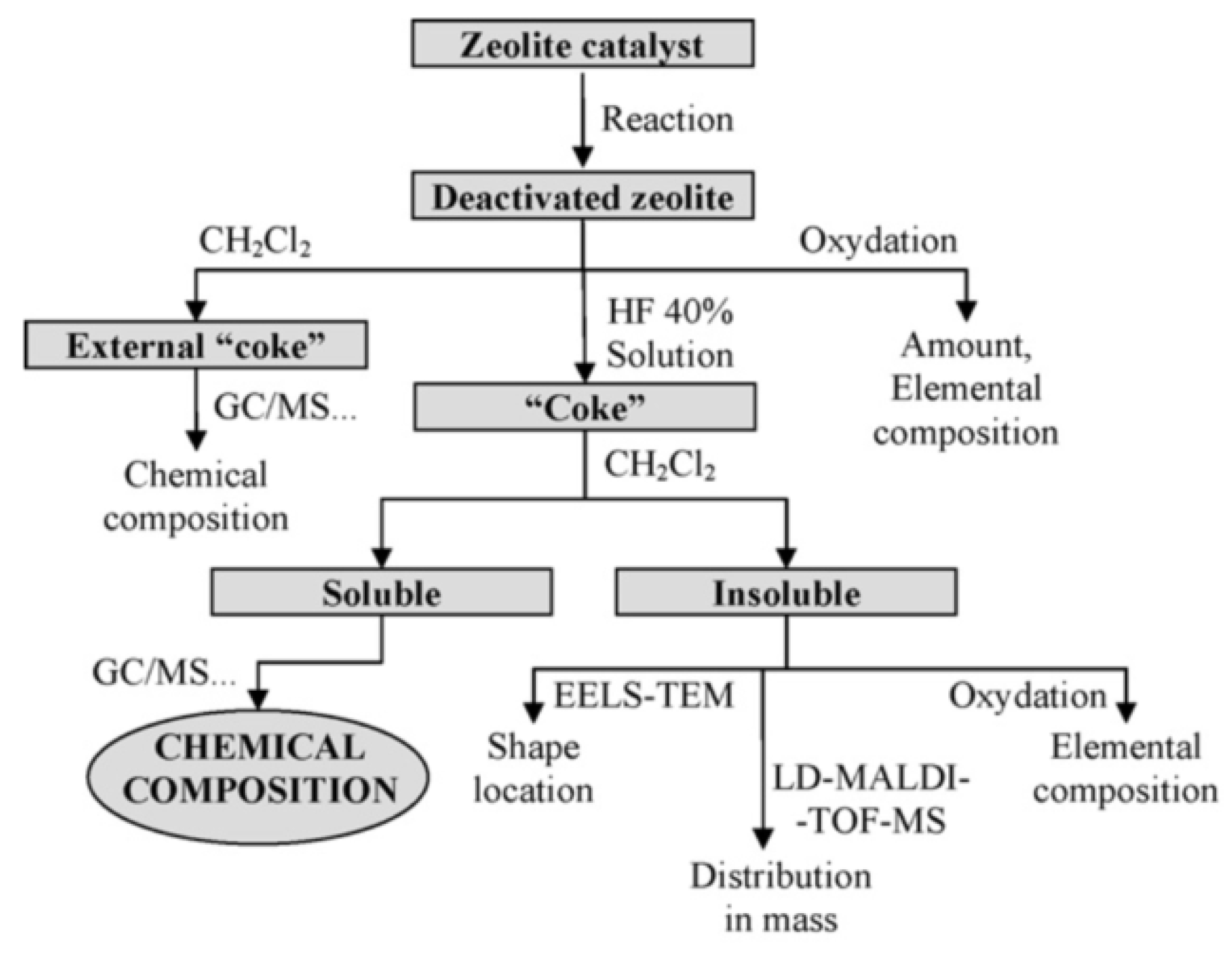
4.2. Coke Analysis
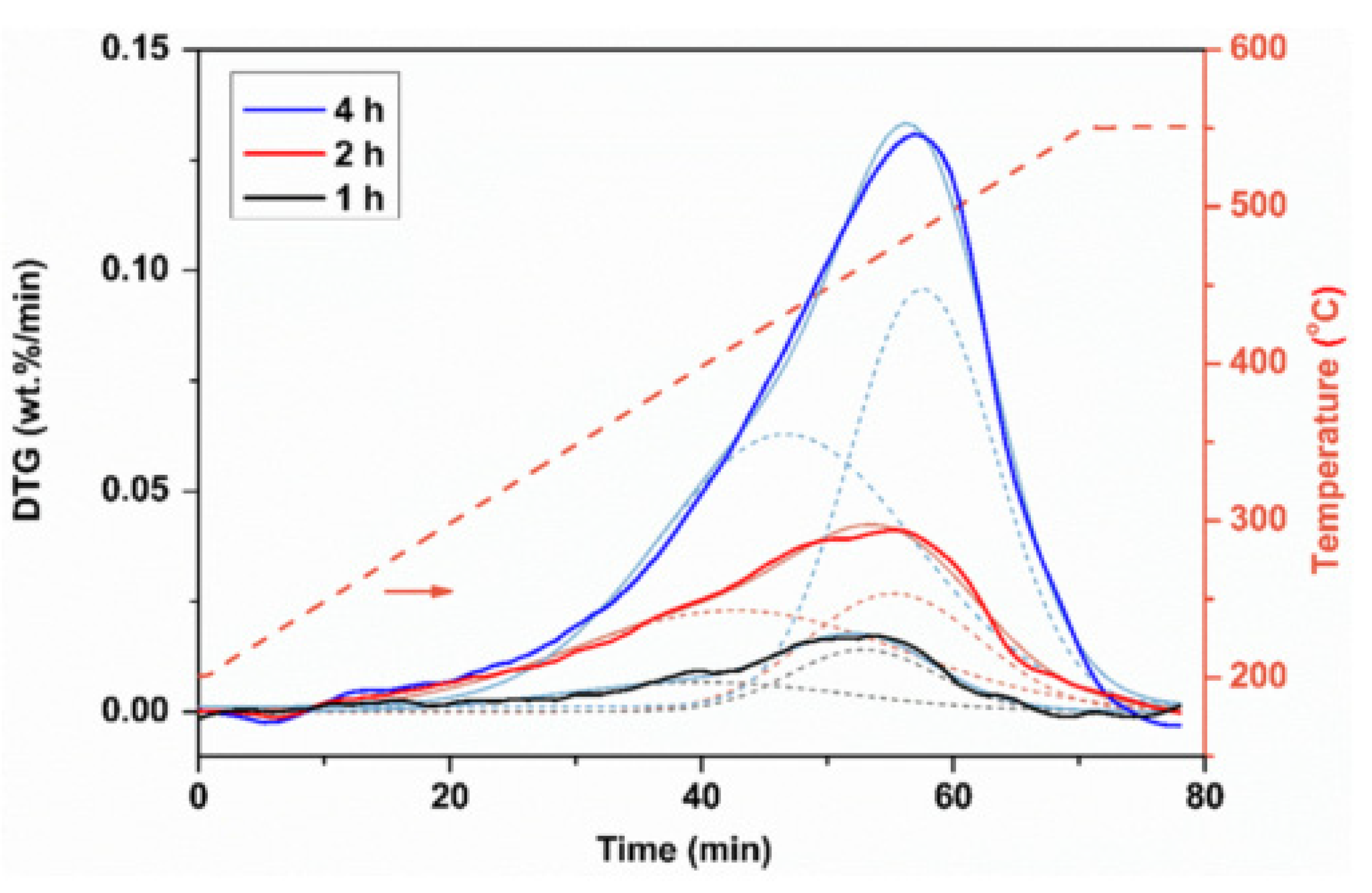
4.2.1. Nature and Composition
4.2.2. Reactivity and Kinetics
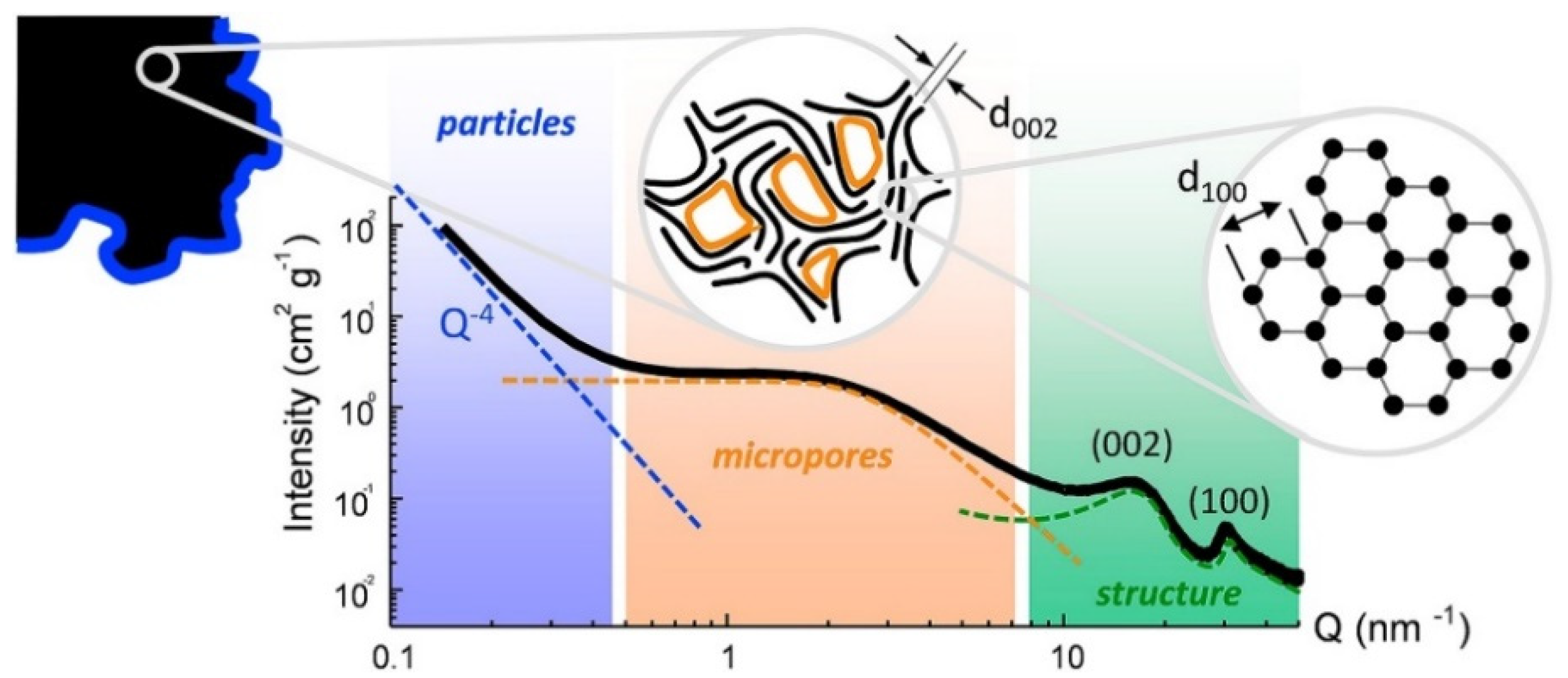
4.3. Particular Focus: Small Angle X-ray Scattering (SAXS)
5. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Anuar Sharuddin, S.D.; Abnisa, F.; Wan Daud, W.M.A.; Aroua, M.K. A review on pyrolysis of plastic wastes. Energy Convers. Manag. 2016, 115, 308–326. [Google Scholar] [CrossRef]
- Baensch-Baltruschat, B.; Brennholt, N.; Kochleus, C.; Reifferscheid, G.; Koschorreck, J. Conference on Plastics in Freshwater Environments. 2017. Available online: https://www.umweltbundesamt.de/sites/default/files/medien/376/publikationen (accessed on 24 June 2021).
- FutureAgenda. Plastic Oceans. Web Page. Available online: https://www.futureagenda.org/foresights/plastic-oceans/ (accessed on 27 May 2021).
- d’Ambrières, W. Plastics Recycling Worldwide: Current Overview and Desirable Changes. Field Actions Sci. Rep. 2019, 12–21. Available online: http://journals.openedition.org/factsreports/5102 (accessed on 23 June 2021).
- Ragaert, K.; Delva, L.; Van Geem, K. Mechanical and chemical recycling of solid plastic waste. Waste Manag. 2017, 69. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Liew, M.S. Tertiary recycling of plastics waste: An analysis of feedstock, chemical and biological degradation methods. J. Mater. Cycles Waste Manag. 2021, 23, 32–43. [Google Scholar] [CrossRef]
- Miandad, R.; Barakat, M.A.; Aburiazaiza, A.S.; Rehan, M.; Nizami, A.S. Catalytic pyrolysis of plastic waste: A review. Process. Saf. Environ. Prot. 2016, 102, 822–838. [Google Scholar] [CrossRef]
- Almeida, D.; Marques, M.d.F. Thermal and catalytic pyrolysis of plastic waste. Polímeros 2016, 26, 44–51. [Google Scholar] [CrossRef]
- Artetxe, M.; Lopez, G.; Amutio, M.; Elordi, G.; Bilbao, J.; Olazar, M. Cracking of High Density Polyethylene Pyrolysis Waxes on HZSM-5 Catalysts of Different Acidity. Ind. Eng. Chem. Res. 2013, 52, 10637–10645. [Google Scholar] [CrossRef]
- Mullen, C.A.; Dorado, C.; Boateng, A.A. Catalytic co-pyrolysis of switchgrass and polyethylene over HZSM-5: Catalyst deactivation and coke formation. J. Anal. Appl. Pyrolysis 2018, 129, 195–203. [Google Scholar] [CrossRef]
- Cocchi, M.; Angelis, D.D.; Mazzeo, L.; Nardozi, P.; Piemonte, V.; Tuffi, R.; Vecchio Ciprioti, S. Catalytic Pyrolysis of a Residual Plastic Waste Using Zeolites Produced by Coal Fly Ash. Catalysts 2020, 10, 1113. [Google Scholar] [CrossRef]
- Quesada, L.; Calero de Hoces, M.; Martín-Lara, M.A.; Luzón, G.; Blázquez, G. Performance of Different Catalysts for the In Situ Cracking of the Oil-Waxes Obtained by the Pyrolysis of Polyethylene Film Waste. Sustainability 2020, 12, 5482. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Ribeiro, F.R. Catalyst Deactivation. In Combinatorial Catalysis and High Throughput Catalyst Design and Testing; Derouane, E.G., Lemos, F., Corma, A., Ribeiro, F.R., Eds.; Springer: Dordrecht, The Netherlands, 2000; pp. 145–173. [Google Scholar] [CrossRef]
- Guisnet, M.; Ribeiro, F.R. Deactivation and Regeneration of Zeolite Catalysts; Imperial College Press: London, UK, 2011. [Google Scholar]
- Bartholomew. Catalyst Deactivation and Regeneration. In Kirk-Othmer Encyclopedia of Chemical Technology; 2003. [Google Scholar] [CrossRef]
- Argyle, M.; Bartholomew, C. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef] [Green Version]
- Ochoa, A.; Bilbao, J.; Gayubo, A.G.; Castaño, P. Coke formation and deactivation during catalytic reforming of biomass and waste pyrolysis products: A review. Renew. Sustain. Energy Rev. 2020, 119, 109600. [Google Scholar] [CrossRef]
- van Sint Annaland, M.; Kuipers, J.A.M.; van Swaaij, W.P.M. A kinetic rate expression for the time-dependent coke formation rate during propane dehydrogenation over a platinum alumina monolithic catalyst. Catal. Today 2001, 66, 427–436. [Google Scholar] [CrossRef]
- Guisnet, M. “Coke” molecules trapped in the micropores of zeolites as active species in hydrocarbon transformations. J. Mol. Catal. A Chem. 2002, 182–183, 367–382. [Google Scholar] [CrossRef]
- Wang, B. Zeolite Deactivation during Hydrocarbon Reactions: Characterisation of Coke Precursors and Acidity, Product Distribution. Doctoral Thesis, UCL (University College London), London, UK, 2008. [Google Scholar]
- Rostami, R.; Ghavipour, M.; Di, Z.; Wang, Y.; Behbahani, R. Study of coke deposition phenomena on the SAPO_34 catalyst and its effects on light olefin selectivity during the methanol to olefin reaction. RSC Adv. 2015, 5, 81965–81980. [Google Scholar] [CrossRef]
- Hou, X.; Zhao, L.; Diao, Z. Roles of Alkenes and Coke Formation in the Deactivation of ZSM-5 Zeolites During n-Pentane Catalytic Cracking. Catal. Lett. 2020, 150, 2716–2725. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, J.; Zhang, J.; Zhang, T.; Ye, M.; Liu, Z. Regeneration of catalysts deactivated by coke deposition: A review. Chin. J. Catal. 2020, 41, 1048–1061. [Google Scholar] [CrossRef]
- PlasticsEurope. Plastics—The Facts 2020. An Analysis of European Latest Plastics Production, Demand and Waste Data. 2020. Available online: https://www.plasticseurope.org/en/resources/publications/4312-plastics-facts-2020 (accessed on 24 June 2021).
- Lackner, M. Bioplastics-Biobased Plastics as Renewable and/or Biodegradable Alternatives to Petroplastics. In Kirk-Othmer Enyclopedia of Chemical Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015. [Google Scholar] [CrossRef]
- van den Oever, M.; Molenveld, K.; van der Zee, M.; Bos, H.T. Bio-Based and Biodegradable Plastics: Facts and Figures: Focus on Food Packaging in the Netherlands; Wageningen Food & Biobased Research: Wageningen, The Netherlands, 2017. [Google Scholar] [CrossRef] [Green Version]
- Yeo, J.; Muiruri, J.; Warintorn, T.; Li, Z.; He, C. Recent advances in the development of biodegradable PHB-based toughening materials: Approaches, advantages and applications. Mater. Sci. Eng. C 2017, 92. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Awasthi, A.K.; Wei, F.; Tan, Q.; Li, J. Single-use plastics: Production, usage, disposal, and adverse impacts. Sci. Total. Environ. 2021, 752, 141772. [Google Scholar] [CrossRef] [PubMed]
- Carroll, C.; Sousa, J. Plastic Debris in the Ocean. Charact. Mar. Plast. Their Environ. Impacts Situat. Anal. Rep. 2014. [Google Scholar] [CrossRef] [Green Version]
- Hidayah, N. Syafrudin, A Review on Landfill Management in the Utilization of Plastic Waste as an Alternative Fuel. E3S Web Conf. 2018, 31, 05013. [Google Scholar] [CrossRef] [Green Version]
- Crippa, M.; De Wilde, B.; Koopmans, R.; Leyssens, J.; Linder, M.; Muncke, J.; Ritschkoff, A.-C.; Van Doorsselaer, K.; Velis, C.; Wagner, M. A circular economy for plastics. In Insights from Research and Innovation to Inform Policy and Funding Decisions; European Commission EC: Brussel, Belgium, 2019. [Google Scholar] [CrossRef]
- Schyns, Z.O.G.; Shaver, M.P. Mechanical Recycling of Packaging Plastics: A Review. Macromol. Rapid Commun. 2021, 42, 2000415. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, I.; Jenks, M.J.F.; Roelands, M.C.P.; White, R.J.; van Harmelen, T.; de Wild, P.; van der Laan, G.P.; Meirer, F.; Keurentjes, J.T.F.; Weckhuysen, B.M. Beyond Mechanical Recycling: Giving New Life to Plastic Waste. Angew. Chem. Int. Ed. 2020, 59, 15402–15423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ügdüler, S.; Van Geem, K.M.; Denolf, R.; Roosen, M.; Mys, N.; Ragaert, K.; De Meester, S. Towards closed-loop recycling of multilayer and coloured PET plastic waste by alkaline hydrolysis. Green Chem. 2020, 22, 5376–5394. [Google Scholar] [CrossRef]
- Hu, L.-C.; Oku, A.; Yamada, E. Alkali-catalyzed methanolysis of polycarbonate. A study on recycling of bisphenol A and dimethyl carbonate. Polymer 1998, 39, 3841–3845. [Google Scholar] [CrossRef]
- Karayannidis, G.P.; Nikolaidis, A.K.; Sideridou, I.D.; Bikiaris, D.N.; Achilias, D.S. Chemical Recycling of PET by Glycolysis: Polymerization and Characterization of the Dimethacrylated Glycolysate. Macromol. Mater. Eng. 2006, 291, 1338–1347. [Google Scholar] [CrossRef]
- Al-Sabagh, A.M.; Yehia, F.Z.; Eshaq, G.; Rabie, A.M.; ElMetwally, A.E. Greener routes for recycling of polyethylene terephthalate. Egypt. J. Pet. 2016, 25, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Grigore, M.E. Methods of Recycling, Properties and Applications of Recycled Thermoplastic Polymers. Recycling 2017, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Zahedifar, P.; Pazdur, L.; Vande Velde, C.M.L.; Billen, P. Multistage Chemical Recycling of Polyurethanes and Dicarbamates: A Glycolysis–Hydrolysis Demonstration. Sustainability 2021, 13, 3583. [Google Scholar] [CrossRef]
- Gamerith, C.; Zartl, B.; Pellis, A.; Guillamot, F.; Marty, A.; Acero, E.H.; Guebitz, G.M. Enzymatic recovery of polyester building blocks from polymer blends. Process Biochem. 2017, 59, 58–64. [Google Scholar] [CrossRef]
- Miandad, R.; Rehan, M.; Barakat, M.A.; Aburiazaiza, A.S.; Khan, H.; Ismail, I.M.I.; Dhavamani, J.; Gardy, J.; Hassanpour, A.; Nizami, A.-S. Catalytic Pyrolysis of Plastic Waste: Moving toward Pyrolysis Based Biorefineries. Front. Energy Res. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Syamsiro, M.; Saptoadi, H.; Norsujianto, T.; Noviasri, P.; Cheng, S.; Alimuddin, Z.; Yoshikawa, K. Fuel Oil Production from Municipal Plastic Wastes in Sequential Pyrolysis and Catalytic Reforming Reactors. Energy Procedia 2014, 47, 180–188. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.-H.; Lee, K.-H.; Shin, D.-H. Investigation of catalytic degradation of high-density polyethylene by hydrocarbon group type analysis. J. Anal. Appl. Pyrolysis 2003, 70, 383–398. [Google Scholar] [CrossRef]
- Lee, K.-H.; Noh, N.-S.; Shin, D.-H.; Seo, Y. Comparison of plastic types for catalytic degradation of waste plastics into liquid product with spent FCC catalyst. Polym. Degrad. Stab. 2002, 78, 539–544. [Google Scholar] [CrossRef]
- Sakata, Y.; Uddin, M.A.; Koizumi, K.; Murata, K. Thermal degradation of polyethylene mixed with poly(vinyl chloride) and poly(ethyleneterephthalate). Polym. Degrad. Stab. 1996, 53, 111–117. [Google Scholar] [CrossRef]
- Chester, A.W.; Derouane, E.G. Zeolite Characterization and Catalysis a Tutorial; Springer: Dordrecht, The Netherlands, 2009. [Google Scholar] [CrossRef]
- Bekkum, H.V. Introduction to Zeolite Science and Practice; Elsevier: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Weitkamp, J. Zeolites and catalysis. Solid State Ion. 2000, 131, 175–188. [Google Scholar] [CrossRef]
- Chen, N.Y.; Garwood, W.E.; Dwyer, F.G. Shape Selective Catalysis in Industrial Applications; M. Dekker: New York, NY, USA, 1989. [Google Scholar]
- Garforth, A.A.; Lin, Y.H.; Sharratt, P.N.; Dwyer, J. Production of hydrocarbons by catalytic degradation of high density polyethylene in a laboratory fluidised-bed reactor. Appl. Catal. A Gen. 1998, 169, 331–342. [Google Scholar] [CrossRef]
- Corma, A. Inorganic Solid Acids and Their Use in Acid-Catalyzed Hydrocarbon Reactions. Chem. Rev. 1995, 95, 559–614. [Google Scholar] [CrossRef]
- Ohkita, H.; Nishiyama, R.; Tochihara, Y.; Mizushima, T.; Kakuta, N.; Morioka, Y.; Ueno, A.; Namiki, Y.; Tanifuji, S. Acid properties of silica-alumina catalysts and catalytic degradation of polyethylene. Ind. Eng. Chem. Res. 1993, 32, 3112–3116. [Google Scholar] [CrossRef]
- Aguado, J.; Serrano, D.P.; Sotelo, J.L.; Van Grieken, R.; Escola, J.M. Influence of the Operating Variables on the Catalytic Conversion of a Polyolefin Mixture over HMCM-41 and Nanosized HZSM-5. Ind. Eng. Chem. Res. 2001, 40, 5696–5704. [Google Scholar] [CrossRef]
- Abdullah, N.A.; Novianti, A.; Hakim, I.I.; Putra, N.; Koestoer, R.A. Influence of temperature on conversion of plastics waste (polystyrene) to liquid oil using pyrolysis process. IOP Conf. Ser. Earth Environ. Sci. 2018, 105, 012033. [Google Scholar] [CrossRef]
- López, A.; de Marco, I.; Caballero, B.M.; Laresgoiti, M.F.; Adrados, A. Influence of time and temperature on pyrolysis of plastic wastes in a semi-batch reactor. Chem. Eng. J. 2011, 173, 62–71. [Google Scholar] [CrossRef]
- Hernandez, M.D.; Gomez, A.; Garcia, A.N.; Agullo, J.; Marcilla, A. Effect of the temperature in the nature and extension of the primary and secondary reactions in the thermal and HZSM-5 catalytic pyrolysis of HDPE. Appl. Catal. A Gen. 2007, 317, 183–194. [Google Scholar] [CrossRef]
- Miandad, R.; Barakat, M.A.; Rehan, M.; Aburiazaiza, A.S.; Ismail, I.M.I.; Nizami, A.S. Plastic waste to liquid oil through catalytic pyrolysis using natural and synthetic zeolite catalysts. Waste Manag. 2017, 69, 66–78. [Google Scholar] [CrossRef]
- Murata, K.; Sato, K.; Sakata, Y. Effect of pressure on thermal degradation of polyethylene. J. Anal. Appl. Pyrolysis 2004, 71, 569–589. [Google Scholar] [CrossRef]
- Lee, K.-H.; Shin, D.-H. Characteristics of liquid product from the pyrolysis of waste plastic mixture at low and high temperatures: Influence of lapse time of reaction. Waste Manag. 2007, 27, 168–176. [Google Scholar] [CrossRef]
- Newalkar, G.; Iisa, K.; D’Amico, A.D.; Sievers, C.; Agrawal, P. Effect of Temperature, Pressure, and Residence Time on Pyrolysis of Pine in an Entrained Flow Reactor. Energy Fuels 2014, 28, 5144–5157. [Google Scholar] [CrossRef]
- Sakata, Y.; Uddin, M.A.; Muto, A. Degradation of polyethylene and polypropylene into fuel oil by using solid acid and non-acid catalysts. J. Anal. Appl. Pyrolysis 1999, 51, 135–155. [Google Scholar] [CrossRef]
- Abbas-Abadi, M.S.; Haghighi, M.N.; Yeganeh, H.; McDonald, A.G. Evaluation of pyrolysis process parameters on polypropylene degradation products. J. Anal. Appl. Pyrolysis 2014, 109, 272–277. [Google Scholar] [CrossRef]
- Renzini, M.S.; Lerici, L.C.; Sedran, U.; Pierella, L.B. Stability of ZSM-11 and BETA zeolites during the catalytic cracking of low-density polyethylene. J. Anal. Appl. Pyrolysis 2011, 92, 450–455. [Google Scholar] [CrossRef]
- Saad, J.M.; Nahil, M.A.; Williams, P.T. Influence of process conditions on syngas production from the thermal processing of waste high density polyethylene. J. Anal. Appl. Pyrolysis 2015, 113, 35–40. [Google Scholar] [CrossRef]
- Chen, Z.; Zhang, X.; Yang, F.; Peng, H.; Zhang, X.; Zhu, S.; Che, L. Deactivation of a Y-zeolite based catalyst with coke evolution during the catalytic pyrolysis of polyethylene for fuel oil. Appl. Catal. A Gen. 2021, 609, 117873. [Google Scholar] [CrossRef]
- Vasile, C.; Pakdel, H.; Mihai, B.; Onu, P.; Darie, H.; Ciocâlteu, S. Thermal and catalytic decomposition of mixed plastics. J. Anal. Appl. Pyrolysis 2001, 57, 287–303. [Google Scholar] [CrossRef]
- Budsaereechai, S.; Hunt, A.J.; Ngernyen, Y. Catalytic pyrolysis of plastic waste for the production of liquid fuels for engines. RSC Adv. 2019, 9, 5844–5857. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.H.; Yen, H.Y. Fluidised bed pyrolysis of polypropylene over cracking catalysts for producing hydrocarbons. Polym. Degrad. Stab. 2005, 89, 101–108. [Google Scholar] [CrossRef]
- Marcilla, A.; Hernández, M.d.R.; García, Á.N. Study of the polymer–catalyst contact effectivity and the heating rate influence on the HDPE pyrolysis. J. Anal. Appl. Pyrolysis 2007, 79, 424–432. [Google Scholar] [CrossRef]
- Isma, M.I.; Ali, S.; Nur, D. Mixed Plastic Wastes Pyrolysis in a Fluidized Bed Reactor for Potential Diesel Production. Int. J. Environ. Sci. Dev. 2015, 6, 606–609. [Google Scholar] [CrossRef] [Green Version]
- Elordi, G.; Olazar, M.; Lopez, G.; Castaño, P.; Bilbao, J. Role of pore structure in the deactivation of zeolites (HZSM-5, Hβ and HY) by coke in the pyrolysis of polyethylene in a conical spouted bed reactor. Appl. Catal. B Environ. 2011, 102, 224–231. [Google Scholar] [CrossRef]
- Olazar, M.; Lopez, G.; Amutio, M.; Elordi, G.; Aguado, R.; Bilbao, J. Influence of FCC catalyst steaming on HDPE pyrolysis product distribution. J. Anal. Appl. Pyrolysis 2009, 85, 359–365. [Google Scholar] [CrossRef]
- Ruckenstein, E.; Dadyburjor, D.B. Sintering and Redispersion in Supported Metal Catalysts. Rev. Chem. Eng. 1983, 1, 251–356. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.; Trimm, D.L. Mechanisms of carbon formation on nickel-containing catalysts. J. Catal. 1977, 48, 155–165. [Google Scholar] [CrossRef]
- Guisnet, M.; Costa, L.; Ribeiro, F.R. Prevention of zeolite deactivation by coking. J. Mol. Catal. A Chem. 2009, 305, 69–83. [Google Scholar] [CrossRef]
- Guisnet, M.; Magnoux, P. Organic chemistry of coke formation. Appl. Catal. A Gen. 2001, 212, 83–96. [Google Scholar] [CrossRef]
- Marcilla, A.; Beltrán, M.I.; Hernández, F.; Navarro, R. HZSM5 and HUSY deactivation during the catalytic pyrolysis of polyethylene. Appl. Catal. A Gen. 2004, 278, 37–43. [Google Scholar] [CrossRef]
- Marcilla, A.; Beltrán, M.I.; Navarro, R. Effect of regeneration temperature and time on the activity of HUSY and HZSM5 zeolites during the catalytic pyrolysis of polyethylene. J. Anal. Appl. Pyrolysis 2005, 74, 361–369. [Google Scholar] [CrossRef]
- López, A.; de Marco, I.; Caballero, B.M.; Adrados, A.; Laresgoiti, M.F. Deactivation and regeneration of ZSM-5 zeolite in catalytic pyrolysis of plastic wastes. Waste Manag. 2011, 31, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Chen, S.-L.; Wei, Y.; Wang, Y.; Sun, W.; Cao, Y.; Zeng, P. Kinetics of coke formation in the dimethyl ether-to-olefins process over SAPO-34 catalyst. Chem. Eng. J. 2017, 326, 528–539. [Google Scholar] [CrossRef]
- Mahamulkar, S.; Yin, K.; Agrawal, P.; Davis, R.; Jones, C.; Malek, A.; Shibata, H. Formation and Oxidation/Gasification of Carbonaceous Deposits: A Review. Ind. Eng. Chem. Res. 2016, 55. [Google Scholar] [CrossRef]
- Ozawa, Y.; Bischoff, K.B. Coke Formation Kinetics on Silica-Alumina Catalyst. Analysis. Ind. Eng. Chem. Process. Des. Dev. 1968, 7, 72–78. [Google Scholar] [CrossRef]
- Wauters, S.; Marin, G.B. Kinetic Modeling of Coke Formation during Steam Cracking. Ind. Eng. Chem. Res. 2002, 41, 2379–2391. [Google Scholar] [CrossRef]
- Moustafa, T.M.; Froment, G.F. Kinetic Modeling of Coke Formation and Deactivation in the Catalytic Cracking of Vacuum Gas Oil. Ind. Eng. Chem. Res. 2003, 42, 14–25. [Google Scholar] [CrossRef]
- Omidkhah, M.; Modarres, J.; Towfighi Darian, J.; Niaei, A. Estimation of Kinetic Parameters of Coking Reaction Rate in Pyrolysis of Naphtha. Int. J. Eng. 2004, 17, 319–332. [Google Scholar]
- Bjørgen, M.; Olsbye, U.; Kolboe, S. Coke precursor formation and zeolite deactivation: Mechanistic insights from hexamethylbenzene conversion. J. Catal. 2003, 215, 30–44. [Google Scholar] [CrossRef]
- Guo, X.; Zheng, Y.; Zhang, B.; Chen, J. Analysis of coke precursor on catalyst and study on regeneration of catalyst in upgrading of bio-oil. Biomass Bioenergy 2009, 33, 1469–1473. [Google Scholar] [CrossRef]
- Ibáñez, M.; Pérez-Uriarte, P.; Sánchez-Contador, M.; Cordero-Lanzac, T.; Aguayo, A.T.; Bilbao, J.; Castaño, P. Nature and Location of Carbonaceous Species in a Composite HZSM-5 Zeolite Catalyst during the Conversion of Dimethyl Ether into Light Olefins. Catalysts 2017, 7, 254. [Google Scholar] [CrossRef] [Green Version]
- Karimi, H.; Olayiwola, B.; Farag, H.; McAuley, K.B. Modelling coke formation in an industrial ethane-cracking furnace for ethylene production. Can. J. Chem. Eng. 2020, 98, 158–171. [Google Scholar] [CrossRef] [Green Version]
- Guisnet, M.; Magnoux, P. Coking and deactivation of zeolites: Influence of the Pore Structure. Appl. Catal. 1989, 54, 1–27. [Google Scholar] [CrossRef]
- Doka Nassionou, G.A.; Magnoux, P.; Guisnet, M. Comparative study of coking and regeneration of HEMT and HFAU zeolites. Microporous Mesoporous Mater. 1998, 22, 389–398. [Google Scholar] [CrossRef]
- Cerqueira, H.S.; Magnoux, P.; Martin, D.; Guisnet, M. Effect of contact time on the nature and location of coke during methylcyclohexane transformation over a USHY zeolite. In Studies in Surface Science and Catalysis; Delmon, B., Froment, G.F., Eds.; Elsevier: Amsterdam, The Netherlands, 1999; pp. 105–112. [Google Scholar] [CrossRef]
- Magnoux, P.; Guisnet, M. Coking, ageing and regeneration of zeolites: VI. Comparison of the Rates of Coke Oxidation of HY, H-Mordenite and HZSM-5. Appl. Catal. 1988, 38, 341–352. [Google Scholar] [CrossRef]
- Uguina, M.A.; Serrano, D.P.; Van Grieken, R.; Vènes, S. Adsorption, acid and catalytic changes induced in ZSM-5 by coking with different hydrocarbons. Appl. Catal. A Gen. 1993, 99, 97–113. [Google Scholar] [CrossRef]
- Bibby, D.M.; Milestone, N.B.; Patterson, J.E.; Aldridge, L.P. Coke formation in zeolite ZSM-5. J. Catal. 1986, 97, 493–502. [Google Scholar] [CrossRef]
- Froment, G.F.; Meyer, J.D.; Derouane, E.G. Deactivation of zeolite catalysts by coke formation. J. Catal. 1990, 124, 391–400. [Google Scholar] [CrossRef]
- Câmara, L.D.T.; Rajagopal, K.; Aranda, D.A.G. The effects of pore structure on catalyst deactivation by coke formation. In Studies in Surface Science and Catalysis; Spivey, J.J., Roberts, G.W., Davis, B.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2001; pp. 61–68. [Google Scholar] [CrossRef]
- Hughes, R.; Parvinian, M. Regeneration kinetics of coked silica-alumina catalyst. Prepr. Pap., Am. Chem. Soc., Div. Fuel Chem 1989, 34. [Google Scholar]
- Santamaría, J.; Monzón, A.; Berbegal, M.; Hughes, R. Regeneration strategies for coked fixed bed reactors. Chem. Eng. Sci. 1991, 46, 11–21. [Google Scholar] [CrossRef]
- Minh, C.L.; Jones, R.A.; Craven, E.I.; Brown, T. Temperature-Programmed Oxidation of Coke Deposited on Cracking Catalysts: Combustion Mechanism Dependence. Energy Fuels 1997, 11, 463–469. [Google Scholar] [CrossRef]
- Minh, C.L.; Li, C.; Brown, T.C. Kinetics of coke combustion during temperature-programmed oxidation of deactivated cracking catalysts. In Studies in Surface Science and Catalysis; Bartholomew, C.H., Fuentes, G.A., Eds.; Elsevier: Amsterdam, The Netherlands, 1997; pp. 383–390. [Google Scholar] [CrossRef]
- Keskitalo, T.J.; Lipiäinen, K.J.T.; Krause, A.O.I. Kinetic Modeling of Coke Oxidation of a Ferrierite Catalyst. Ind. Eng. Chem. Res. 2006, 45, 6458–6467. [Google Scholar] [CrossRef]
- Nováková, J.; Dolejšek, Z. A comment on the oxidation of coke deposited on zeolites. Zeolites 1990, 10, 189–192. [Google Scholar] [CrossRef]
- Morley, K.; de Lasa, H.I. On the determination of kinetic parameters for the regeneration of cracking catalyst. Can. J. Chem. Eng. 1987, 65, 773–777. [Google Scholar] [CrossRef]
- Ngomo, H.M.; Susu, A.A. The Influence of nature of coke on kinetics of catalyst regeneration. Pet. Sci. Technol. 2002, 20, 671–691. [Google Scholar] [CrossRef]
- Querini, C.A.; Fung, S.C. Temperature-programmed oxidation technique: Kinetics of coke-O2 reaction on supported metal catalysts. Appl. Catal. A Gen. 1994, 117, 53–74. [Google Scholar] [CrossRef]
- Hashimoto, K.; Takatani, K.; Iwasa, H.; Masuda, T. A multiple-reaction model for burning regeneration of coked catalysts. Chem. Eng. J. 1983, 27, 177–186. [Google Scholar] [CrossRef]
- Nakasaka, Y.; Tago, T.; Konno, H.; Okabe, A.; Masuda, T. Kinetic study for burning regeneration of coked MFI-type zeolite and numerical modeling for regeneration process in a fixed-bed reactor. Chem. Eng. J. 2012, 207–208, 368–376. [Google Scholar] [CrossRef]
- Richardson, J.T. Experimental Determination of Catalyst Fouling Parameters. Carbon Profiles. Ind. Eng. Chem. Process. Des. Dev. 1972, 11, 8–11. [Google Scholar] [CrossRef]
- Müller, N.; Moos, R.; Jess, A. In situ Monitoring of Coke Deposits during Coking and Regeneration of Solid Catalysts by Electrical Impedance-based Sensors. Chem. Eng. Technol. 2010, 33, 103–112. [Google Scholar] [CrossRef]
- Kern, C.; Jess, A. Regeneration of coked catalysts—modelling and verification of coke burn-off in single particles and fixed bed reactors. Chem. Eng. Sci. 2005, 60, 4249–4264. [Google Scholar] [CrossRef]
- Dubinets, O.V.; Davletova, M.R.; Islamova, G.I.; Gubaydullin, I.M.; Khannanova, G.D.; Akhmetov, I.V. Simulation of the coke burning process on the catalyst grain. J. Phys. Conf. Ser. 2019, 1368, 042068. [Google Scholar] [CrossRef]
- Pinard, L.; Astafan, A.; Sachse, A.; Dupeyrat, C. Impact of the Framework Type on the Regeneration of Coked Zeolites by Non-Thermal Plasma in a Fixed Bed Dielectric Barrier Reactor. Catalysts 2019, 9, 985. [Google Scholar] [CrossRef] [Green Version]
- Moljord, K.; Magnoux, P.; Guisnet, M. Evidence for a participation of zeolite acid sites in the coke removal through oxidative treatment. Catal. Lett. 1994, 25, 141–147. [Google Scholar] [CrossRef]
- Pieck, C.L.; Vera, C.R.; Querini, C.A.; Parera, J.M. Differences in coke burning-off from Pt–Sn/Al2O3 catalyst with oxygen or ozone. Appl. Catal. A Gen. 2005, 278, 173–180. [Google Scholar] [CrossRef]
- Dong, Q.N.; Anderson, J.R.; Mole, T.; Chang, Y.F.; Western, R.J. Reaction of benzene and toluene in the presence of oxygen over H-ZSM5 zeolite Aromatic oxygenates in the product. Appl. Catal. 1991, 72, 99–107. [Google Scholar] [CrossRef]
- Adachi, K.; Hirabayashi, K.; Hoshino, N.; Inoue, S.; Kondoh, T.; Murakami, T.; Shibata, S.; Ueda, M. Process for the Regeneration of Coke-Deposited, Crystalline Silicate Catalyst. U.S. Patent 5,396,682, 26 April 1994. [Google Scholar]
- Jong, S.-J.; Pradhan, A.R.; Wu, J.-F.; Tsai, T.-C.; Liu, S.-B. On the Regeneration of Coked H-ZSM-5 Catalysts. J. Catal. 1998, 174, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Koves, W.J. Apparatus for Coke Burning in Regeneration of Hydrocarbon Conversion Catalyst. U.S. Patent US4880604A, 14 November 1989. [Google Scholar]
- Nazarova, G.; Ivashkina, E.; Shafran, T.; Oreshina, A.; Seitenova, G. Prediction of residue coke content and operating modes of regenerator in the catalytic cracking technology. Pet. Sci. Technol. 2020, 1–9. [Google Scholar] [CrossRef]
- Sano, T.; Yamashita, N.; Iwami, Y.; Takeda, K.; Kawakami, Y. Estimation of dealumination rate of ZSM-5 zeolite by adsorption of water vapor. Zeolites 1996, 16, 258–264. [Google Scholar] [CrossRef]
- Sano, T.; Ikeya, H.; Kasuno, T.; Wang, Z.B.; Kawakami, Y.; Soga, K. Influence of Crystallinity of HZSM-5 Zeolite on Its Dealumination Rate. Zeolites 1997, 19, 80–86. [Google Scholar] [CrossRef]
- Nikolopoulos, A.A.; Kogelbauer, A.; Goodwin, J.G.; Marcelin, G. Effect of dealumination on the catalytic activity of acid zeolites for the gas phase synthesis of MTBE. Appl. Catal. A Gen. 1994, 119, 69–81. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Aguayo, A.T.; Atutxa, A.; Prieto, R.; Bilbao, J. Deactivation of a HZSM-5 Zeolite Catalyst in the Transformation of the Aqueous Fraction of Biomass Pyrolysis Oil into Hydrocarbons. Energy Fuels 2004, 18, 1640–1647. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V.; Holmes, S.; Siddiqui, M.R.H. Coking and regeneration of H3PW12O40/SiO2 catalysts. Appl. Catal. A Gen. 2001, 214, 47–58. [Google Scholar] [CrossRef]
- Pieck, C.L.; Verderone, R.J.; Jablonski, E.L.; Parera, J.M. Burning of coke on Pt Re/Al2O3 catalyst: Activation energy and oxygen reaction order. Appl. Catal. 1989, 55, 1–10. [Google Scholar] [CrossRef]
- Kassargy, C.; Awad, S.; Burnens, G.; Upreti, G.; Kahine, K.; Tazerout, M. Study of the effects of regeneration of USY zeolite on the catalytic cracking of polyethylene. Appl. Catal. B Environ. 2019, 244, 704–708. [Google Scholar] [CrossRef]
- Lu, P.; Huang, Q.; Chi, Y.; Yan, J. Coking and Regeneration of Nickel Catalyst for the Cracking of Toluene As a Tar Model Compound. Energy Fuels 2017, 31, 8283–8290. [Google Scholar] [CrossRef]
- Furusawa, T.; Miura, Y.; Kori, Y.; Sato, M.; Suzuki, N. The cycle usage test of Ni/MgO catalyst for the steam reforming of naphthalene/benzene as model tar compounds of biomass gasification. Catal. Commun. 2009, 10, 552–556. [Google Scholar] [CrossRef]
- Parsons, S. Advanced Oxidation Processes for Water and Wastewater Treatment; IWA Publishing: London, UK, 2005. [Google Scholar] [CrossRef]
- Deng, Y.; Zhao, R. Advanced Oxidation Processes (AOPs) in Wastewater Treatment. Curr. Pollut. Rep. 2015, 1, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Brodu, N.; Manero, M.-H.; Andriantsiferana, C.; Pic, J.-S.; Valdés, H. Role of Lewis acid sites of ZSM-5 zeolite on gaseous ozone abatement. Chem. Eng. J. 2013, 231, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Alejandro, S.; Valdés, H.; Manéro, M.-H.; Zaror, C.A. Oxidative regeneration of toluene-saturated natural zeolite by gaseous ozone: The influence of zeolite chemical surface characteristics. J. Hazard. Mater. 2014, 274, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alejandro-Martín, S.; Valdés, H.; Manero, M.-H.; Zaror, C.A. Catalytic Ozonation of Toluene Using Chilean Natural Zeolite: The Key Role of Brønsted and Lewis Acid Sites. Catalysts 2018, 8, 211. [Google Scholar] [CrossRef] [Green Version]
- Lenntech. Ozone Reaction Mechanisms. 2020. Available online: https://www.lenntech.com/library/ozone/reaction/ozone-reaction-mechanisms.htm (accessed on 15 May 2021).
- Rekhate, C.V.; Srivastava, J.K. Recent advances in ozone-based advanced oxidation processes for treatment of wastewater—A review. Chem. Eng. J. Adv. 2020, 3, 100031. [Google Scholar] [CrossRef]
- Rakovsky, S.; Anachkov, M.; Zaikov, G. Fields of Ozone Applications. Chem. Chem. Technol. 2009. Available online: http://vlp.com.ua/files/10_65.pdf (accessed on 24 June 2021).
- Oyama, S.T. Chemical and Catalytic Properties of Ozone. Catal. Rev. 2000, 42, 279–322. [Google Scholar] [CrossRef]
- Vijayan, T.; Patil, J.G. High concentration ozone generation in the laboratory for various applications. Int. J. Sci. Technol. Educ. Res. 2010. [Google Scholar] [CrossRef]
- Benson, S.W.; Axworthy, A.E., Jr. Mechanism of the Gas Phase, Thermal Decomposition of Ozone. J. Chem. Phys. 1957, 26, 1718–1726. [Google Scholar] [CrossRef]
- Li, W.; Gibbs, G.V.; Oyama, S.T. Mechanism of Ozone Decomposition on a Manganese Oxide Catalyst. 1. In Situ Raman Spectroscopy and Ab Initio Molecular Orbital Calculations. J. Am. Chem. Soc. 1998, 120, 9041–9046. [Google Scholar] [CrossRef]
- Monneyron, P.; Mathé, S.; Manero, M.H.; Foussard, J.N. Regeneration of High Silica Zeolites via Advanced Oxidation Processes—A Preliminary Study About Adsorbent Reactivity Toward Ozone. Chem. Eng. Res. Des. 2003, 81, 1193–1198. [Google Scholar] [CrossRef] [Green Version]
- Brodu, N.; Manero, M.-H.; Andriantsiferana, C.; Pic, J.-S.; Valdés, H. Gaseous ozone decomposition over high silica zeolitic frameworks. Can. J. Chem. Eng. 2018, 96, 1911–1918. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Li, W. Destruction of toluene by ozone-enhanced photocatalysis: Performance and mechanism. Appl. Catal. B Environ. 2011, 102, 449–453. [Google Scholar] [CrossRef]
- Mariey, L.; Lamotte, J.; Chevreau, T.; Lavalley, J.C. FT-IR study of coked HY zeolite regeneration using oxygen or ozone. React. Kinet. Catal. Lett. 1996, 59, 241–246. [Google Scholar] [CrossRef]
- Copperthwaite, R.G.; Hutchings, G.J.; Johnston, P.; Orchard, S.W. Regeneration of pentasil zeolite catalysts using ozone and oxygen. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1986, 82, 1007–1017. [Google Scholar] [CrossRef]
- Hutchings, G.J.; Copperthwaite, R.G.; Themistocleous, T.; Foulds, G.A.; Bielovitch, A.S.; Loots, B.J.; Nowitz, G.; Van Eck, P. A comparative study of reactivation of zeolite Y using oxygen and ozone/oxygen mixtures. Appl. Catal. 1987, 34, 153–161. [Google Scholar] [CrossRef]
- Khangkham, S.; Julcour-Lebigue, C.; Damronglerd, S.; Ngamcharussrivichai, C.; Manero, M.-H.; Delmas, H. Regeneration of coked zeolite from PMMA cracking process by ozonation. Appl. Catal. B Environ. 2013, 140–141, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Richard, R.; Julcour, C.; Manéro, M. Towards a New Oxidation Process Using Ozone to Regenerate Coked Catalysts. Ozone Sci. Eng. 2017, 39, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Querini, C.A. Isobutane/butene alkylation: Regeneration of solid acid catalysts. Catal. Today 2000, 62, 135–143. [Google Scholar] [CrossRef]
- Jia, L.Y.; Farouha, A.; Pinard, L.; Hedan, S.; Comparot, J.D.; Dufour, A.; Ben Tayeb, K.; Vezin, H.; Batiot-Dupeyrat, C. New routes for complete regeneration of coked zeolite. Appl. Catal. B Environ. 2017, 219, 82–91. [Google Scholar] [CrossRef]
- HafezKhiabani, N.; Fathi, S.; Shokri, B.; Hosseini, S.I. A novel method for decoking of Pt–Sn/Al2O3 in the naphtha reforming process using RF and pin-to-plate DBD plasma systems. Appl. Catal. A Gen. 2015, 493, 8–16. [Google Scholar] [CrossRef]
- Astafan, A.; Batiot-Dupeyrat, C.; Pinard, L. Mechanism and Kinetic of Coke Oxidation by Nonthermal Plasma in Fixed-Bed Dielectric Barrier Reactor. J. Phys. Chem. C 2019, 123, 9168–9175. [Google Scholar] [CrossRef]
- Pinard, L.; Ayoub, N.; Batiot-Dupeyrat, C. Regeneration of a Coked Zeolite via Nonthermal Plasma Process: A Parametric Study. Plasma Chem. Plasma Process. 2019, 39, 929–936. [Google Scholar] [CrossRef]
- Braschi, I.; Blasioli, S.; Buscaroli, E.; Montecchio, D.; Martucci, A. Physicochemical regeneration of high silica zeolite Y used to clean-up water polluted with sulfonamide antibiotics. J. Environ. Sci. 2016, 43, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Díez, A.M.; Sanromán, M.A.; Pazos, M. Fenton-based processes for the regeneration of catalytic adsorbents. Catal. Today 2018, 313, 122–127. [Google Scholar] [CrossRef]
- Morales, M.V.; Góra-Marek, K.; Musch, H.; Pineda, A.; Murray, B.; Stefanidis, S.; Falco, L.; Tarach, K.; Ponomareva, E.; Marsman, J.H.; et al. Advanced oxidation process for coke removal: A systematic study of hydrogen peroxide and OH-derived-Fenton radicals of a fouled zeolite. Appl. Catal. A Gen. 2018, 562, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Lin, Q.; Xue, Z.; Munson, R.; Magneschi, G. Sinopec Zhongyuan Oil Field Company Refinery CCS-EOR Project. Energy Procedia 2017, 114, 5869–5873. [Google Scholar] [CrossRef]
- Hunt, J.; Ferrari, A.; Lita, A.; Crosswhite, M.; Ashley, B.; Stiegman, A.E. Microwave-Specific Enhancement of the Carbon–Carbon Dioxide (Boudouard) Reaction. J. Phys. Chem. C 2013, 117, 26871–26880. [Google Scholar] [CrossRef]
- Triantafillidis, C.S.; Vlessidis, A.G.; Evmiridis, N.P. Dealuminated H−Y Zeolites: Influence of the Degree and the Type of Dealumination Method on the Structural and Acidic Characteristics of H−Y Zeolites. Ind. Eng. Chem. Res. 2000, 39, 307–319. [Google Scholar] [CrossRef]
- Santos, L.T.d.; Santos, F.M.; Silva, R.S.; Gomes, T.S.; Esteves, P.M.; Pimenta, R.D.M.; Menezes, S.M.C.; Chamberlain, O.R.; Lam, Y.L.; Pereira, M.M. Mechanistic insights of CO2-coke reaction during the regeneration step of the fluid cracking catalyst. Appl. Catal. A Gen. 2008, 336, 40–47. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, G.; Gao, S.; Xu, G. Regeneration Kinetics of Spent FCC Catalyst via Coke Gasification in a Micro Fluidized Bed. Procedia Eng. 2015, 102, 1758–1765. [Google Scholar] [CrossRef] [Green Version]
- Tian, G.; Wang, G.; Xu, C.; Gao, J. Gasification of the Coke on Spent-Residue-Pretreating Catalysts with Steam and Steam–O2 Mixtures. Energy Fuels 2014, 28, 1372–1379. [Google Scholar] [CrossRef]
- Ferella, F.; D’Adamo, I.; Leone, S.; Innocenzi, V.; De Michelis, I.; Vegliò, F. Spent FCC E-Cat: Towards a Circular Approach in the Oil Refining Industry. Sustainability 2019, 11, 113. [Google Scholar] [CrossRef] [Green Version]
- Bauer, F.; Ernst, H.; Geidel, E.; Schödel, R. Reactivation of Coked H-ZSM-5 by Treatment with Hydrogen and Alkanes. J. Catal. 1996, 164, 146–151. [Google Scholar] [CrossRef]
- Marecot*, P.; Peyrovi, S.; Bahloul, D.; Barbier, J. Regeneration by hydrogen treatment of bifunctional catalysts deactivated by coke deposition. Appl. Catal. 1990, 66, 181–190. [Google Scholar] [CrossRef]
- Walker, P.L.; Matsumoto, S.; Hanzawa, T.; Muira, T.; Ismail, I.M.K. Catalysis of gasification of coal-derived cokes and chars. Fuel 1983, 62, 140–149. [Google Scholar] [CrossRef]
- Josl, R.; Klingmann, R.; Traa, Y.; Gläser, R.; Weitkamp, J. Regeneration of zeolite catalysts deactivated in isobutane/butene alkylation: An in situ FTIR investigation at elevated H2 pressure. Catal. Commun. 2004, 5, 239–241. [Google Scholar] [CrossRef]
- Klingmann, R.; Josl, R.; Traa, Y.; Gläser, R.; Weitkamp, J. Hydrogenative regeneration of a Pt/La-Y zeolite catalyst deactivated in the isobutane/n-butene alkylation. Appl. Catal. A Gen. 2005, 281, 215–223. [Google Scholar] [CrossRef]
- Aguayo, A.T.; Gayubo, A.G.; Ereña, J.; Atutxa, A.; Bilbao, J. Coke Aging and Its Incidence on Catalyst Regeneration. Ind. Eng. Chem. Res. 2003, 42, 3914–3921. [Google Scholar] [CrossRef]
- Gnep, N.S.; de Armando, M.L.M.; Guisnet, M. Toluene Disproportionation and Coke Formation over Fluorired alumina and hydrogen-mordenite. React. Kinet. Catal. Lett. 1980, 13, 183–189. [Google Scholar] [CrossRef]
- Alvarez, A.G.; Viturro, H.; Bonetto, R.D. Structural changes on deactivation of ZSM-5. A study by X-ray powder diffraction. Mater. Chem. Phys. 1992, 32, 135–140. [Google Scholar] [CrossRef]
- Schmidt, L.D.; Wang, T.; Vacquez, A. Electron microscopy of catalyst particles. Ultramicroscopy 1982, 8, 175–184. [Google Scholar] [CrossRef]
- Datye, A.K.; Smith, D.J. The Study of Heterogeneous Catalysts by High-Resolution Transmission Electron MicroscoDV. Catal. Rev. 1992, 34, 129–178. [Google Scholar] [CrossRef]
- Ajibola, A.A.; Omoleye, J.A.; Efeovbokhan, V.E. Catalytic cracking of polyethylene plastic waste using synthesised zeolite Y from Nigerian kaolin deposit. Appl. Petrochem. Res. 2018, 8, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Zaera, F. Characterization of Heterogeneous Catalysts. Surface and Nanomolecular Catalysis. 2006, 1–37. [Google Scholar] [CrossRef]
- Guisnet, M.; Pinard, L. Catalyse Hétérogène: Désactivation et Régénération des Catalyseurs. Techniques de l’ingénieur Catalyse et procédés Catalytiques, 2014. Base Documentaire: TIB325DUO(ref. article: j1265). Available online: https://www.techniques-ingenieur.fr/base-documentaire/procedes-chimie-bio-agro-th2/catalyse-et-procedes-catalytiques-42325210/catalyse-heterogene-desactivation-et-regeneration-des-catalyseurs-j1265/ (accessed on 20 November 2020).
- Engtrakul, C.; Mukarakate, C.; Starace, A.K.; Magrini, K.A.; Rogers, A.K.; Yung, M.M. Effect of ZSM-5 acidity on aromatic product selectivity during upgrading of pine pyrolysis vapors. Catal. Today 2016, 269, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Goetze, J.; Weckhuysen, B.M. Spatiotemporal coke formation over zeolite ZSM-5 during the methanol-to-olefins process as studied with operando UV-vis spectroscopy: A comparison between H-ZSM-5 and Mg-ZSM-5. Catal. Sci. Technol. 2018, 8, 1632–1644. [Google Scholar] [CrossRef] [Green Version]
- Westermann, A. Élimination Sélective d’un Mélange D’hydrocarbures Imbrûlés Diesel par Adsorption sur des Matériaux Zéolithiques. 2013. Thèse de Doctorat Chimie Université de Lorraine. Available online: http://www.theses.fr/2013LORR0275/document (accessed on 24 June 2021).
- Wang, B.; Manos, G. Acid Site Characterization of Coked USHY Zeolite Using Temperature Programmed Desorption with a Component-Nonspecific Detector. Ind. Eng. Chem. Res. 2007, 46, 7977–7983. [Google Scholar] [CrossRef]
- Khangkham, S. Catalytic Degradation of Poly(Methyl Methacrylate) by Zeolites and Regeneration of Used Zeolites via Ozonation. 2012. Thèse de Doctorat Génie des Procédés et Environnement Toulouse, INPT 2012. Available online: http://www.theses.fr/2012INPT0109/document (accessed on 24 June 2021).
- Weitkamp, J. Critical Evaluation of Catalytic Testing of Zeolites. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1988; pp. 515–534. [Google Scholar] [CrossRef]
- van Donk, S.; Bus, E.; Broersma, A.; Bitter, J.H.; de Jong, K.P. Butene skeletal isomerization over H-ferrierite: A TEOM and in situ IR study on the role of carbonaceous deposits and the location of Brønsted acid sites. Appl. Catal. A Gen. 2002, 237, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Gomm, S.; Gläser, R.; Weitkamp, J. In situ Observation of Coke Deposition on Zeolite Catalysts Using a Tapered Element Oscillating Microbalance. Chem. Eng. Technol. 2002, 25, 962–966. [Google Scholar] [CrossRef]
- Bellare, A.; Dadyburjor, D.B. Evaluation of Modes of Catalyst Deactivation by Coking for Cumene Cracking over Zeolites. J. Catal. 1993, 140, 510–525. [Google Scholar] [CrossRef]
- Zhang, Y.; Williams, P.T. Carbon nanotubes and hydrogen production from the pyrolysis catalysis or catalytic-steam reforming of waste tyres. J. Anal. Appl. Pyrolysis 2016, 122, 490–501. [Google Scholar] [CrossRef] [Green Version]
- Chu, B.; Hsiao, B.S. Small-Angle X-ray Scattering of Polymers. Chem. Rev. 2001, 101, 1727–1762. [Google Scholar] [CrossRef]
- McDonald, M.J.; Smith, J.W.H.; Dahn, J.R. A study of small angle X-ray scattering from impregnated activated carbons. Carbon 2014, 68, 452–461. [Google Scholar] [CrossRef]
- Du, X.; Wu, E. Porosity of microporous zeolites A, X and ZSM-5 studied by small angle X-ray scattering and nitrogen adsorption. J. Phys. Chem. Solids 2007, 68, 1692–1699. [Google Scholar] [CrossRef]
- Härk, E.; Ballauff, M. Carbonaceous Materials Investigated by Small-Angle X-ray and Neutron Scattering. J. Carbon Res. 2020, 6, 82. [Google Scholar] [CrossRef]
- Saurel, D.; Segalini, J.; Jauregui, M.; Pendashteh, A.; Daffos, B.; Simon, P.; Casas-Cabanas, M. A SAXS outlook on disordered carbonaceous materials for electrochemical energy storage. Energy Storage Mater. 2019, 21, 162–173. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Light coke combustion Hydrogen oxidation | (5) | |
| Heavy coke combustion Carbon oxidation | (6) | |
| (7) | ||
| (8) | ||
| (9) | ||
| (10) |
| Stoichiometry | Kinetics | Global Reaction Rates | |
|---|---|---|---|
| (11) | [Hydrogen] [Carbon] | ||
| (12) | |||
| (13) | |||
| Oxidant | Redox Couple | Redox Potential (V) |
|---|---|---|
| Oxygen molecule | O2/H2O | 1.23 |
| Hydrogen peroxide | H2O2/H2O | 1.78 |
| Ozone | O3/O2 | 2.07 |
| Atomic oxygen | O•/H2O | 2.42 |
| Hydroxyl radical | HO•/H2O | 2.80 |
| Ozonation Conditions | Deactivated Catalyst | Deactivating Reaction | Ref. |
|---|---|---|---|
| O2 + O3 flow 120 cm3.min−1 (ratio not given), 137 °C. | HY and HYS zeolite | Exposition to cyclohexene at 347 °C. | [145] |
| O3/O2 mole ratio 0.05–0.06, 150 °C, 90 min. | HZSM-5 zeolites (Si/Al ratio 35 and 70) | Methanol conversion to hydrocarbons and o-xylene isomerization. | [146] |
| O3/O2 mole ratio 0.05, 200 °C, 4h (bed agitation). | HY zeolite | Exposition to various alkanes and alkenes at 500 °C. | [147] |
| O3 conc. from 16 to 50 g/m3, 20–150 °C, 0.5–4 h. | ZSM-5 zeolite | PMMA cracking process at 250–300 °C. | [148] |
| O3 conc. from 4 to 25 g/m3, 50–200 °C, 2–8.5 h. | Undefined zeolite | Not given. | [149] |
| O3/O2 mole ratio 0.01, 125 °C, 4 h + H2 regeneration | Y-zeolite (UOP, Y-54) | Isobutane alkylation in liquid phase (25–80 °C). | [150] |
| CO2 gasification | C(s) + CO2(g) → 2CO(g) | +172 kJ/mol | (24) |
| Steam gasification | C(s) + H2O(g) → CO(g) + H2(g) | +131 kJ/mol | (25) |
| Analytical Technique | Related Information |
|---|---|
| X-ray Diffraction (XRD) + SAXS | Crystalline structure: zeolite type, PSD. |
| Electron microscopy (SEM, TEM) | Surface aspect image, particle size. |
| X-ray Fluorescence spectrometry (XRF) | Elemental analysis: Si/Al ratio for zeolites. |
| Physisorption of N2 (or other inert molecule) | Porosimetry analysis: surface area, pore volume |
| Chemisorption of NH3 (or other probe molecule) | Surface acidity: concentration, strength, type. |
| Analytical Technique | Related Information |
|---|---|
| Elemental analysis | C, H, N, S contents H/C ratio |
| Temperature-Programmed Oxidation (TPO) | Global coke content, H/C ratio Coke reactivity and optimal oxidation temperature |
| FTIR, Raman | Coke nature: aliphatic, aromatic Coke effect on active sites |
| UV-Vis | Coke nature: insaturated compounds |
| NMR and XRD | Coke nature and location Structural degradation |
| Coke extraction + analysis (see Figure 22) | Chemical nature of coke Distribution of coke components. |
| Tapered Element Oscillating Microbalance (TEOM) | Coking and deactivation kinetics |
| Thermogravimetric Analysis (TGA) | Coke amount and thermal degradation products |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daligaux, V.; Richard, R.; Manero, M.-H. Deactivation and Regeneration of Zeolite Catalysts Used in Pyrolysis of Plastic Wastes—A Process and Analytical Review. Catalysts 2021, 11, 770. https://doi.org/10.3390/catal11070770
Daligaux V, Richard R, Manero M-H. Deactivation and Regeneration of Zeolite Catalysts Used in Pyrolysis of Plastic Wastes—A Process and Analytical Review. Catalysts. 2021; 11(7):770. https://doi.org/10.3390/catal11070770
Chicago/Turabian StyleDaligaux, Vivien, Romain Richard, and Marie-Hélène Manero. 2021. "Deactivation and Regeneration of Zeolite Catalysts Used in Pyrolysis of Plastic Wastes—A Process and Analytical Review" Catalysts 11, no. 7: 770. https://doi.org/10.3390/catal11070770
APA StyleDaligaux, V., Richard, R., & Manero, M.-H. (2021). Deactivation and Regeneration of Zeolite Catalysts Used in Pyrolysis of Plastic Wastes—A Process and Analytical Review. Catalysts, 11(7), 770. https://doi.org/10.3390/catal11070770