
Improving the Conversion of Biomass in Catalytic Pyrolysis via Intensification of Biomass—Catalyst Contact by Co-Pressing



Abstract
:
1. Introduction
2. Results and Discussion
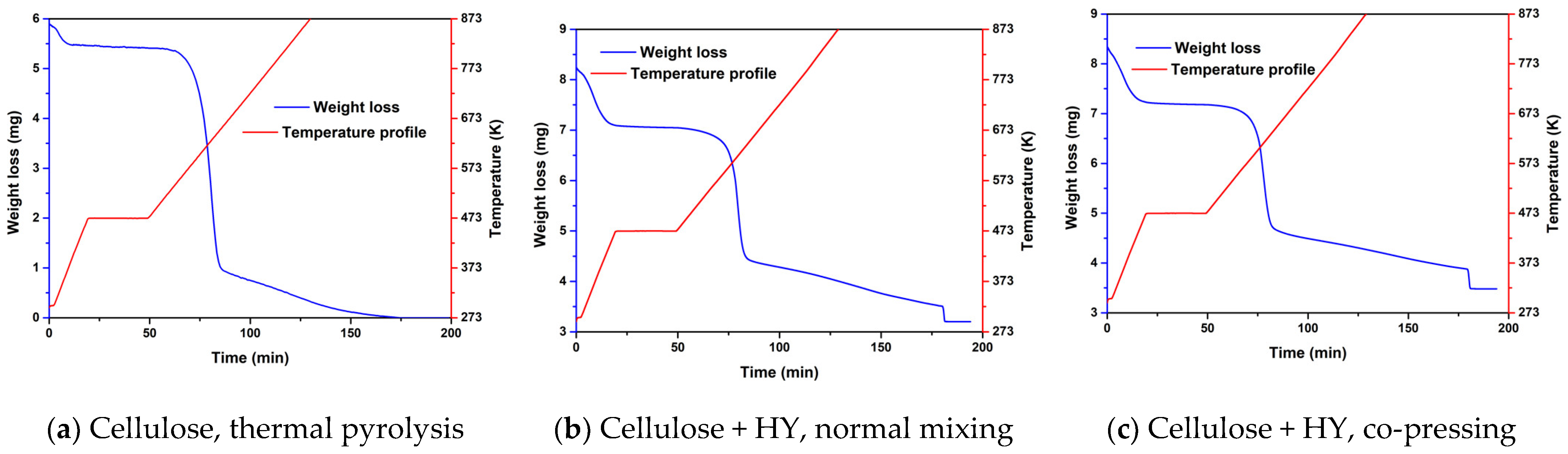
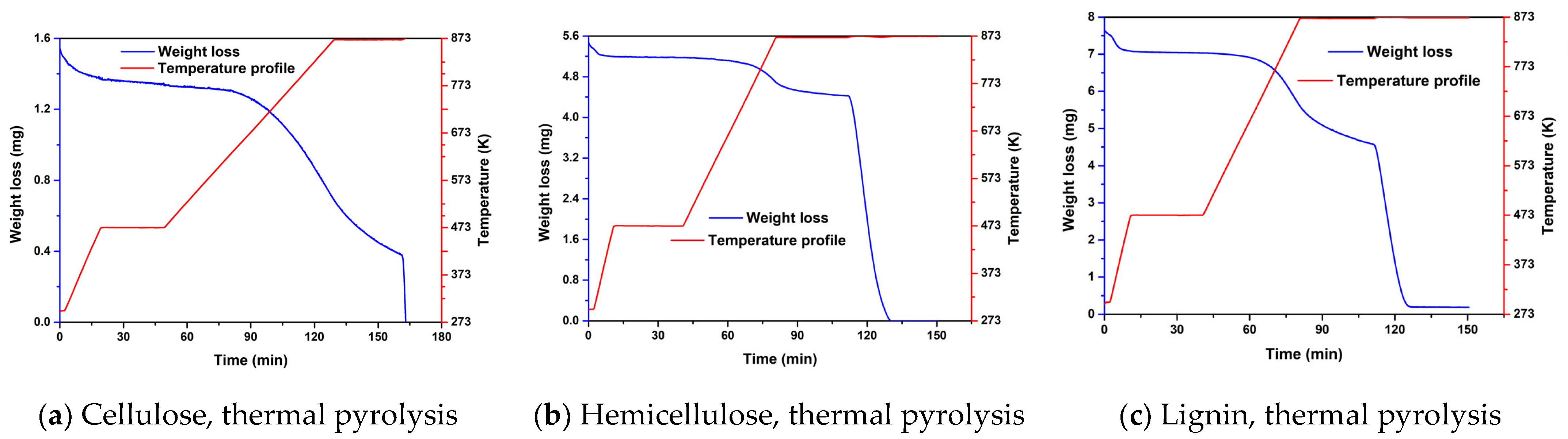
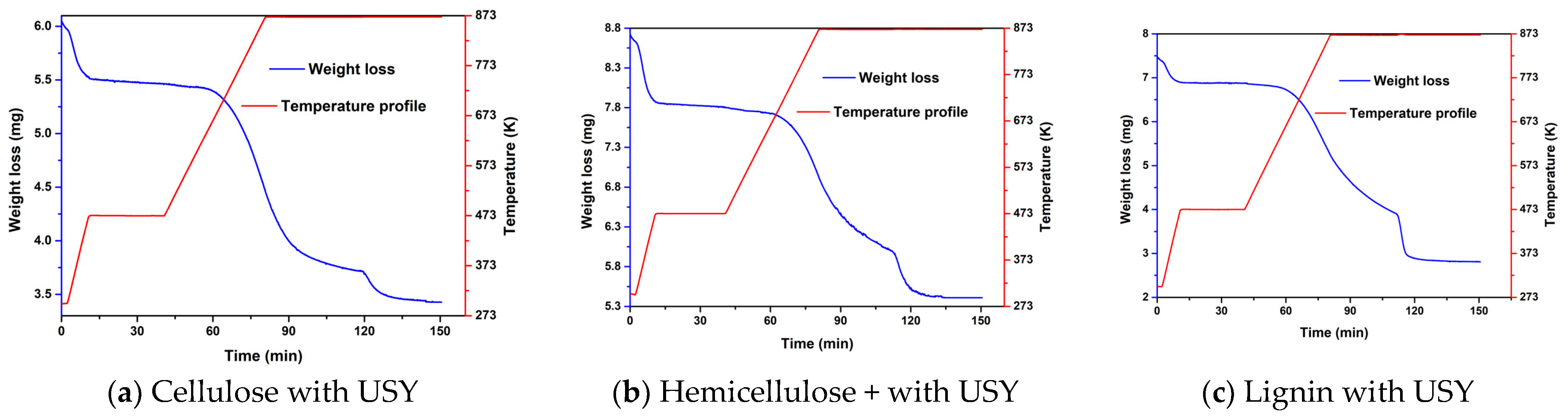
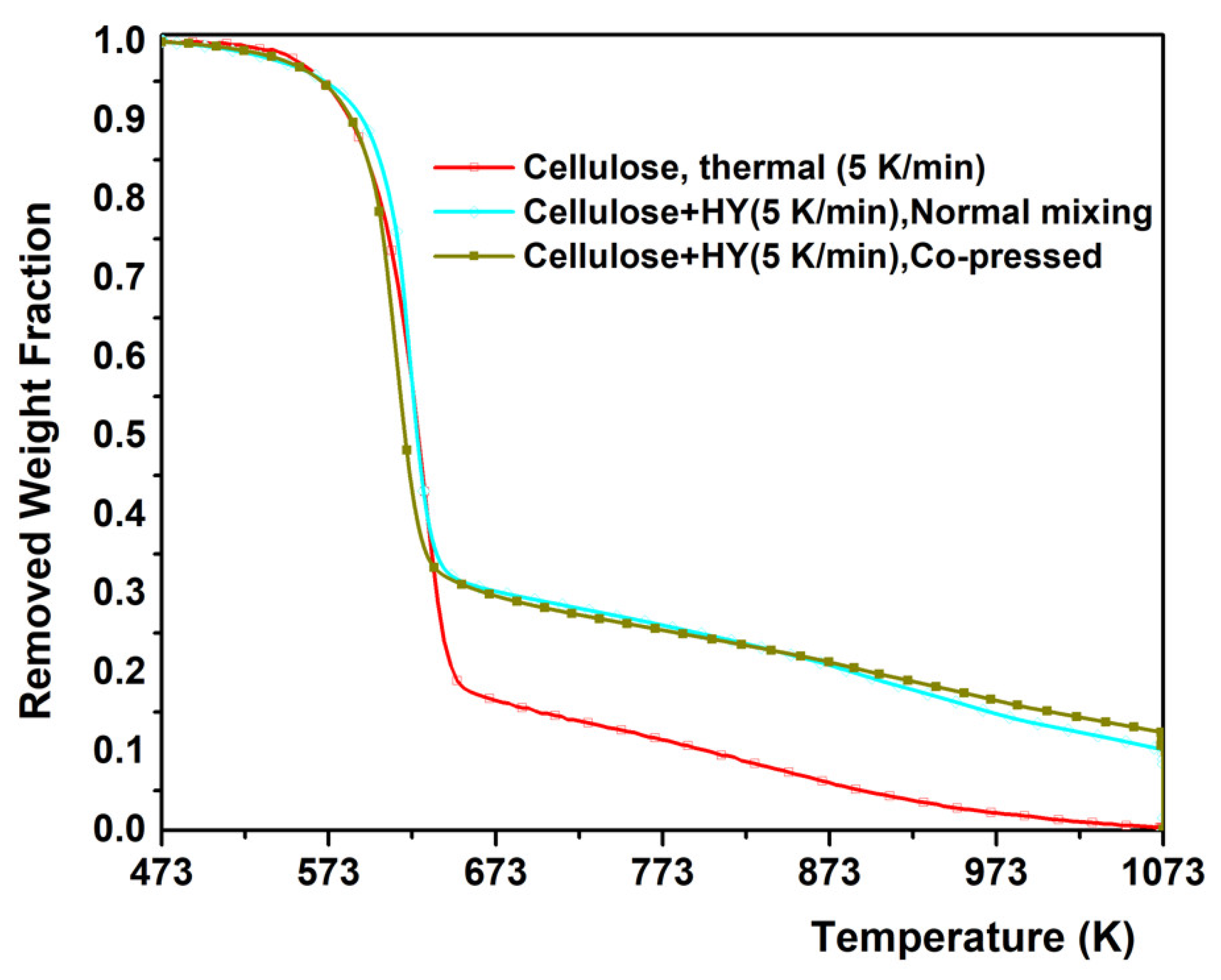
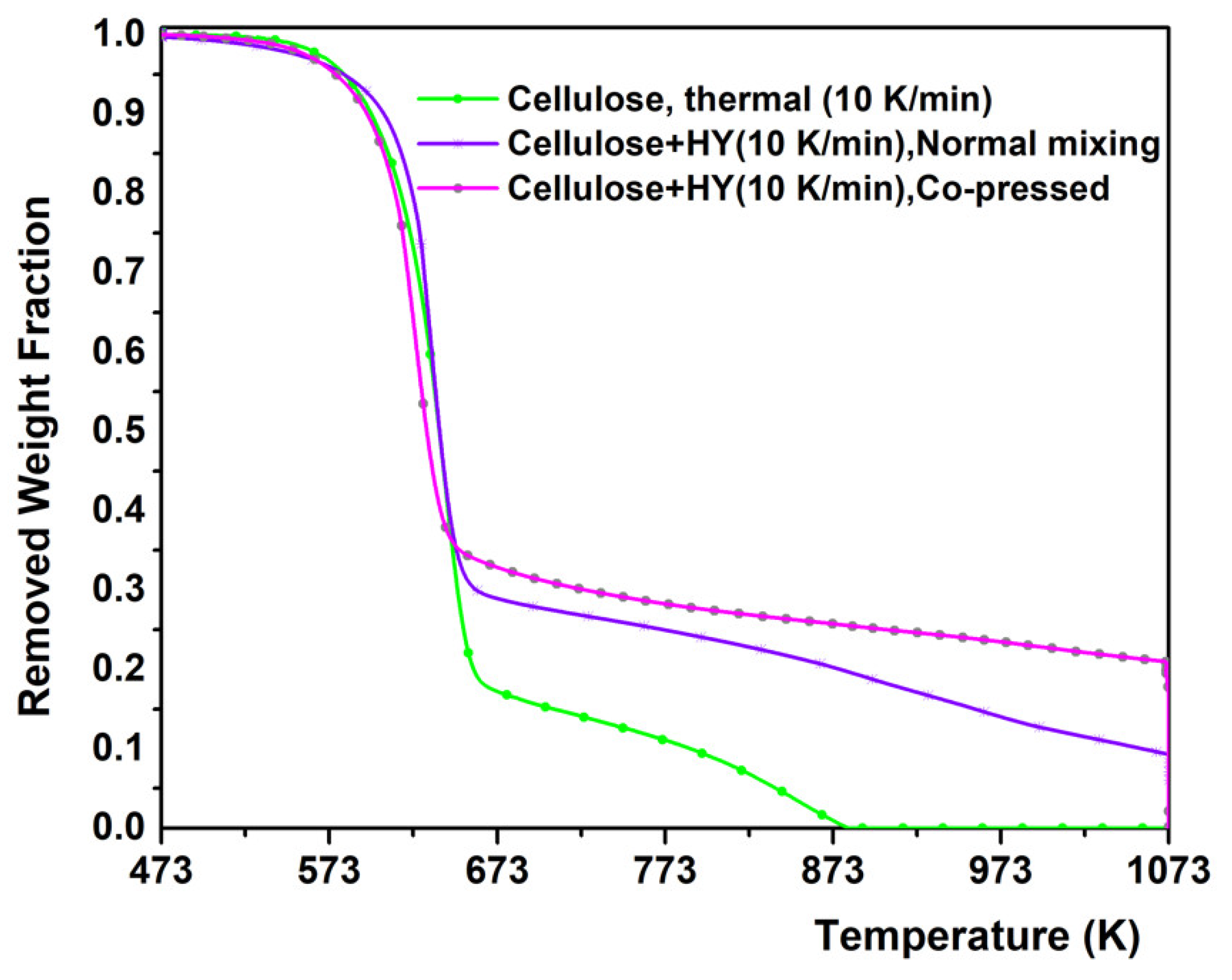
2.1. Experimental Results from TGA of Thermal and Catalytic Degradation of Cellulose
2.2. Pyrolysis Reactor Experiments
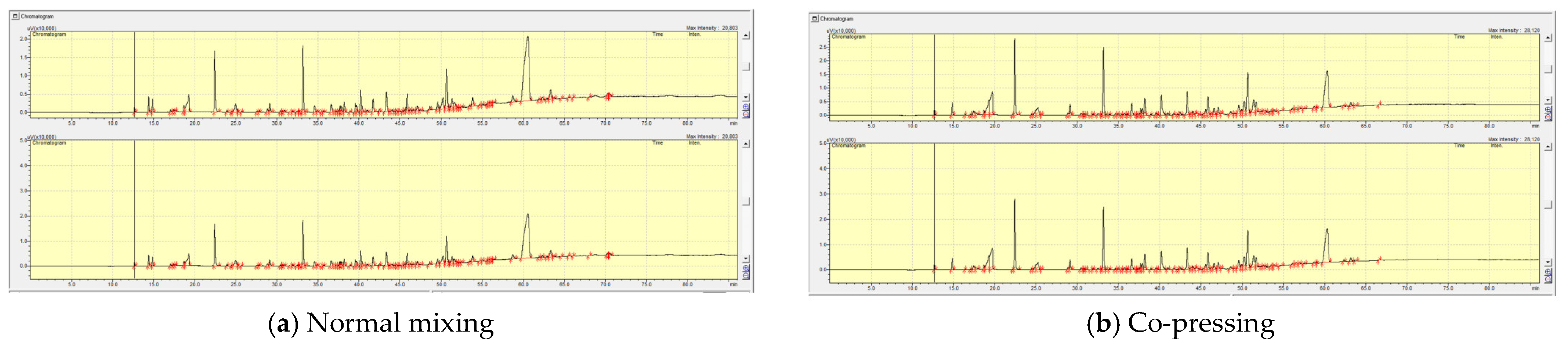
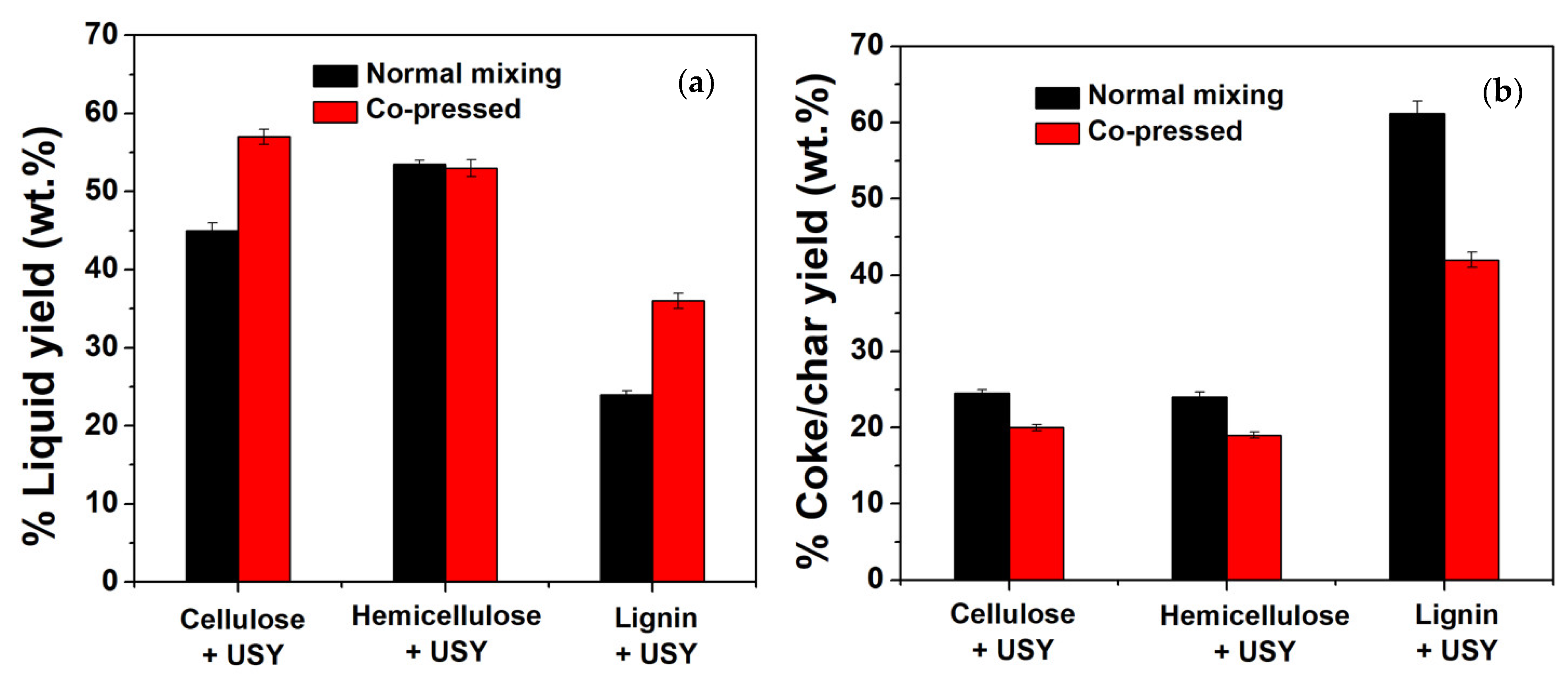
2.2.1. The Effect of Co-Pressing Pre-Treatment Method on Liquid Product Yields
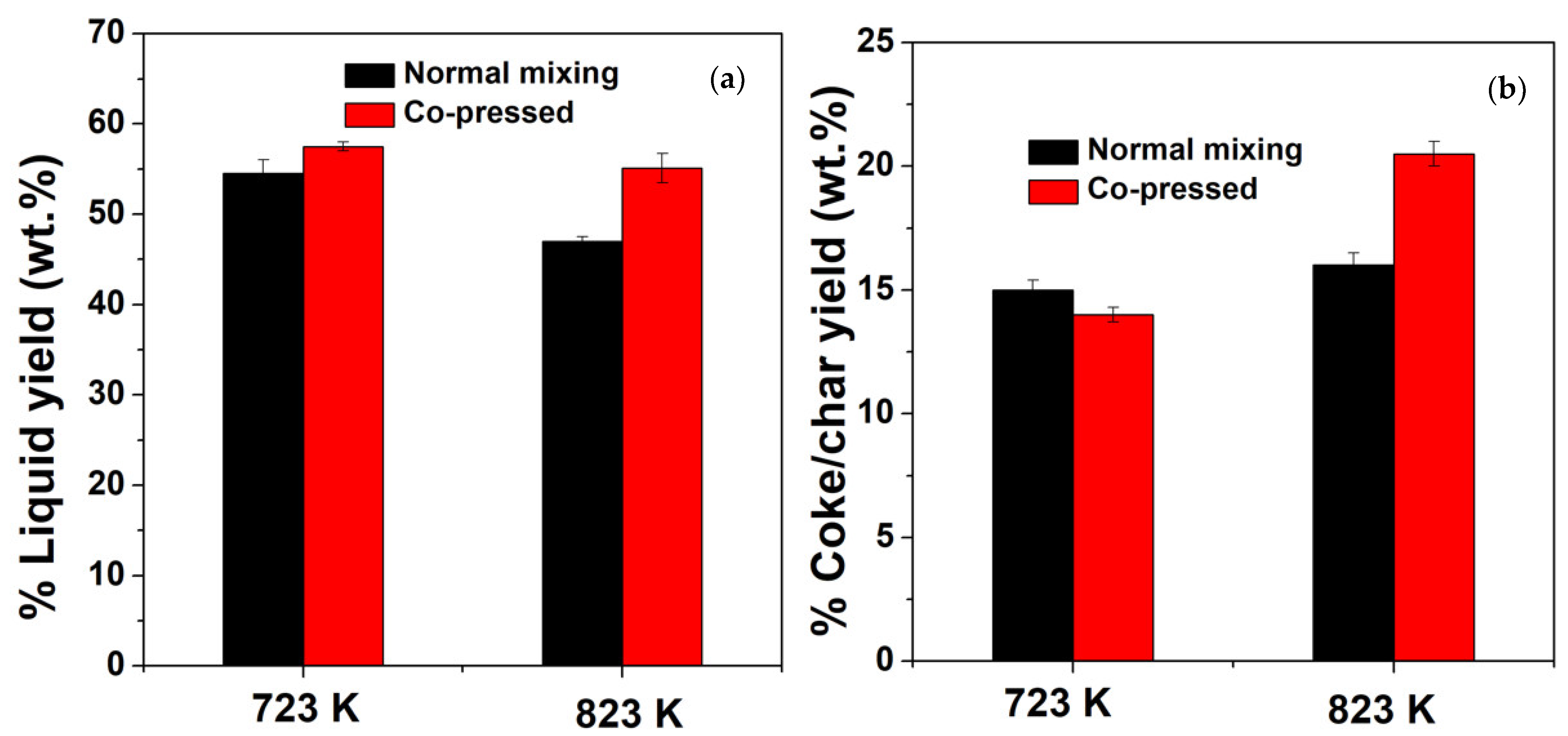
2.2.2. The Effect of the Reaction Temperature on the Product Yields of the Normal Mixing and Co-Pressed Samples
2.3. Liquid Product Characterisation
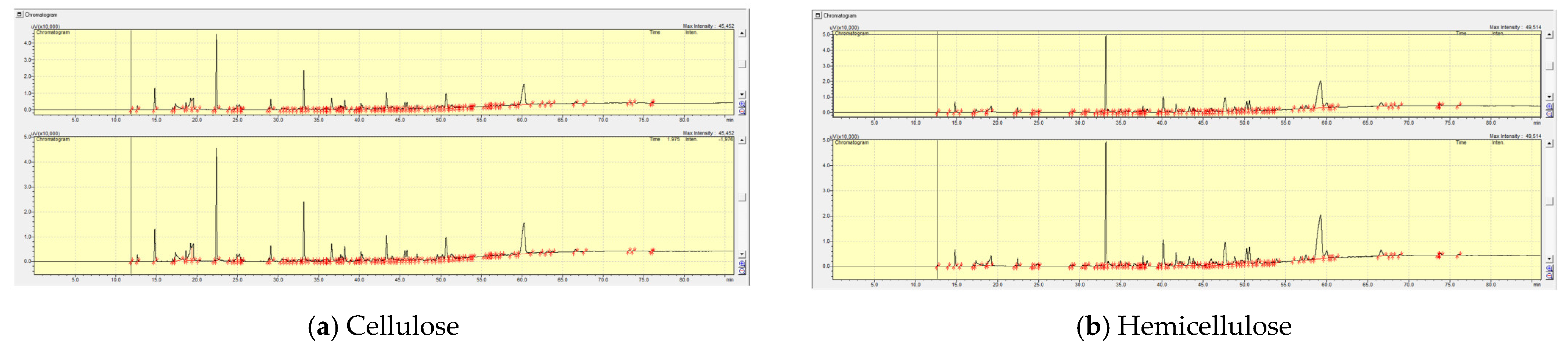
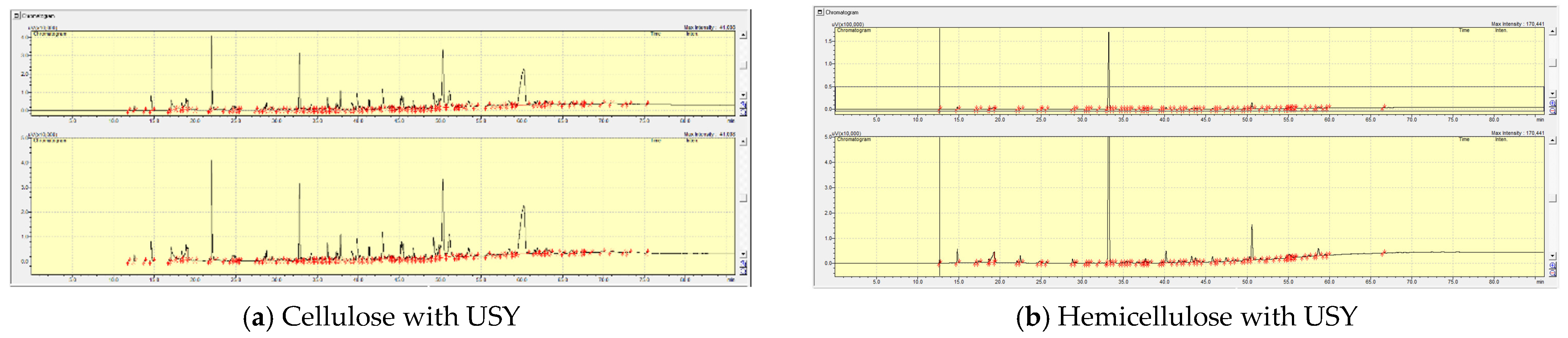
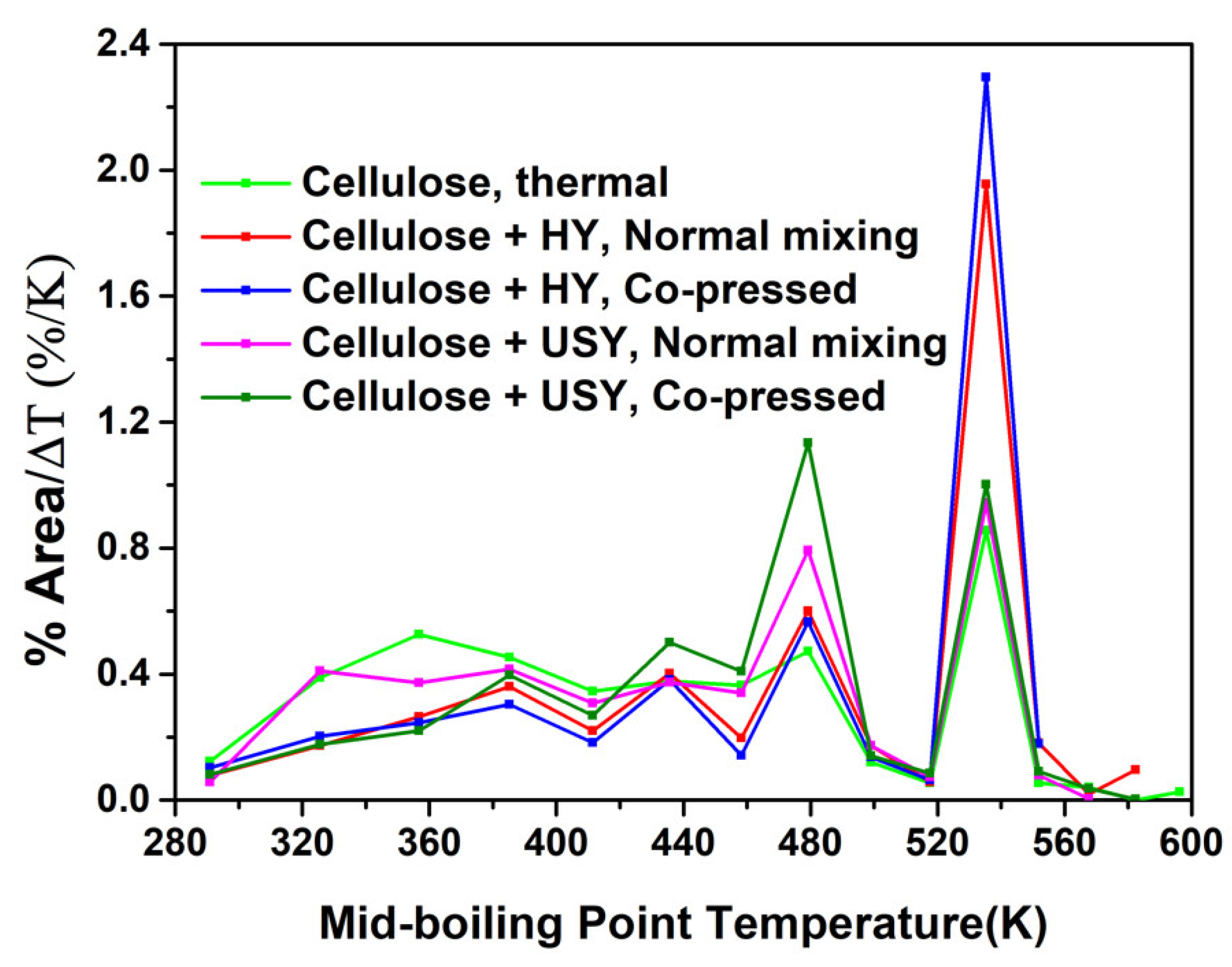
2.3.1. The Effect of Pre-Treatment (Co-Pressing Method) on the Boiling Point Distribution of the Bio-Oil from Catalytic Pyrolysis of Cellulose with HY and USY Catalysts
2.3.2. The Effect of Pre-Treatment (Co-Pressing Method) on the Boiling Point Distribution of the Bio-Oil from Catalytic Pyrolysis of Hemicellulose on HY and USY Catalysts
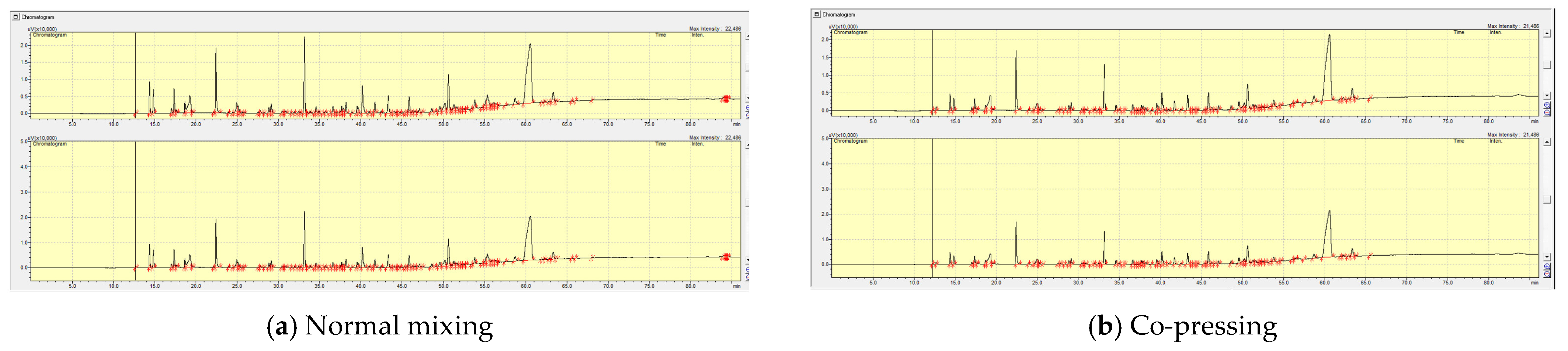
2.3.3. The Effect of Pre-Treatment (Co-Pressing Method) on the Boiling Point Distribution of the Bio-Oil from Catalytic Pyrolysis of Cellulose at Different Reaction Temperatures
2.4. Coked/Charred Catalyst Characterization
3. Experimental
3.1. Materials
3.2. Experimental Setup
3.3. Pre-Treatment Process
3.3.1. Normal Mixing
3.3.2. Co-Pressing
3.4. Experimental Calculations
3.5. Liquid Sample Analysis
3.6. Thermal Gravimetric Analysis
Coke/Char Catalyst Characterisation and Calculation
4. Conclusions
- Co-pressing method has improved the catalytic degradation of cellulose by further lowering the decomposition temperature.
- All the three biomass components produce a better result using the co-pressing method, where the liquid yields increased and coke/char yields decreased.
- The co-pressed cellulose with HY catalyst had further decreased the amount of lighter oxygenated fraction from cellulose thermal cracking.
- The application of USY catalyst has altered the selectivity in the boiling point distribution with the majority of the product within the middle fraction.
- The co-pressed cellulose with USY catalyst has further increased the amount of the middle fraction predominantly within the gasoline range fraction.
- The co-pressed hemicellulose with HY catalyst showed higher selectivity to middle fraction compared to the heavy fraction from the thermal cracking.
- The co-pressed hemicellulose with USY catalyst has produced higher amount lighter fraction compared to the heavy fraction from the thermal cracking.
- The improved catalytic effect of the co-pressed cellulose and USY catalyst has enhanced the formation of hard coke/char.
- The higher catalytic effect of the co-pressed sample at lower reaction temperatures leads to lower concentration of coke/char on the catalyst.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Replicates Values | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Experiment Type | Liquid Yields | Coke/Char Yields | ||||||||
| 1 | 2 | 3 | Mean | SD | 1 | 2 | 3 | Mean | SD | |
| Cellulose: USY/Normal | 44.00 | 46.00 | 45.00 | 45.00 | 1.00 | 24.50 | 24.00 | 25.00 | 24.50 | 0.50 |
| Cellulose: USY/Pressed | 57.00 | 56.00 | 58.00 | 57.00 | 1.00 | 20.00 | 20.40 | 19.60 | 20.00 | 0.40 |
| Hemicellulose: SY/Normal | 53.00 | 53.50 | 54.00 | 53.50 | 0.50 | 23.30 | 24.00 | 24.70 | 24.00 | 0.70 |
| Hemicellulose: SY/Pressed | 54.10 | 53.00 | 51.90 | 53.00 | 1.10 | 18.60 | 19.40 | 19.00 | 19.00 | 0.40 |
| Lignin: USY/Normal | 24.00 | 24.50 | 23.50 | 24.00 | 0.50 | 62.80 | 61.20 | 59.60 | 61.20 | 1.60 |
| Lignin: USY/Pressed | 37.00 | 35.00 | 36.00 | 36.00 | 1.00 | 43.00 | 41.00 | 42.00 | 42.00 | 1.00 |
| Cellulose: HY/Normal,723K | 56.00 | 54.50 | 53.00 | 54.50 | 1.50 | 14.60 | 15.40 | 15.00 | 15.00 | 0.40 |
| Cellulose: HY/Pressed,723K | 57.50 | 57.00 | 58.00 | 57.50 | 0.50 | 13.70 | 14.00 | 14.30 | 14.00 | 0.30 |
| Cellulose: HY/Normal,823K | 46.50 | 47.00 | 47.50 | 47.00 | 0.50 | 15.50 | 16.50 | 16.00 | 16.00 | 0.50 |
| Cellulose: HY/Pressed,823K | 55.10 | 56.70 | 53.50 | 55.10 | 1.60 | 20.50 | 20.00 | 21.00 | 20.50 | 0.50 |
References
- Chen, W.H.; Eng, C.F.; Lin, Y.Y.; Bach, Q.V. Independent parallel pyrolysis kinetics of cellulose, hemicelluloses and lignin at various heating rates analyzed by evolutionary computation. Energy Convers. Manag. 2020, 221, 113165. [Google Scholar] [CrossRef]
- Du, S.; Valla, J.A.; Bollas, G.M. Characteristics and origin of char and coke from fast and slow, catalytic and thermal pyrolysis of biomass and relevant model compounds. Green Chem. 2013, 15, 3214–3229. [Google Scholar] [CrossRef]
- Alonso, D.M.; Bond, J.Q.; Dumesic, J.A. Catalytic conversion of biomass to fuel. Green Chem. 2010, 12, 1493–1513. [Google Scholar] [CrossRef]
- Kan, T.; Strezov, V.; Evans, T.; He, J.; Kumar, R.; Lu, Q. Catalytic pyrolysis of lignocellulosic biomass: A review of variations in process factors and system structure. Renew. Sustain. Energy Rev. 2020, 134, 110305. [Google Scholar] [CrossRef]
- Zhu, L.; Zhong, Z. Effects of cellulose, hemicellulose and lignin on biomass pyrolysis kinetics. Korean J. Chem. Eng. 2020, 37, 1660–1668. [Google Scholar] [CrossRef]
- Zheng, Y.; Tao, L.; Yang, X.; Huang, Y.; Liu, C.; Zheng, Z. Comparative study on pyrolysis and catalytic pyrolysis upgrading of biomass model compounds: Thermochemical behaviors, kinetics, and aromatic hydrocarbon formation. J. Energy Inst. 2019, 92, 1348–1363. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Zheng, C.; Lee, D.H.; Liang, D.T. In-depth investigation of biomass pyrolysis based on three major components: Hemicellulose, cellulose and lignin. Energy Fuels 2006, 20, 388–393. [Google Scholar] [CrossRef]
- Huang, J.; He, C.; Wu, L.; Tong, H. Theoretical studies on thermal decomposition mechanism of arabinofuranose. J. Energy Inst. 2017, 90, 372–381. [Google Scholar] [CrossRef]
- Wang, S.R.; Ru, B.; Lin, H.Z.; Luo, Z.Y. Degradation mechanism of monosaccharides and xylan under pyrolytic conditions with theoretic modeling on the energy profiles. Bioresour. Technol. 2013, 143, 378–383. [Google Scholar] [CrossRef]
- Zhou, X.; Li, W.; Mabon, R.; Broadbelt, L.J. A critical review on hemicellulose pyrolysis. Energy Technol. 2017, 5, 52–79. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Parihar, A.; Bhattacharya, S. Cellulose fast pyrolysis for platform chemicals: Assessment of potential targets and suitable reactor technology. Biofuels Bioprod. Biorefin. 2020, 14, 446–468. [Google Scholar] [CrossRef]
- Brebu, M.; Tamminen, T.; Spiridon, I. Thermal degradation of various lignins by TG-MS/FTIR and Py-GC-MS. J. Anal. Appl. Pyrolysis 2013, 104, 531–539. [Google Scholar] [CrossRef]
- Bu, Q.; Lei, H.; Zacher, A.H.; Wang, L.; Ren, S.; Liang, J.; Wei, Y.; Liu, Y.; Tang, J.; Zhang, Q.; et al. A review of catalytic hydrodeoxygenation of lignin-derived phenols from biomass pyrolysis. Bioresour. Technol. 2012, 124, 470–477. [Google Scholar] [CrossRef]
- Chen, W.-H.; Peng, J.; Bi, X.T. A state-of-the-art review of biomass torrefaction, densification and applications. Renew. Sustain. Energy Rev. 2015, 44, 847–866. [Google Scholar] [CrossRef]
- Zadeh, Z.E.; Abdulkhani, A.; Aboelazayem, O.; Saha, B. Recent insights into lignocellulosic biomass pyrolysis: A critical review on pretreatment, characterization, and products upgrading. Processes 2020, 8, 799. [Google Scholar] [CrossRef]
- Siyal, A.A.; Mao, X.; Liu, Y.; Ran, C.; Fu, J.; Kang, Q.; Ao, W.; Zhang, R.; Dai, J.; Liu, G. Torrefaction subsequent to pelletization: Characterization and analysis of furfural residue and sawdust pellets. Waste Manag. 2020, 113, 210–224. [Google Scholar] [CrossRef]
- Chen, D.; Gao, A.; Ma, Z.; Fei, D.; Chang, Y.; Shen, C. In-depth study of rice husk torrefaction: Characterization of solid, liquid and gaseous products, oxygen migration and energy yield. Bioresour. Technol. 2018, 253, 148–153. [Google Scholar] [CrossRef]
- Ukaew, S.; Schoenborn, J.; Klemetsrud, B.; Shonnard, D.R. Effects of torrefaction temperature and acid pretreatment on the yield and quality of fast pyrolysis bio-oil from rice straw. J. Anal. Appl. Pyrolysis 2018, 129, 112–122. [Google Scholar] [CrossRef]
- Kumar, R.; Strezov, V.; Weldekidan, H.; He, J.; Singh, S.; Kan, T.; Dastjerdi, B. Lignocellulose biomass pyrolysis for bio-oil production: A review of biomass pre-treatment methods for production of drop-in fuels. Renew. Sustain. Energy Rev. 2020, 123, 109763. [Google Scholar] [CrossRef]
- Tumuluru, J.S.; Wright, C.T.; Hess, J.R.; Kenney, K.L. A review of biomass densification systems to develop uniform feedstock commodities for bioenergy application. Biofuels Bioprod. Biorefin. 2011, 5, 683–707. [Google Scholar] [CrossRef]
- Hoover, A.N.; Tumuluru, J.S.; Teymouri, F.; Moore, J.; Gresham, G. Effect of pelleting process variables on physical properties and sugar yields of ammonia fiber expansion pretreated corn stover. Bioresour. Technol. 2014, 164, 128–135. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.; Tabil, L.G.; Sokhansanj, S. Effects of compressive force, particle size and moisture content on mechanical properties of biomass pellets from grasses. Biomass Bioenergy 2006, 30, 648–654. [Google Scholar] [CrossRef]
- Pang, Y.; Wu, D.; Chen, Y.; Xu, J.; Wu, J.; Zhai, M. Pyrolysis of pine pellets catalyzed by blast furnace gas ash. Chem. Eng. Process. Process. Intensif. 2020, 156, 108094. [Google Scholar] [CrossRef]
- Shi, Y.; Xing, E.; Wu, K.; Wang, J.; Yang, M.; Wu, Y. Recent progress on upgrading of bio-oil to hydrocarbons over metal/zeolite bifunctional catalysts. Catal. Sci. Technol. 2017, 7, 2385–2415. [Google Scholar] [CrossRef]
- Adjaye, J.; Bakhshi, N. Production of hydrocarbons by catalytic upgrading of a fast pyrolysis bio-oil. Part I: Conversion over various catalysts. Fuel Process. Technol. 1995, 45, 161–183. [Google Scholar] [CrossRef]
- Hernando, H.; Hernández-Giménez, A.M.; Ochoa-Hernández, C.; Bruijnincx, P.C.A.; Houben, K.; Baldus, M.; Pizarro, P.; Coronado, J.M.; Fermoso, J.; Čejka, J.; et al. Engineering the acidity and accessibility of the zeolite ZSM-5 for efficient bio-oil upgrading in catalytic pyrolysis of lignocellulose. Green Chem. 2018, 20, 3499–3511. [Google Scholar] [CrossRef]
- Richardson, Y.; Eibner, S.; Tanoh, S.; Broust, F.; Blin, J.; Julbe, A. The catalyst/biomass integration concept for the direct thermo-catalytic conversion of biomass into either syngas or added-value molecules. ISGC 2015. Available online: https://agritrop.cirad.fr/579912/ (accessed on 29 June 2021).
- Lee, H.W.; Kim, Y.-M.; Lee, B.; Kim, S.; Jae, J.; Jung, S.-C.; Kim, T.-W.; Park, Y.-K. Catalytic copyrolysis of torrefied cork oak and high density polyethylene over a mesoporous HY catalyst. Catal. Today 2018, 307, 301–307. [Google Scholar] [CrossRef]
- Kabakcı, S.B.; Hacıbektaşoğlu, S. Catalytic Pyrolysis of Biomass, Pyrolysis, Mohamed Samer. In Pyrolysis; Samer, M., Ed.; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef] [Green Version]
- Adjaye, J.; Bakhshi, N. Production of hydrocarbons by catalytic upgrading of a fast pyrolysis bio-oil. Part II: Comparative catalyst performance and reaction pathways. Fuel Process. Technol. 1995, 45, 185–202. [Google Scholar] [CrossRef]
- Cheah, S.; Jablonski, W.S.; Olstad, J.L.; Carpenter, D.; Barthelemy, K.D.; Robichaud, D.J.; Andrews, J.C.; Black, S.K.; Oddo, M.D.; Westover, T. Effects of thermal pretreatment and catalyst on biomass gasification efficiency and syngas composition. Green Chem. 2016, 18, 6291–6304. [Google Scholar] [CrossRef]
- Jin, Q.; Wang, X.; Li, S.; Mikulčić, H.; Bešenić, T.; Deng, S.; Vujanović, M.; Tan, H.; Kumfer, B. Synergistic effects during co-pyrolysis of biomass and plastic: Gas, tar, soot, char products and thermogravimetric study. J. Energy Inst. 2019, 92, 108–117. [Google Scholar] [CrossRef]
- Mishra, R.K.; Mohanty, K. Thermal and catalytic pyrolysis of pine sawdust (Pinus ponderosa) and Gulmohar seed (Delonix regia) towards production of fuel and chemicals. Mater. Sci. Energy Technol. 2019, 2, 139–149. [Google Scholar] [CrossRef]
- Gunasee, S.D.; Danon, B.; Gorgens, J.; Mohee, R. Co-pyrolysis of LDPE and cellulose: Synergies during devolatilization and condensation. J. Anal. Appl. Pyrolysis 2017, 126, 307–314. [Google Scholar] [CrossRef]
- Patwardhan, P.R.; Dalluge, D.L.; Shanks, B.H.; Brown, R.C. Distinguishing primary and secondary reactions of cellulose pyrolysis. Bioresour. Technol. 2011, 102, 5265–5269. [Google Scholar] [CrossRef]
- Scheirs, J.; Camino, G.; Tumiatti, W. Overview of water evolution during the thermal degradation of cellulose. Eur. Polym. J. 2001, 37, 933–942. [Google Scholar] [CrossRef]
- Koufopanos, C.A.; Papayannakos, N.; Maschio, G.; Lucchesi, A. Modelling of the pyrolysis of biomass particles. Studies on kinetics, thermal and heat transfer effects. Can. J. Chem. Eng. 1991, 69, 907–915. [Google Scholar] [CrossRef]
- Milosavljevic, I.; Oja, A.V.; Suuberg, E.M. Thermal Effects in Cellulose Pyrolysis: Relationship to Char Formation Processes. Ind. Eng. Chem. Res. 1996, 35, 653–662. [Google Scholar] [CrossRef]
- Fermoso, J.; Mašek, O. Thermochemical decomposition of coffee ground residues by TG-MS: A kinetic study. J. Anal. Appl. Pyrolysis 2018, 130, 358–367. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhang, H.; Li, J.; Su, L.; Zuo, J.; Komarneni, S.; Wang, Y. Improving the aromatic production in catalytic fast pyrolysis of cellulose by co-feeding low-density polyethylene. Appl. Catal. A Gen. 2013, 455, 114–121. [Google Scholar] [CrossRef]
- Muhammad, I.; Manos, G. Simultaneous pretreatment and catalytic conversion of polyolefins into hydrocarbon fuels over acidic zeolite catalysts. Process. Saf. Environ. Prot. 2021, 146, 702–717. [Google Scholar] [CrossRef]
- Manos, G.; Garforth, A.; Dwyer, J. Catalytic Degradation of High-Density Polyethylene over Different Zeolitic Structures. Ind. Eng. Chem. Res. 2000, 39, 1198–1202. [Google Scholar] [CrossRef]
- Kassargy, C.; Awad, S.; Burnens, G.; Kahine, K.; Tazerout, M. Experimental study of catalytic pyrolysis of polyethylene and polypropylene over USY zeolite and separation to gasoline and diesel-like fuels. J. Anal. Appl. Pyrolysis 2017, 127, 31–37. [Google Scholar] [CrossRef]
- Kubička, D.; Kikhtyanin, O. Opportunities for zeolites in biomass upgrading—Lessons from the refining and petrochemical industry. Catal. Today 2015, 243, 10–22. [Google Scholar] [CrossRef]
- Jeong, J.; Lee, H.W.; Jang, S.H.; Ryu, S.; Kim, Y.-M.; Park, R.-S.; Jung, S.-C.; Jeon, J.-K.; Park, Y.-K. Park In-Situ Catalytic Fast Pyrolysis of Pinecone over HY Catalysts. Catalysts 2019, 9, 1034. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Z.; Wang, Y.; Syed-Hassan, S.S.A.; Hu, X.; Han, H.; Su, S.; Xu, K.; Jiang, L.; Guo, J.; Berthold, E.E.S.; et al. Effects of heating rate on the evolution of bio-oil during its pyrolysis. Energy Convers. Manag. 2018, 163, 420–427. [Google Scholar] [CrossRef]
- Liu, X.; Yao, Z.; Zhao, L.; Song, J.; Jia, J. Torrefaction of Sorghum Straw Pellets in a Stationary Reactor with a Feeding Screw. Energy Fuels 2020, 34, 5997–6007. [Google Scholar] [CrossRef]
- Wang, G.; Li, W.; Li, B.; Chen, H. TG study on pyrolysis of biomass and its three components under syngas. Fuel 2008, 87, 552–558. [Google Scholar] [CrossRef]
- Kim, B.-S.; Kim, Y.-M.; Lee, H.W.; Jae, J.; Kim, D.H.; Jung, S.-C.; Watanabe, C.; Park, Y.-K. Catalytic Copyrolysis of Cellulose and Thermoplastics over HZSM-5 and HY. ACS Sustain. Chem. Eng. 2016, 4, 1354–1363. [Google Scholar] [CrossRef]
- Xue, J.; Zhuo, J.; Liu, M.; Chi, Y.; Zhang, D.; Yao, Q. Synergetic Effect of Co-pyrolysis of Cellulose and Polypropylene over an All-Silica Mesoporous Catalyst MCM-41 Using Thermogravimetry–Fourier Transform Infrared Spectroscopy and Pyrolysis–Gas Chromatography–Mass Spectrometry. Energy Fuels 2017, 31, 9576–9584. [Google Scholar] [CrossRef]
- Balat, M. Mechanisms of Thermochemical Biomass Conversion Processes. Part 1: Reactions of Pyrolysis. Energy Sources 2008, 30, 620–635. [Google Scholar] [CrossRef]
- Ranzi, E.; Cuoci, A.; Faravelli, T.; Frassoldati, A.; Migliavacca, G.; Pierucci, S.; Sommariva, S. Chemical Kinetics of Biomass Pyrolysis. Energy Fuels 2008, 22, 4292–4300. [Google Scholar] [CrossRef]
- El Mansouri, N.-E.; Salvadó, J. Structural characterization of technical lignins for the production of adhesives: Application to lignosulfonate, kraft, soda-anthraquinone, organosolv and ethanol process lignins. Ind. Crop. Prod. 2006, 24, 8–16. [Google Scholar] [CrossRef]
- Jaea, J.; Tompsett, G.A.; Foster, A.J.; Hammond, K.D.; Auerbachac, S.M.; Lobo, R.F.; Huber, G.W. Investigation into the shape selectivity of zeolite catalysts for biomass conversion. J. Catal. 2011, 279, 257–268. [Google Scholar] [CrossRef]
- Muhammad, I.; Manos, G. Intensification of co-pyrolysis of plastic with biomass via pretreatment. Process. Saf. Environ. Prot. 2021, 146, 586–598. [Google Scholar] [CrossRef]
- Paulsen, A.D.; Mettler, M.S.; Dauenhauer, P.J. The Role of Sample Dimension and Temperature in Cellulose Pyrolysis. Energy Fuels 2013, 27, 2126–2134. [Google Scholar] [CrossRef]
- Mettler, M.S.; Mushrif, S.H.; Paulsen, A.D.; Javadekar, A.D.; Vlachos, D.; Dauenhauer, P.J. Revealing pyrolysis chemistry for biofuels production: Conversion of cellulose to furans and small oxygenates. Energy Environ. Sci. 2012, 5, 5414–5424. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Wang, H.; Lakshmikandan, M.; Wang, S.; Wang, Q.; He, Z.; Abomohra, A.E.-F. Catalytic co-pyrolysis of seaweeds and cellulose using mixed ZSM-5 and MCM-41 for enhanced crude bio-oil production. J. Therm. Anal. Calorim. 2020, 1–16. [Google Scholar] [CrossRef]
- Zhang, H.; Xiao, R.; Wang, D.; Zhong, Z.; Song, M.; Pan, Q.; He, G. Catalytic Fast Pyrolysis of Biomass in a Fluidized Bed with Fresh and Spent Fluidized Catalytic Cracking (FCC) Catalysts. Energy Fuels 2009, 23, 6199–6206. [Google Scholar] [CrossRef]
- Liu, Z.; Shi, C.; Wu, D.; He, S.; Ren, B. A Simple Method of Preparation of High Silica Zeolite Y and Its Performance in the Catalytic Cracking of Cumene. J. Nanotechnol. 2016, 2016, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Meng, B.; Ren, S.; Liu, X.; Zhang, L.; Hu, Q.; Wang, J.; Guo, Q.; Shen, B. Synthesis of USY Zeolite with a High Mesoporous Content by Introducing Sn and Enhanced Catalytic Performance. Ind. Eng. Chem. Res. 2020, 59, 5712–5719. [Google Scholar] [CrossRef]
- Zhang, L.; Bao, Z.; Xia, S.; Lu, Q.; Walters, K.B. Catalytic Pyrolysis of Biomass and Polymer Wastes. Catalysts 2018, 8, 659. [Google Scholar] [CrossRef] [Green Version]
- Niwa, M.; Suzuki, K.; Isamoto, K.; Katada, N. Identification and Measurements of Strong Brønsted Acid Site in Ultrastable Y (USY) Zeolite. J. Phys. Chem. B 2006, 110, 264–269. [Google Scholar] [CrossRef]
- Tang, B.; Li, S.; Song, W.C.; Yang, E.C.; Zhao, X.J.; Guan, N.; Li, L. Hierarchical FAU-Type Hafnosilicate Zeolite as a Robust Lewis Acid Catalyst for Catalytic Transfer Hydrogenation. ACS Sustain. Chem. Eng. 2019, 7, 16329–16343. [Google Scholar] [CrossRef]
- Marcilly, C. Acido-Basic Catalysis Application to Refining and Petrochemistry. Technip 2006, 2, 689–717. [Google Scholar]
- Corma, A.; Orchillés, A. Current views on the mechanism of catalytic cracking. Microporous Mesoporous Mater. 2000, 35, 21–30. [Google Scholar] [CrossRef]
- Lee, H.W.; Kim, Y.-M.; Jae, J.; Jeon, J.-K.; Jung, S.-C.; Kim, S.C.; Park, Y.-K. Production of aromatic hydrocarbons via catalytic co-pyrolysis of torrefied cellulose and polypropylene. Energy Convers. Manag. 2016, 129, 81–88. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Lee, H.W.; Jae, J.; Bin Jung, K.; Jung, S.-C.; Watanabe, A.; Park, Y.-K. Catalytic co-pyrolysis of biomass carbohydrates with LLDPE over Al-SBA-15 and mesoporous ZSM-5. Catal. Today 2017, 298, 46–52. [Google Scholar] [CrossRef]
- Park, Y.-K.; Siddiqui, M.Z.; Kang, Y.; Watanabe, A.; Lee, H.W.; Jeong, S.J.; Kim, S.; Kim, Y.-M. Increased Aromatics Formation by the Use of High-Density Polyethylene on the Catalytic Pyrolysis of Mandarin Peel over HY and HZSM-5. Catalysts 2018, 8, 656. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Li, J.; Zhou, G.; Feng, Y.; Wang, Y.; Yu, G.; Deng, S.; Huang, J.; Wang, B. Enhancing the production of renewable petrochemicals by co-feeding of biomass with plastics in catalytic fast pyrolysis with ZSM-5 zeolites. Appl. Catal. A Gen. 2014, 481, 173–182. [Google Scholar] [CrossRef]
- Bertero, M.; García, J.R.; Falco, M.; Sedran, U. Equilibrium FCC catalysts to improve liquid products from biomass pyrolysis. Renew. Energy 2019, 132, 11–18. [Google Scholar] [CrossRef]
- Patwardhan, P.R.; Brown, R.C.; Shanks, B.H. Product Distribution from the Fast Pyrolysis of Hemicellulose. ChemSusChem 2011, 4, 636–643. [Google Scholar] [CrossRef]
- Corma, A.; Huber, G.W.; Sauvanaud, L.; O’Connor, P. Processing biomass-derived oxygenates in the oil refinery: Catalytic cracking (FCC) reaction pathways and role of catalyst. J. Catal. 2007, 247, 307–327. [Google Scholar] [CrossRef]
- Wang, K.; Kim, K.H.; Brown, R.C. Catalytic pyrolysis of individual components of lignocellulosic biomass. Green Chem. 2014, 16, 727–735. [Google Scholar] [CrossRef]
- Räisänen, U.; Pitkänen, I.; Halttunen, H.; Hurtta, M. Formation of the main degradation compounds from arabinose, xylose, mannose and arabinitol during pyrolysis. J. Therm. Anal. Calorim. 2003, 72, 481–488. [Google Scholar] [CrossRef]
- Wang, S.; Liu, Q.; Wang, K.; Guo, X.; Luo, Z.; Cen, K.; Fransson, T. Study on Catalytic Pyrolysis of Manchurian Ash for Production of Bio-Oil. Int. J. Green Energy 2010, 7, 300–309. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, Y.; Liang, T.; Guo, X. Catalytic pyrolysis of mannose as a model compound of hemicellulose over zeolites. Biomass Bioenergy 2013, 57, 106–112. [Google Scholar] [CrossRef]
- Guo, X.; Wang, S.; Zhou, Y.; Luo, Z. Catalytic pyrolysis of xylan-based hemicellulose over zeolites. Int. J. Energy Env. 2011, 5, 137–142. [Google Scholar] [CrossRef]
- Lee, H.W.; Farooq, A.; Jang, S.-H.; Kwon, E.E.; Jae, J.; Lam, S.S.; Jung, S.-C.; Park, Y.-K. Enhanced bioaromatics synthesis via catalytic co-pyrolysis of cellulose and spent coffee ground over microporous HZSM-5 and HY. Environ. Res. 2020, 184, 109311. [Google Scholar] [CrossRef]
- Wong, S.L.; Ngadi, N.; Abdullah, T.; Inuwa, I. Conversion of low density polyethylene (LDPE) over ZSM-5 zeolite to liquid fuel. Fuel 2017, 192, 71–82. [Google Scholar] [CrossRef]
- Carlson, T.R.; Jae, J.; Lin, Y.-C.; Tompsett, G.A.; Huber, G.W. Catalytic fast pyrolysis of glucose with HZSM-5: The combined homogeneous and heterogeneous reactions. J. Catal. 2010, 270, 110–124. [Google Scholar] [CrossRef]
- French, R.; Czernik, S. Catalytic pyrolysis of biomass for biofuels production. Fuel Process. Technol. 2010, 91, 25–32. [Google Scholar] [CrossRef]
- Zhang, H.; Cheng, Y.-T.; Vispute, T.P.; Xiao, R.; Huber, G.W. Catalytic conversion of biomass-derived feedstocks into olefins and aromatics with ZSM-5: The hydrogen to carbon effective ratio. Energy Environ. Sci. 2011, 4, 2297–2307. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Brown, R.C. Catalytic pyrolysis of microalgae for production of aromatics and ammonia. Green Chem. 2013, 15, 675–681. [Google Scholar] [CrossRef]
- Rezaei, P.S.; Shafaghat, H.; Daud, W.M.A.W. Suppression of coke formation and enhancement of aromatic hydrocarbon production in catalytic fast pyrolysis of cellulose over different zeolites: Effects of pore structure and acidity. RSC Adv. 2015, 5, 65408–65414. [Google Scholar] [CrossRef]
- Wang, S.; Dai, G.; Yang, H.; Luo, Z. Lignocellulosic biomass pyrolysis mechanism: A state-of-the-art review. Prog. Energy Combust. Sci. 2017, 62, 33–86. [Google Scholar] [CrossRef]
- Ma, Z.; Van Bokhoven, J.A. Deactivation and Regeneration of H-USY Zeolite during Lignin Catalytic Fast Pyrolysis. ChemCatChem 2012, 4, 2036–2044. [Google Scholar] [CrossRef]
- Zong, P.; Jiang, Y.; Tian, Y.; Li, J.; Yuan, M.; Ji, Y.; Chen, M.; Li, D.; Qiao, Y. Pyrolysis behavior and product distributions of biomass six group components: Starch, cellulose, hemicellulose, lignin, protein and oil. Energy Convers. Manag. 2020, 216, 112777. [Google Scholar] [CrossRef]
- Wang, S.; Ru, B.; Lin, H.; Sun, W. Pyrolysis behaviors of four O-acetyl-preserved hemicelluloses isolated from hardwoods and softwoods. Fuel 2015, 150, 243–251. [Google Scholar] [CrossRef]
- Chew, J.J.; Doshi, V. Recent advances in biomass pretreatment—Torrefaction fundamentals and technology. Renew. Sustain. Energy Rev. 2011, 15, 4212–4222. [Google Scholar] [CrossRef]
- Shuping, Z.; Yulong, W.; Mingde, Y.; Chun, L.; Junmao, T. Pyrolysis characteristics and kinetics of the marine microalgae Dunaliella tertiolecta using thermogravimetric analyzer. Bioresour. Technol. 2010, 101, 359–365. [Google Scholar] [CrossRef]
- Huang, Y.; Kudo, S.; Masek, O.; Norinaga, K.; Hayashi, J.-I. Simultaneous Maximization of the Char Yield and Volatility of Oil from Biomass Pyrolysis. Energy Fuels 2013, 27, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Johnston, P.; Brown, R.C. An experimental study of the competing processes of evaporation and polymerization of levoglucosan in cellulose pyrolysis. J. Anal. Appl. Pyrolysis 2013, 99, 130–136. [Google Scholar] [CrossRef]
- Gobin, K.; Manos, G. Polymer degradation to fuels over microporous catalysts as a novel tertiary plastic recycling method. Polym. Degrad. Stab. 2004, 83, 267–279. [Google Scholar] [CrossRef]
- Akpanudoh, N.S.; Gobin, K.; Manos, G. Catalytic degradation of plastic waste to liquid fuel over commercial cracking catalysts: Effect of polymer to catalyst ratio/acidity content. J. Mol. Catal. A Chem. 2005, 235, 67–73. [Google Scholar] [CrossRef]
- Gulab, H.; Jan, M.R.; Shah, J.; Manos, G. Plastic catalytic pyrolysis to fuels as tertiary polymer recycling method: Effect of process conditions. J. Environ. Sci. Health Part A 2010, 45, 908–915. [Google Scholar] [CrossRef]
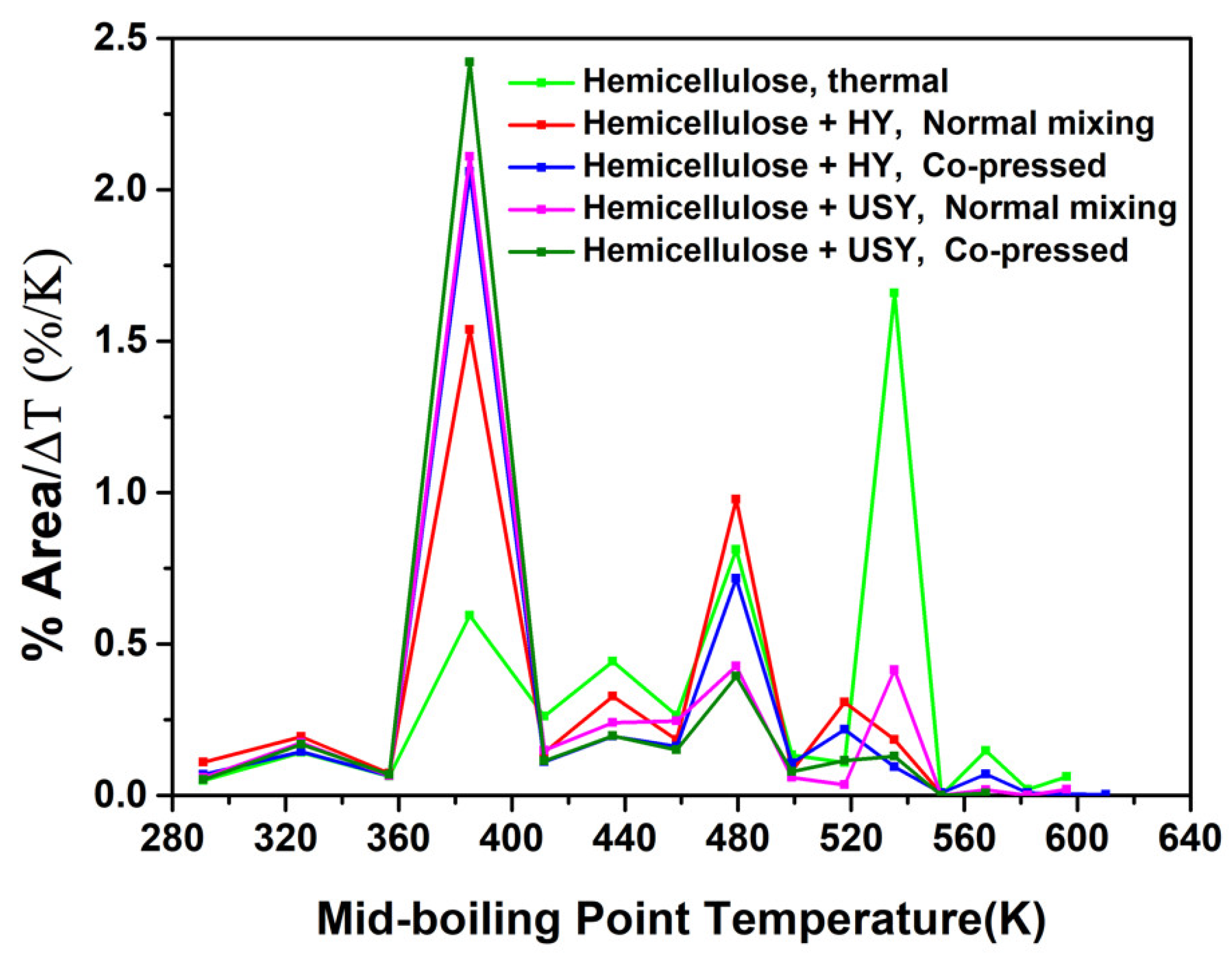

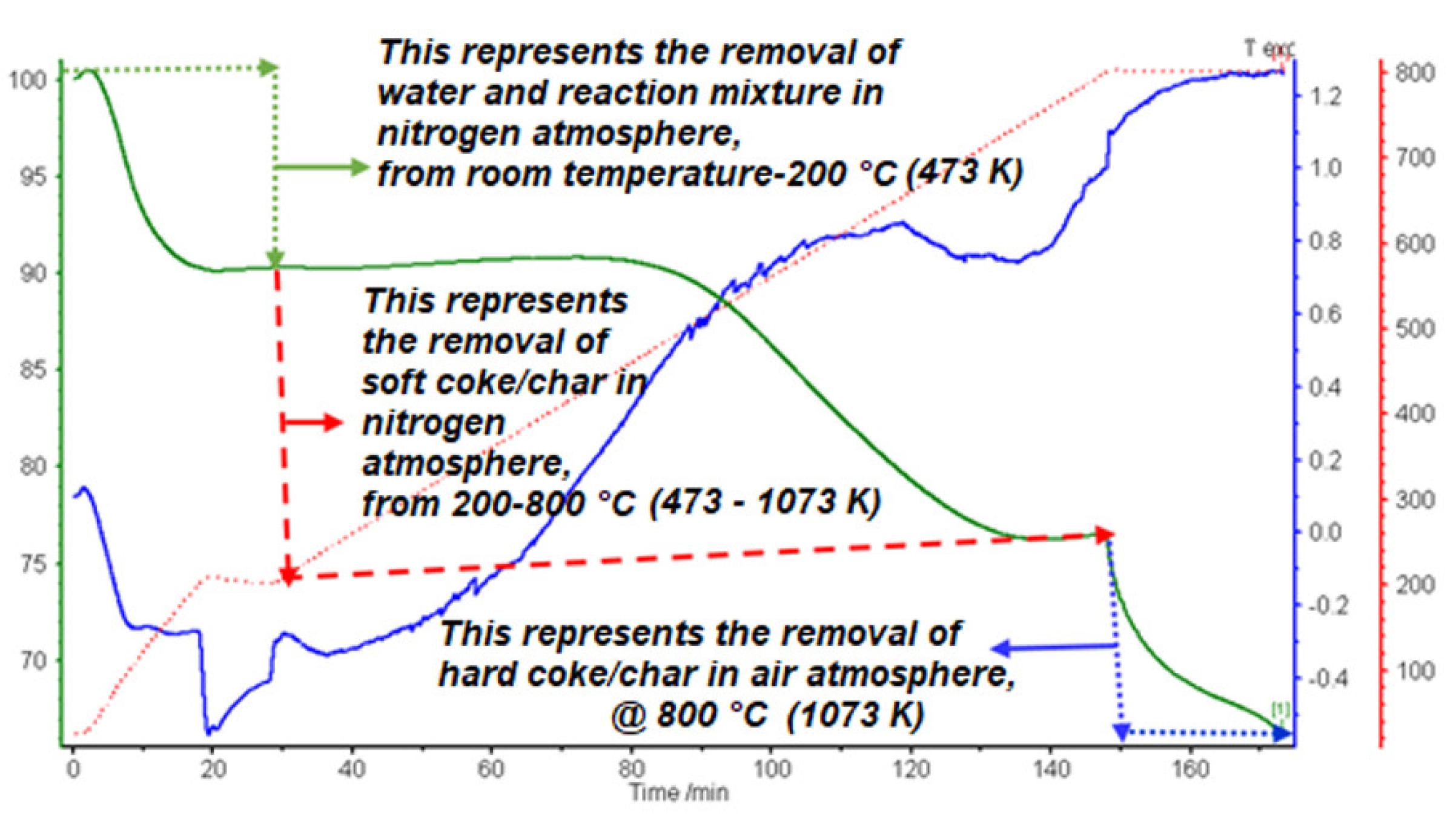
| Sample | Experiment Type | % Soft Coke/Char | % Hard Coke/Char | Coke/Char Concentration |
|---|---|---|---|---|
| Cellulose | Thermal | 72.0 | 28.0 | - |
| Cellulose + USY | Catalytic (Normal mixing) | 78.0 | 22.0 | 0.59 |
| Cellulose + USY | Catalytic (Co-pressed) | 71.0 | 29.0 | 0.51 |
| Hemicellulose | Thermal | 14.5 | 85.5 | - |
| Hemicellulose + USY | Catalytic (Normal mixing) | 75.5 | 24.5 | 0.45 |
| Lignin | Thermal | 36.0 | 64.0 | - |
| Lignin + USY | Catalytic (Normal mixing) | 73.0 | 27.0 | 1.45 |
| Catalyst | Properties | ||||
|---|---|---|---|---|---|
| BET (m2/g) | Micropore Volume (cm3/g) | Micropore Surface Area (m2/g) | External Surface Area (m2/g) | Si/Al Ratio | |
| HY | 590.00 ± 23.50 | 0.26 | 532.40 | 72.58 | 2.5 |
| USY | 433.60 ± 14.08 | 0.19 | 361.02 | 72.58 | 5.7 (framework) |
| Group of Carbon Atom | Boiling Point (K) | Group of Carbon Atom | Average Boiling Point (K) | ΔT(K) | Retention Time (min) |
|---|---|---|---|---|---|
| C4H10 | 272.70 | C4H10–C5H12 | 290.95 | 36.50 | - |
| C5H12 | 309.20 | C5H12–C6H14 | 325.55 | 32.70 | 16.03 |
| C6H14 | 341.90 | C6H14–C7H16 | 356.75 | 29.70 | 20.94 |
| C7H16 | 371.60 | C7H16–C8H18 | 385.20 | 27.20 | 27.16 |
| C8H18 | 398.80 | C8H18–C9H20 | 411.40 | 25.20 | 33.26 |
| C9H20 | 424.00 | C9H20–C10H22 | 435.65 | 23.30 | 38.64 |
| C10H22 | 447.30 | C10H22–C11H24 | 458.20 | 21.80 | 43.37 |
| C11H24 | 469.10 | C11H24–C12H26 | 479.30 | 20.40 | 47.61 |
| C12H26 | 489.50 | C12H26–C13H28 | 499.05 | 19.10 | 51.49 |
| C13H28 | 508.60 | C13H28–C14H30 | 517.65 | 18.10 | 55.05 |
| C14H30 | 526.70 | C14H30–C15H32 | 535.25 | 17.10 | 58.51 |
| C15H32 | 543.80 | C15H32–C16H34 | 551.90 | 16.20 | 61.91 |
| C16H34 | 560.00 | C16H34–C17H36 | 567.60 | 15.20 | 65.26 |
| C17H36 | 575.20 | C17H36–C18H38 | 582.35 | 14.30 | 68.71 |
| C18H38 | 589.50 | C18H38–C19H40 | 596.30 | 13.60 | 72.43 |
| C19H40 | 603.10 | C19H40–C20H42 | 610.05 | 13.90 | 76.60 |
| C20H42 | 617.00 | C20H42+ | 617.00 | - | 81.45 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhammad, I.; Manos, G. Improving the Conversion of Biomass in Catalytic Pyrolysis via Intensification of Biomass—Catalyst Contact by Co-Pressing. Catalysts 2021, 11, 805. https://doi.org/10.3390/catal11070805
Muhammad I, Manos G. Improving the Conversion of Biomass in Catalytic Pyrolysis via Intensification of Biomass—Catalyst Contact by Co-Pressing. Catalysts. 2021; 11(7):805. https://doi.org/10.3390/catal11070805
Chicago/Turabian StyleMuhammad, Ishaka, and George Manos. 2021. "Improving the Conversion of Biomass in Catalytic Pyrolysis via Intensification of Biomass—Catalyst Contact by Co-Pressing" Catalysts 11, no. 7: 805. https://doi.org/10.3390/catal11070805
APA StyleMuhammad, I., & Manos, G. (2021). Improving the Conversion of Biomass in Catalytic Pyrolysis via Intensification of Biomass—Catalyst Contact by Co-Pressing. Catalysts, 11(7), 805. https://doi.org/10.3390/catal11070805