
Alternative Aqueous Phase Synthesis of a PtRu/C Electrocatalyst for Direct Methanol Fuel Cells


Abstract
:
1. Introduction
2. Results and Discussion
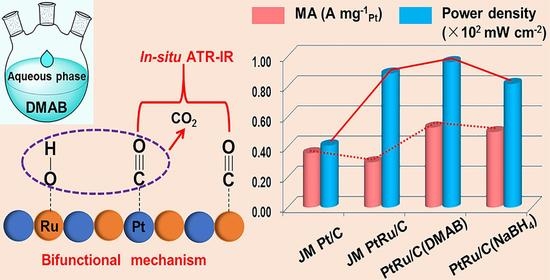
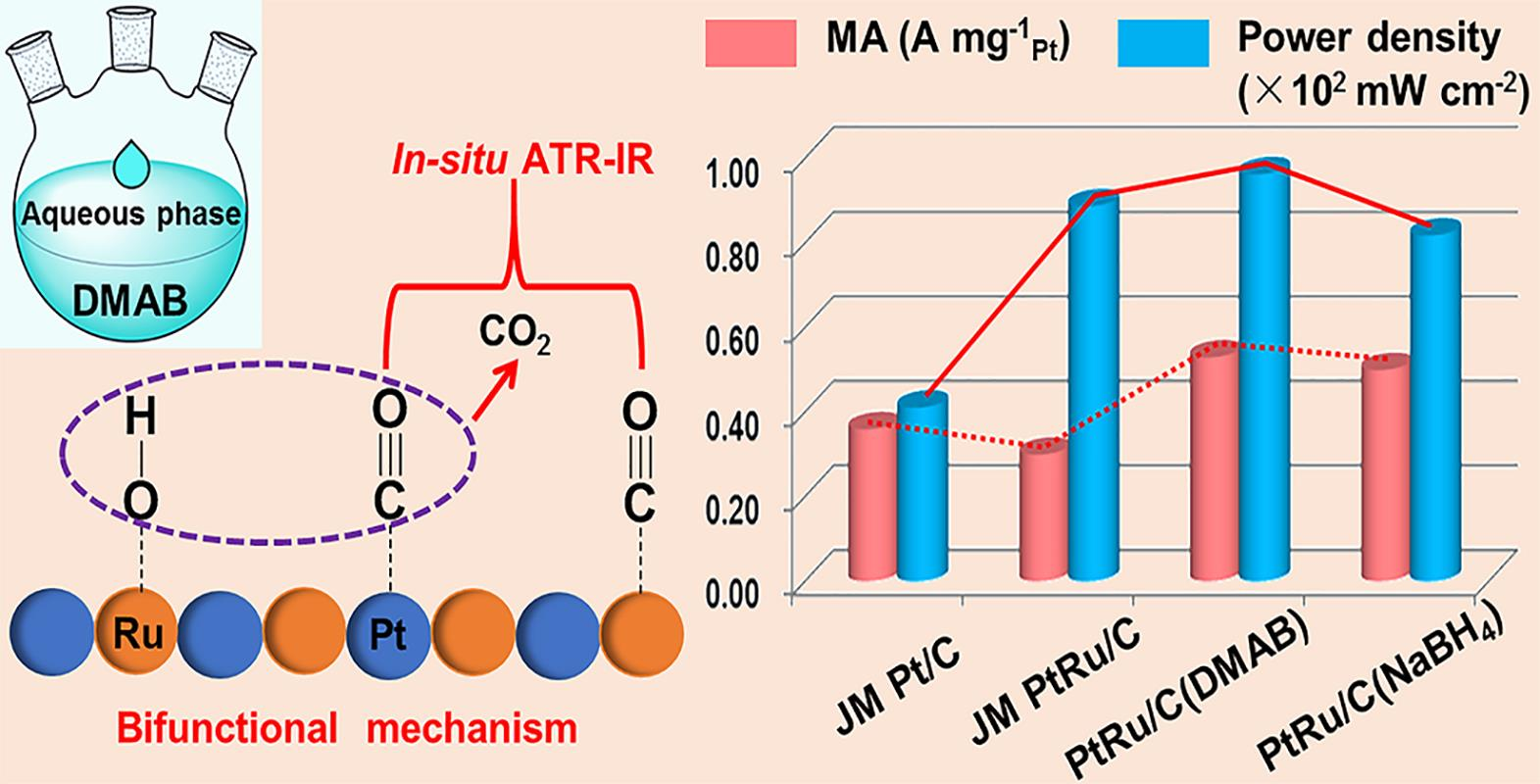
2.1. Synthesis Mechanism of PtRu/C(DMAB) Nanoparticles
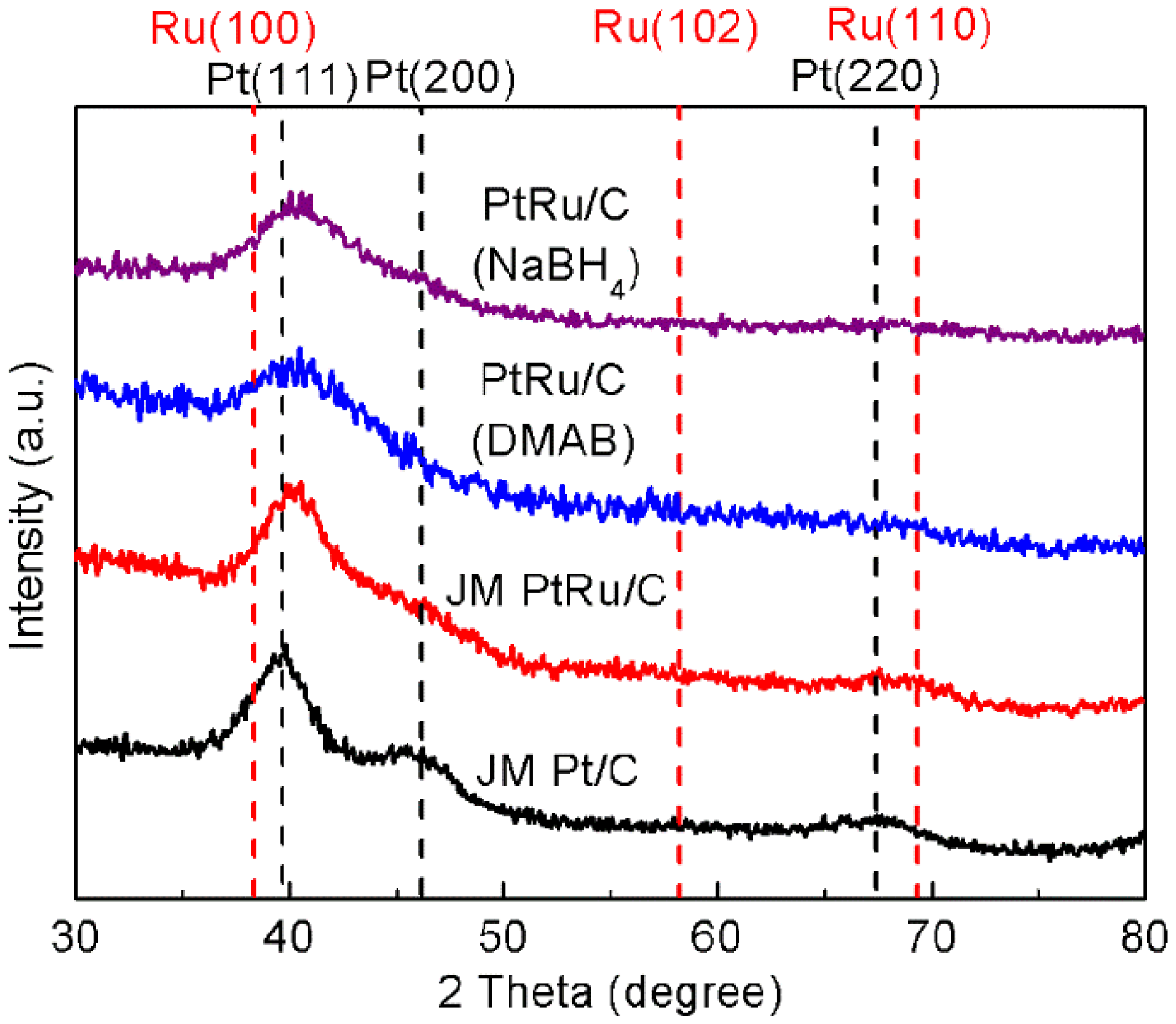
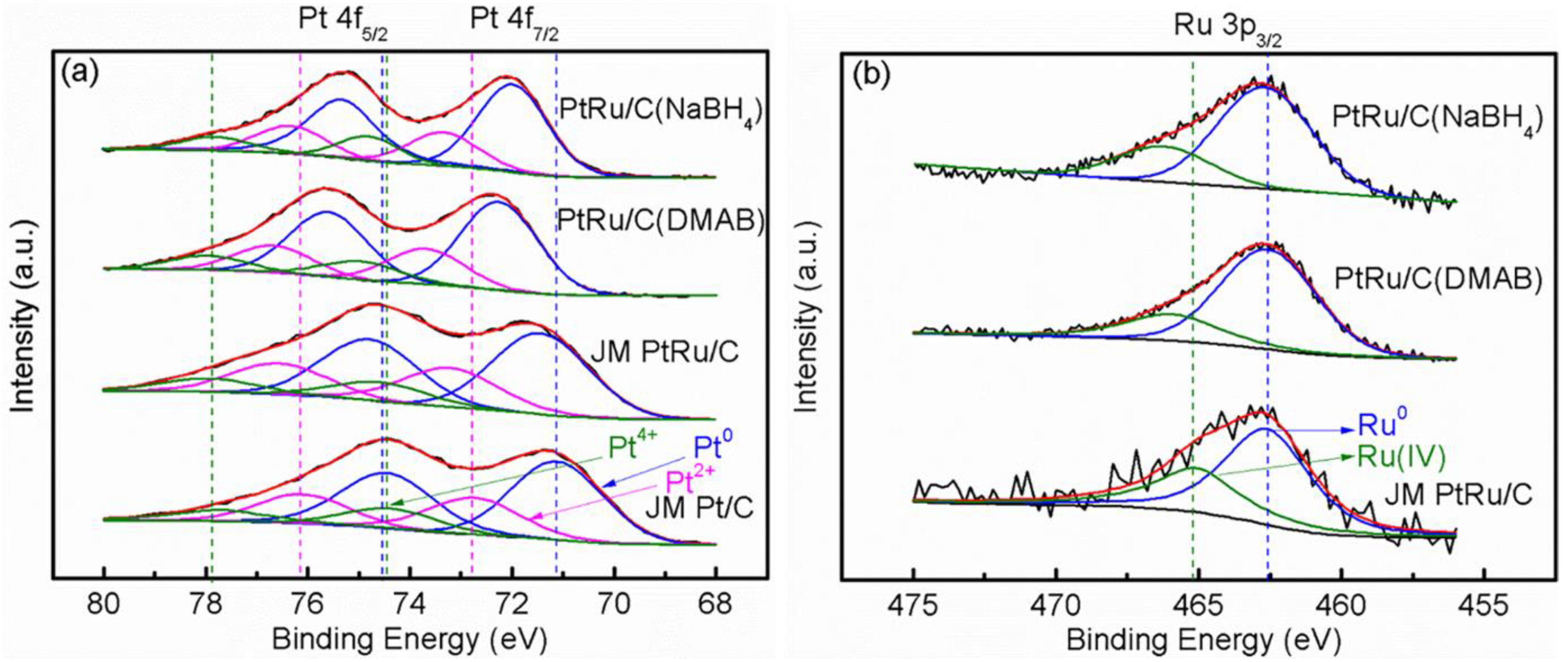
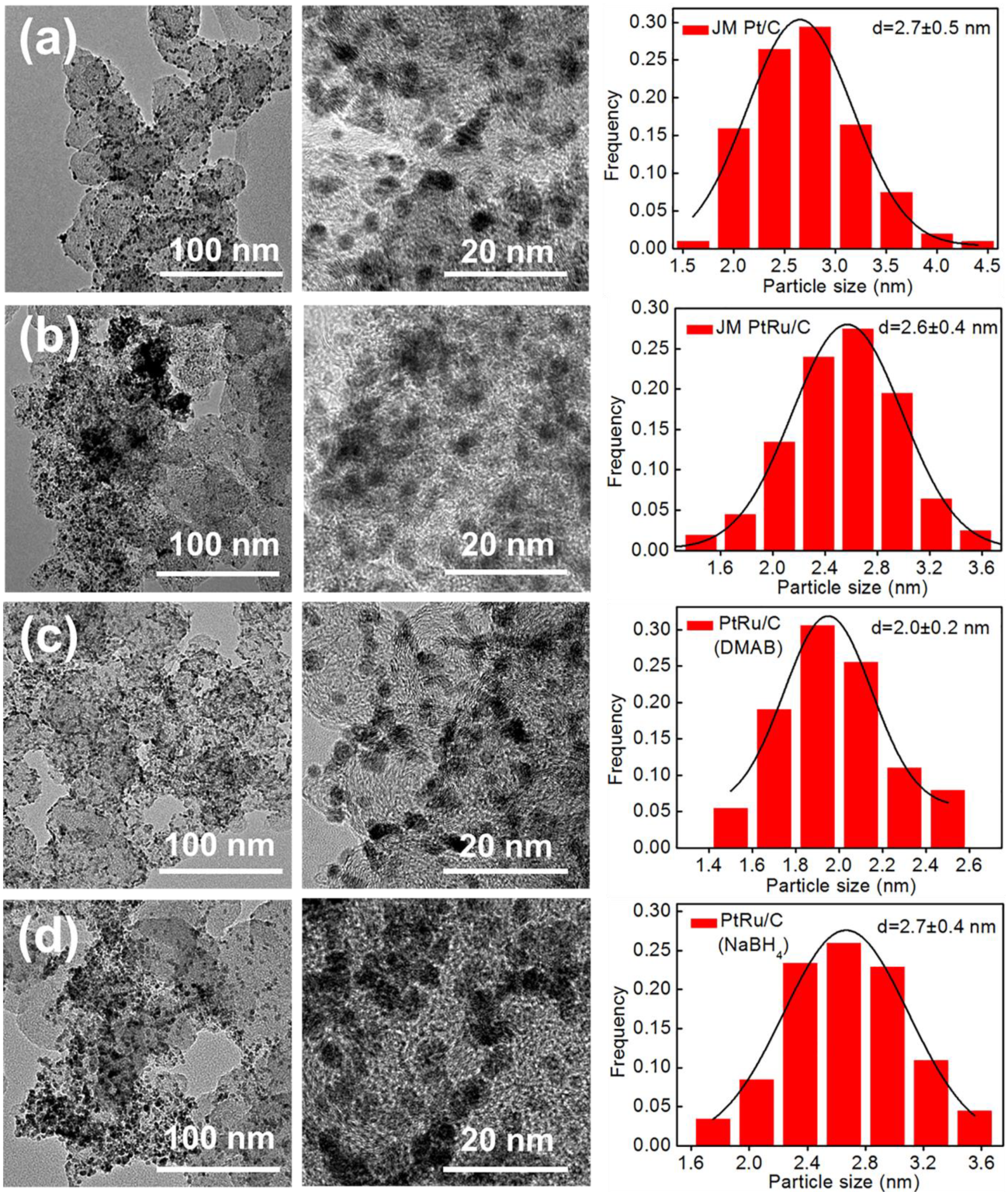
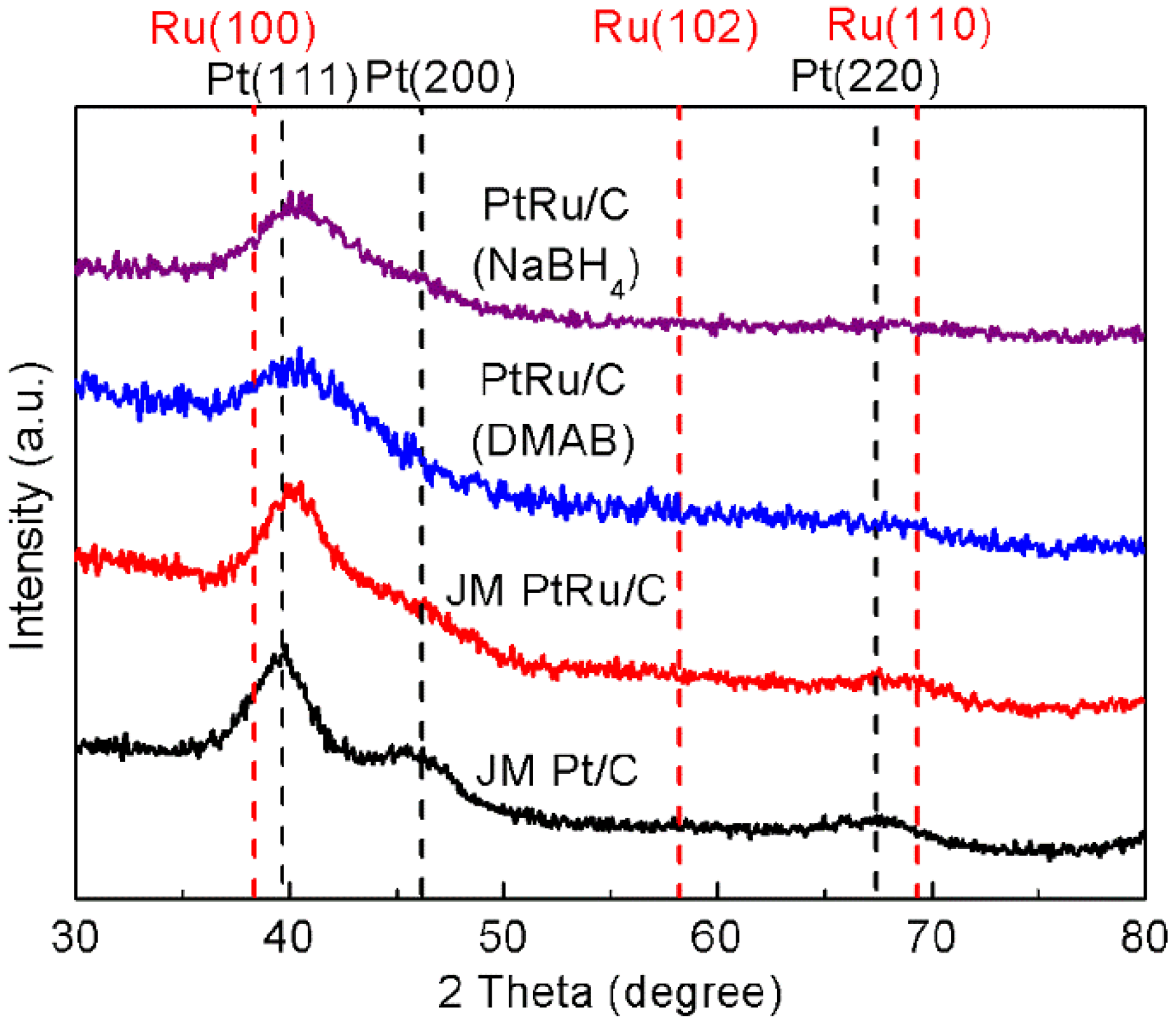
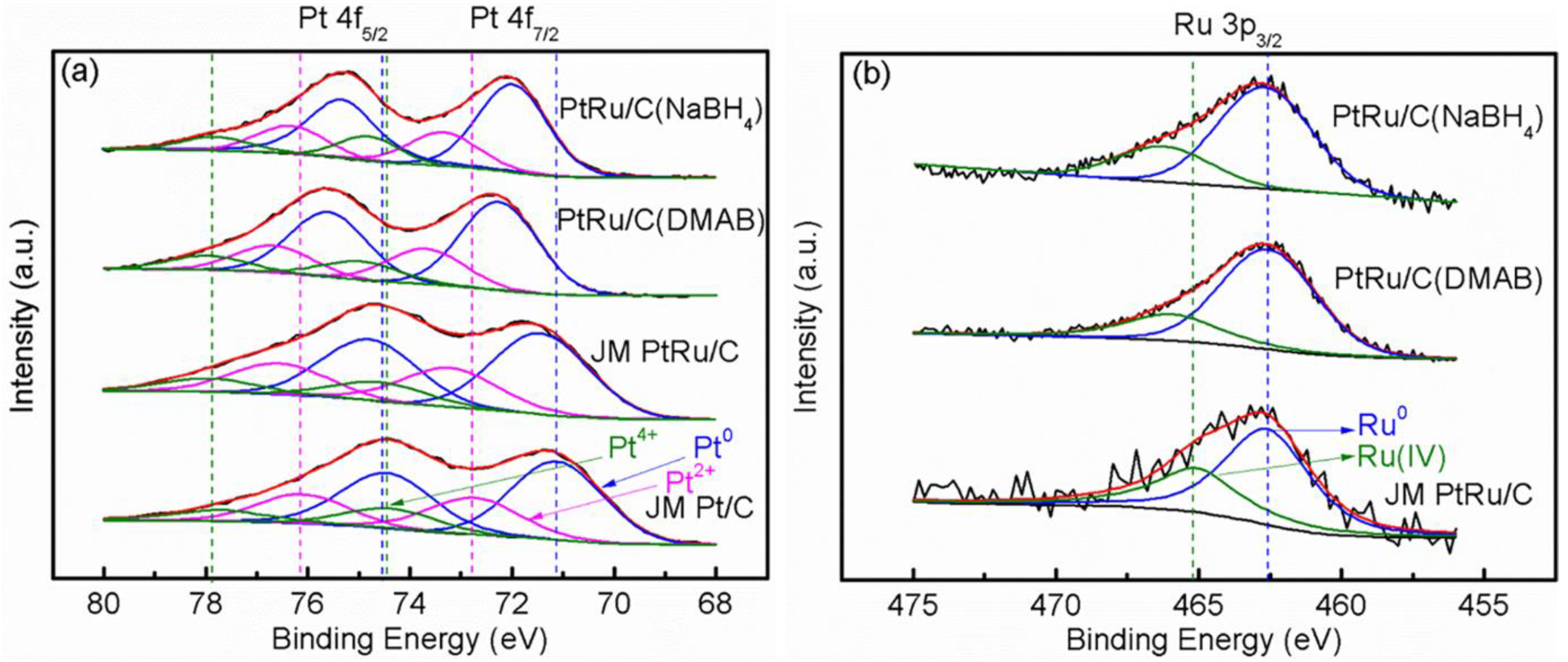
2.2. Physicochemical Characterization
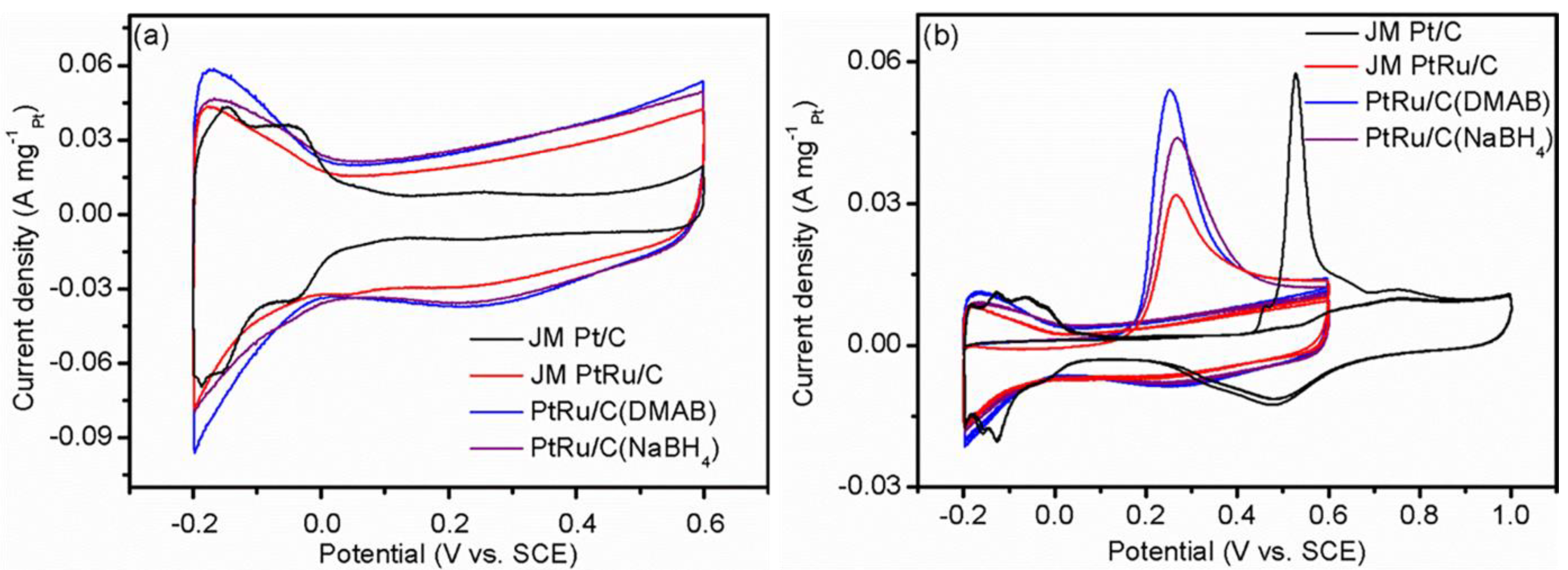
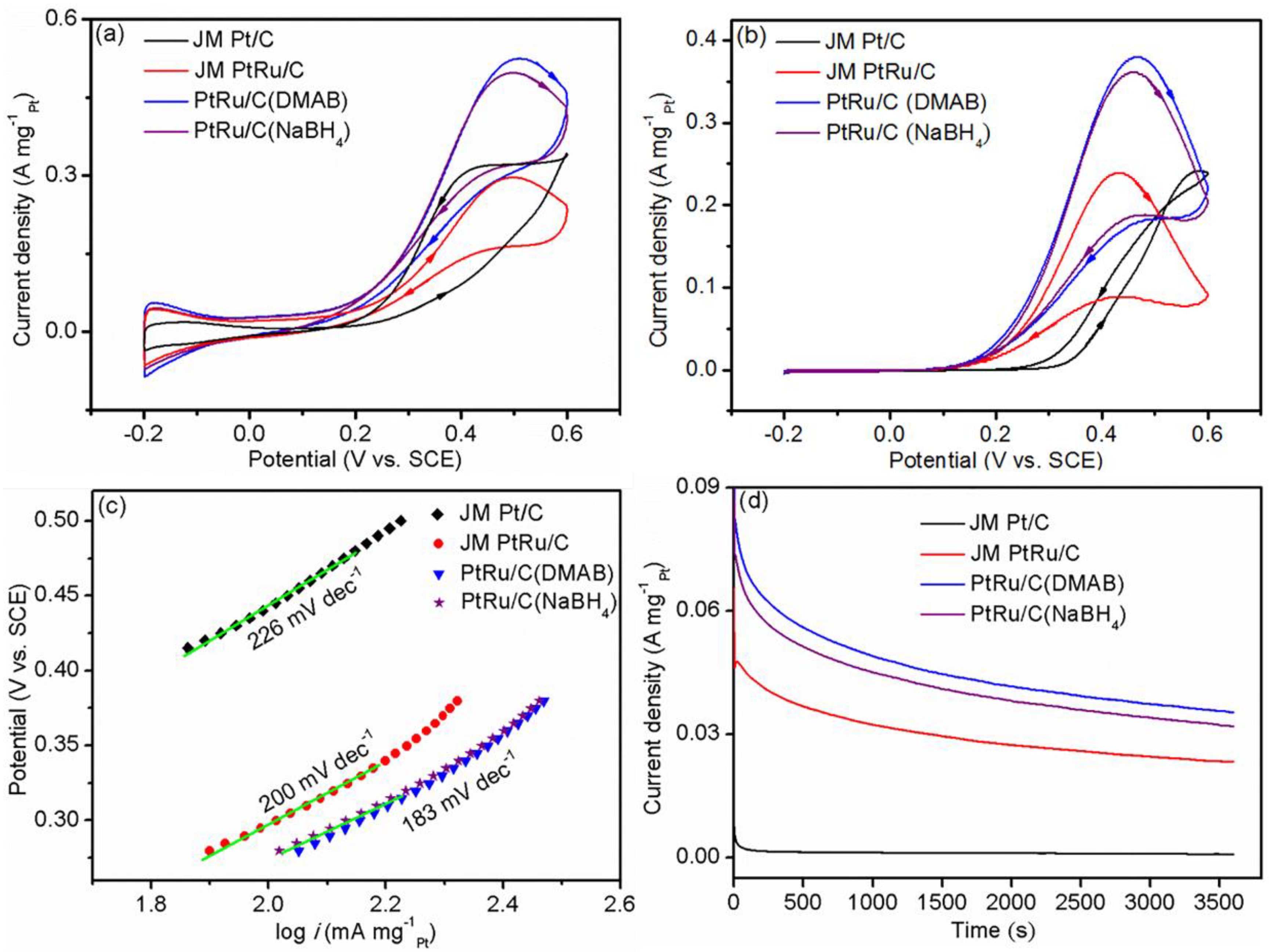
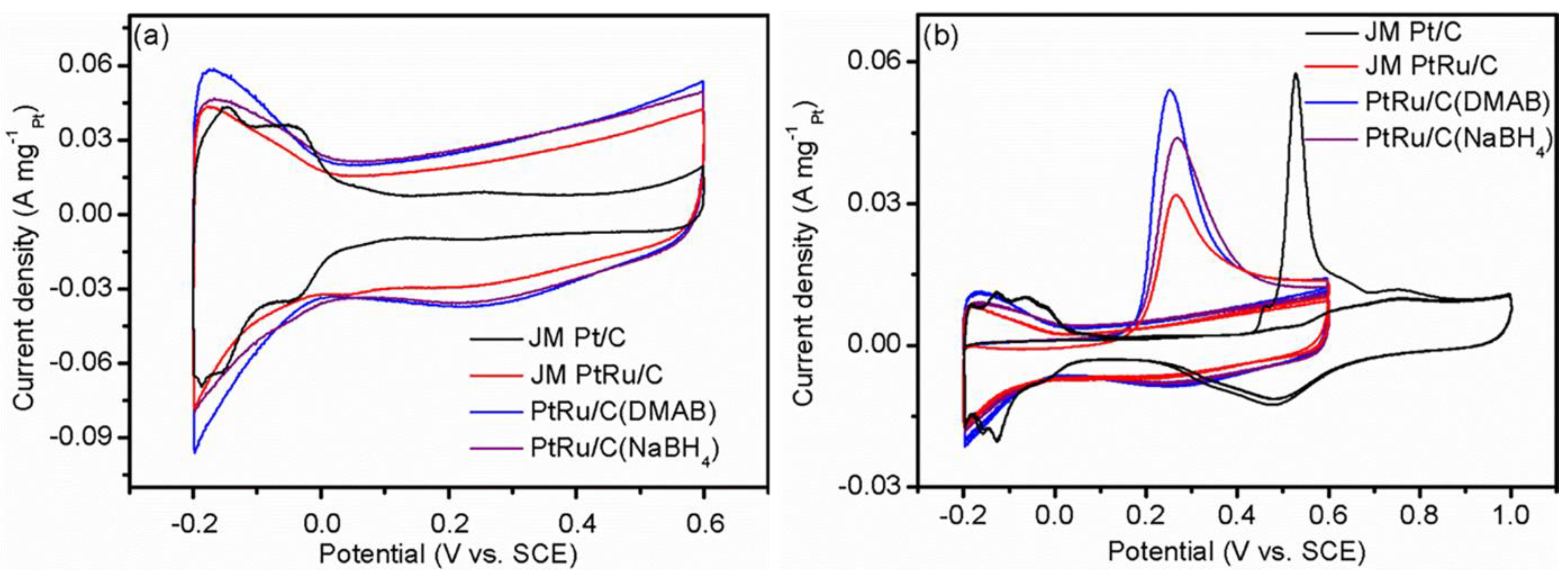
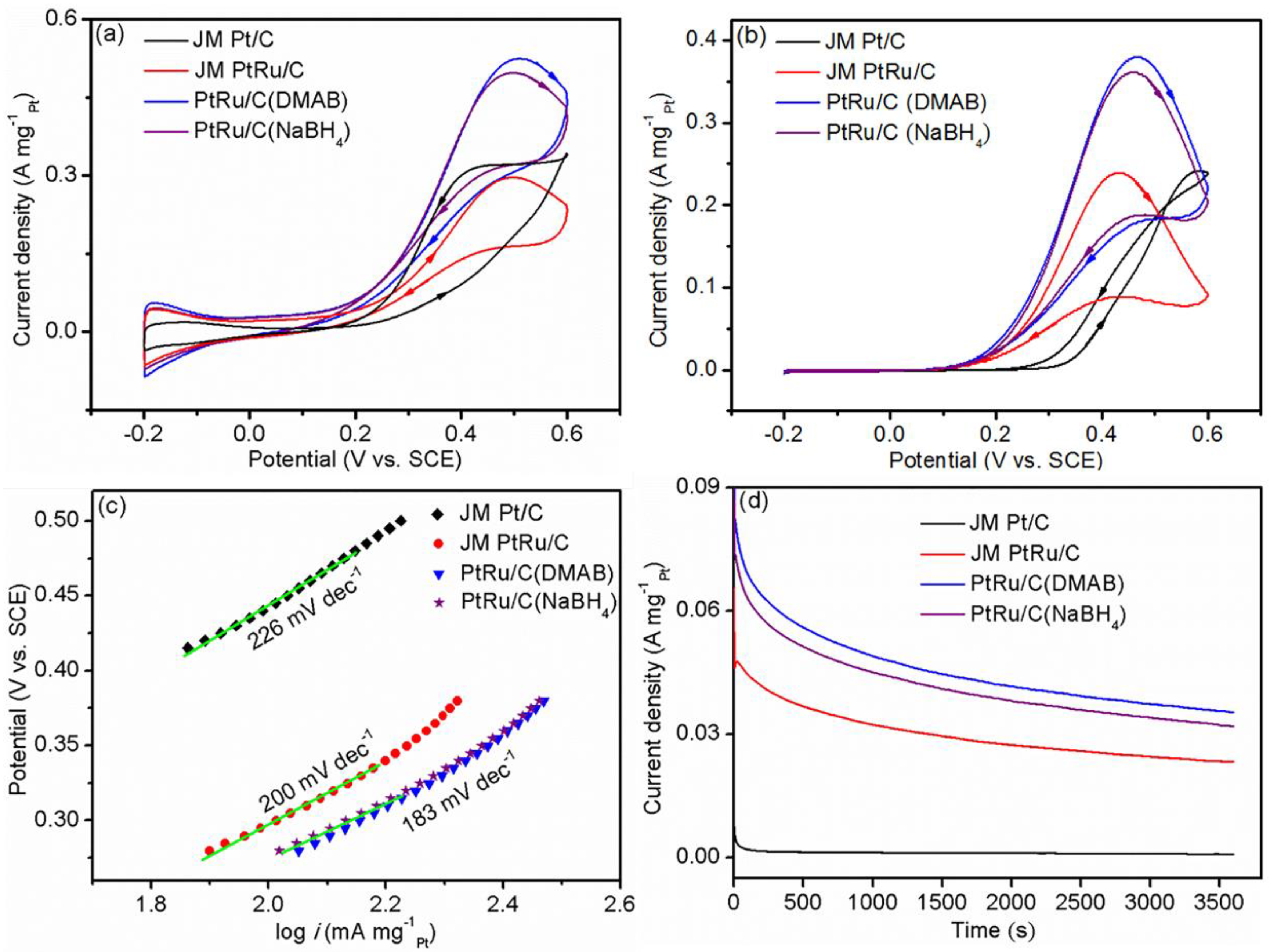
2.3. Electrochemical Performance
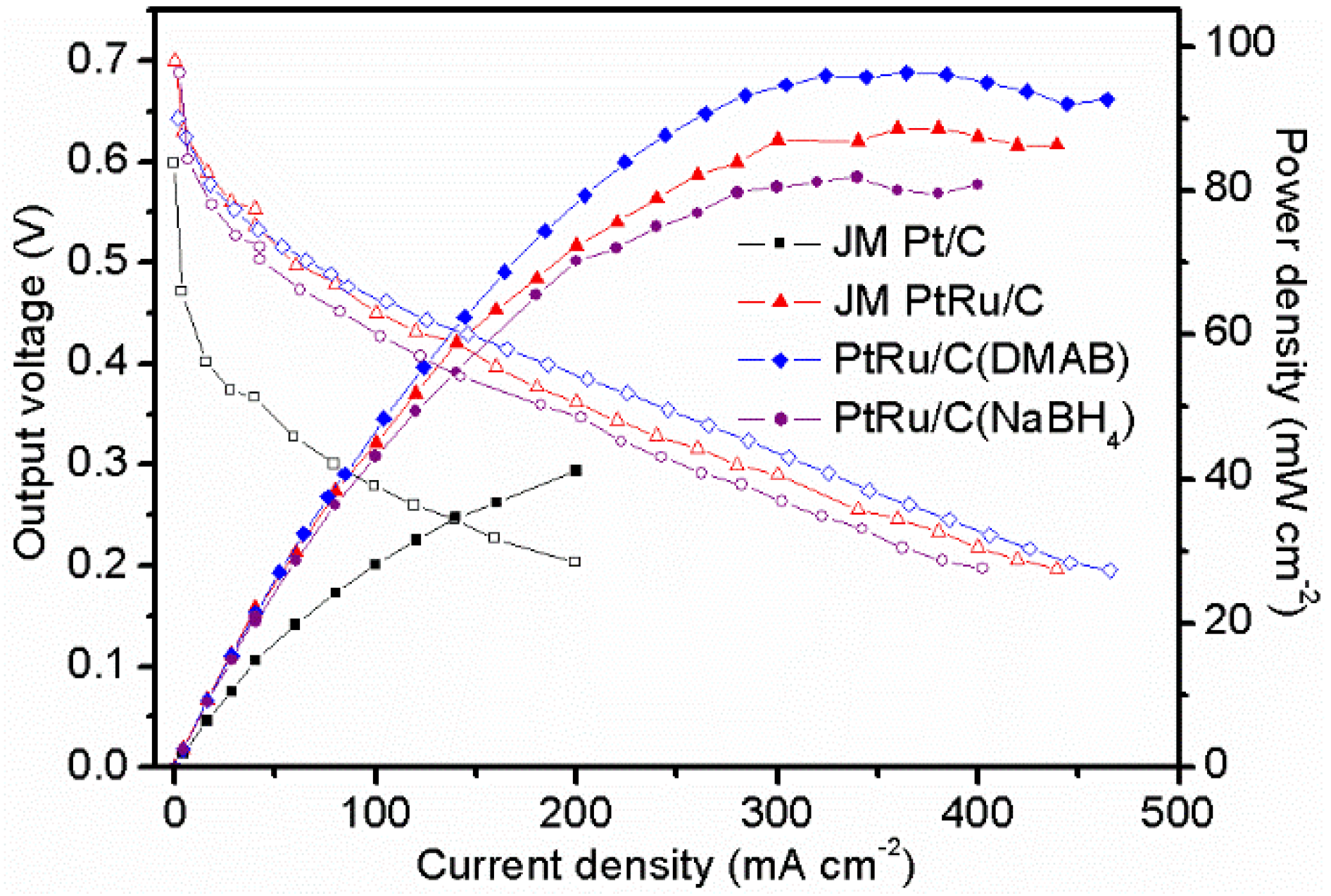
2.4. Preliminary DMFC Test
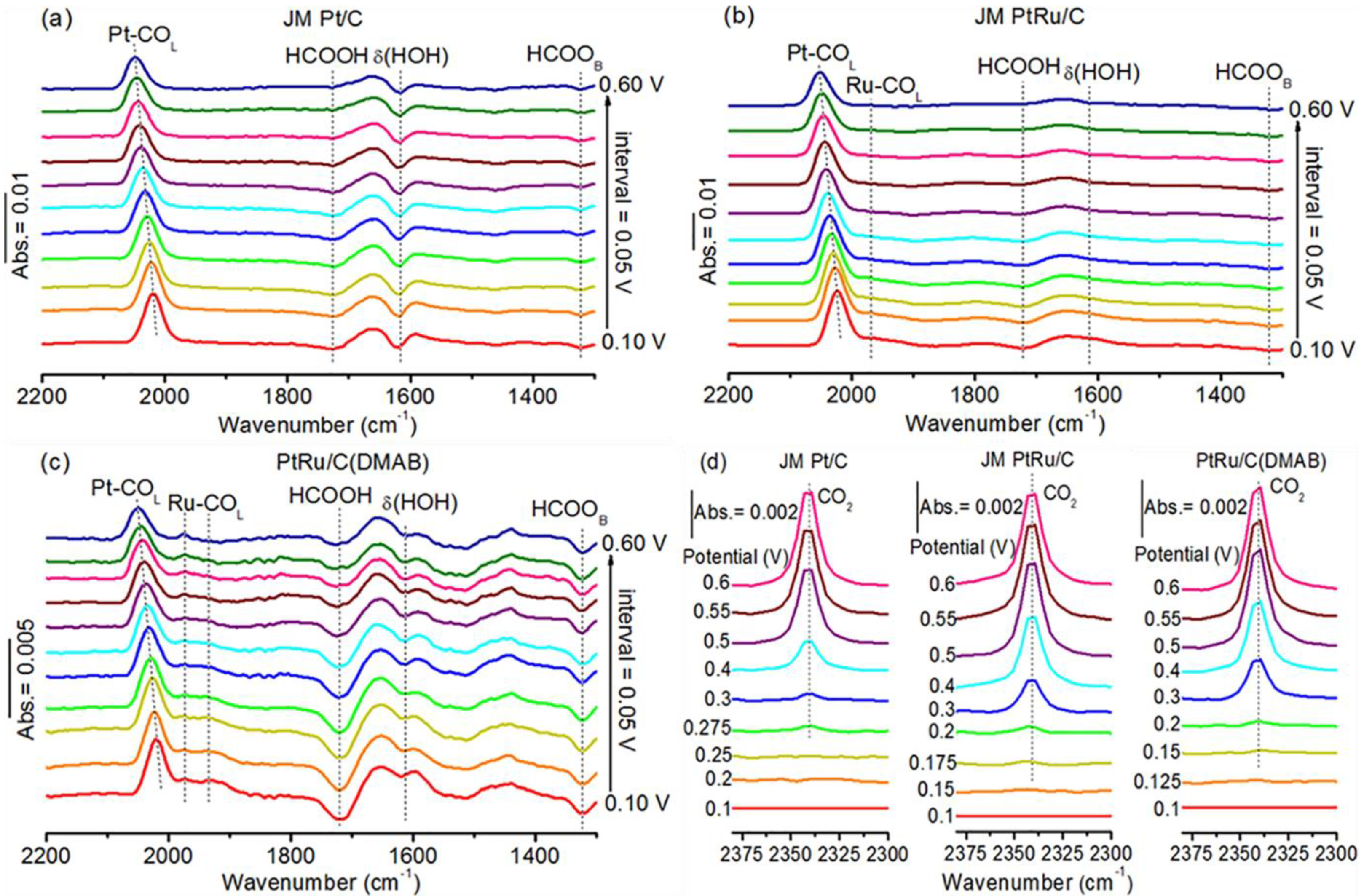
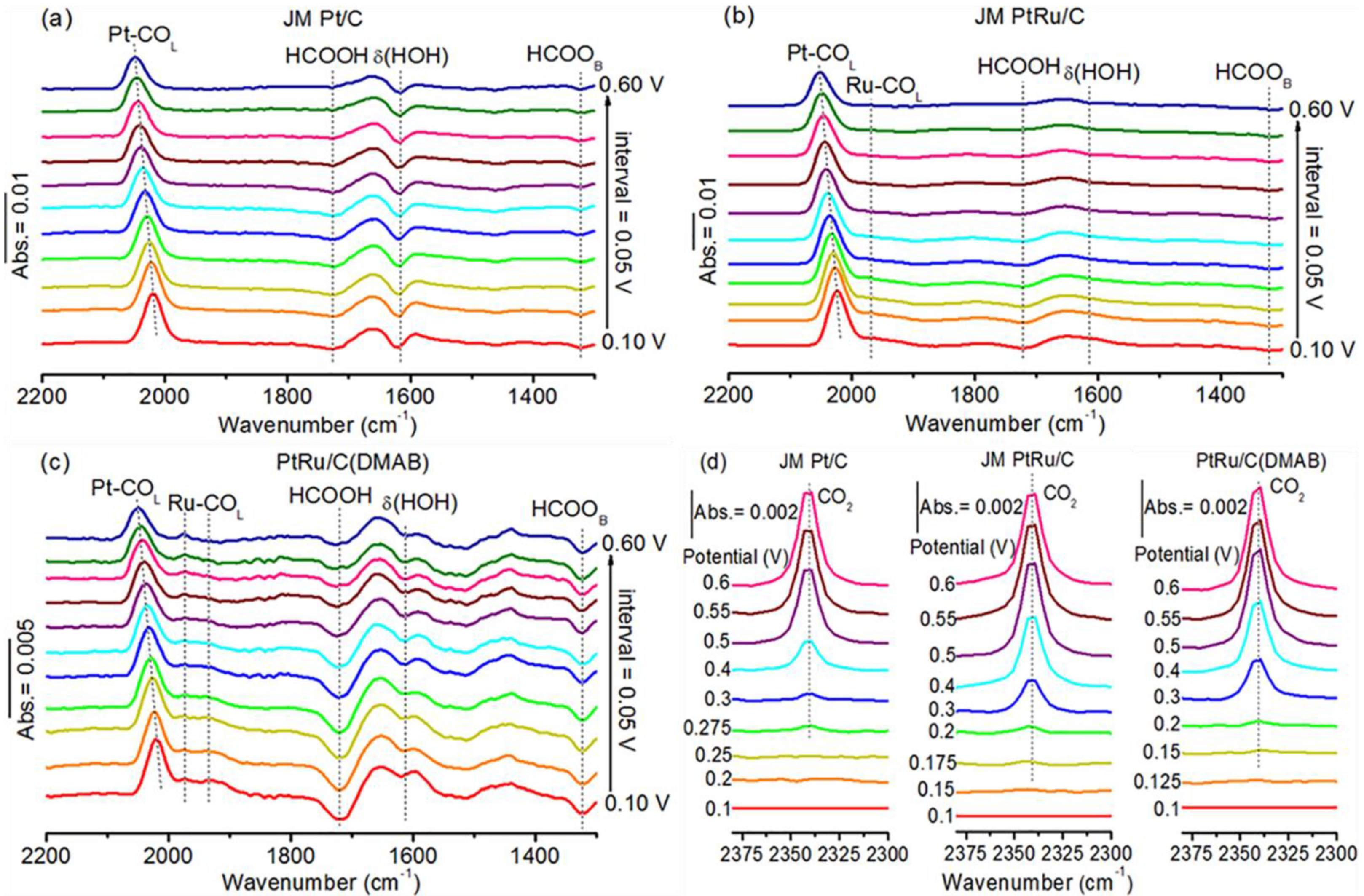
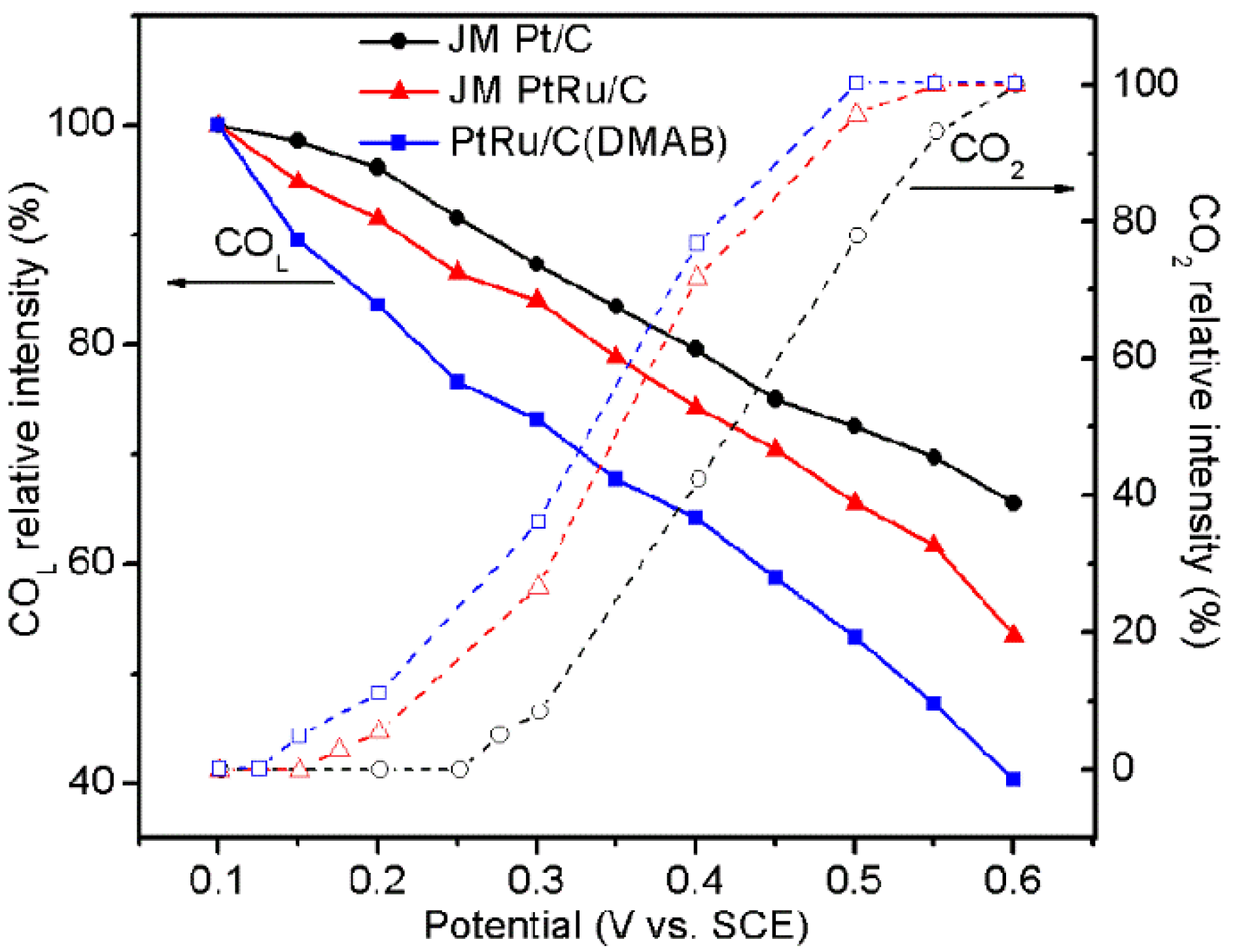
2.5. Interfacial Species Evolved during MOR
3. Materials and Methods
3.1. Reagents
3.2. Preparation of Catalysts
3.3. Material Characterization
3.4. Electrochemical Measurements
3.5. Fuel Cell Performance Test
3.6. In-Situ ATR-IR Measurement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gong, L.; Yang, Z.; Li, K.; Xing, W.; Liu, C.; Ge, J. Recent Development of Methanol Electrooxidation Catalysts for Direct Methanol Fuel Cell. J. Energy Chem. 2018, 27, 1618–1628. [Google Scholar] [CrossRef]
- Yang, Z.; Shi, Y.; Wang, X.; Zhang, G.; Cui, P. Boron as a Superior Activator for Pt Anode Catalyst in Direct Alcohol Fuel Cell. J. Power Sources 2019, 431, 125–134. [Google Scholar] [CrossRef]
- Zhou, Z.-Y.; Shang, S.-J.; Tian, N.; Wu, B.-H.; Zheng, N.-F.; Xu, B.-B.; Chen, C.; Wang, H.-H.; Xiang, D.-M.; Sun, S.-G. Shape Transformation from Pt Nanocubes to Tetrahexahedra with Size near 10 nm. Electrochem. Commun. 2012, 22, 61–64. [Google Scholar] [CrossRef]
- Chen, J.; Luo, S.; Liu, Y.; Chen, S. Theoretical Analysis of Electrochemical Formation and Phase Transition of Oxygenated Adsorbates on Pt(111). ACS Appl. Mater. Interfaces 2016, 8, 20448–20458. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, X.-F.; Chen, X.-L.; Wang, A.-J.; Han, D.-M.; Wang, Z.-G.; Feng, J.-J. Facile Solvothermal Synthesis of Pt71Co29 Lamellar Nanoflowers as an Efficient Catalyst for Oxygen Reduction and Methanol Oxidation Reactions. J. Colloid Interface Sci. 2019, 536, 556–562. [Google Scholar] [CrossRef]
- Zhao, H.; Qi, W.; Zhou, X.; Wu, H.; Li, Y. Composition-controlled Synthesis of Platinum and Palladium Nanoalloys as Highly Active Electrocatalysts for Methanol Oxidation. Chin. J. Catal. 2018, 39, 342–349. [Google Scholar] [CrossRef]
- Liu, H.; Song, C.; Zhang, L.; Zhang, J.; Wang, H.; Wilkinson, D.P. A Review of Anode Catalysis in the Direct Methanol Fuel Cell. J. Power Sources 2006, 155, 95–110. [Google Scholar] [CrossRef]
- Watanabe, M.; Motoo, S. Electrocatalysis by Ad-atoms.2. Enhancement of Oxidation of Methanol on Platinum by Ruthenium Ad-atoms. J. Electroanal. Chem. 1975, 60, 267–273. [Google Scholar] [CrossRef]
- Demirci, U.B. Theoretical Means for Searching Bimetallic Alloys as Anode Electrocatalysts for Direct Liquid-feed Fuel Cells. J. Power Sources 2007, 173, 11–18. [Google Scholar] [CrossRef]
- Liu, J.; Jiang, L. Electrostatic Self-assembly of Pt Nanoparticles on Hexagonal Tungsten Oxide as an Active CO-tolerant Hydrogen Oxidation Electrocatalyst. Int. J. Hydrog. Energy 2018, 43, 8944–8952. [Google Scholar] [CrossRef]
- Wang, Z.-B.; Yin, G.-P.; Lin, Y.-G. Synthesis and Characterization of PtRuMo/C Nanoparticle Electrocatalyst for Direct Ethanol Fuel Cell. J. Power Sources 2007, 170, 242–250. [Google Scholar] [CrossRef]
- Wakisaka, M.; Mitsui, S.; Hirose, Y.; Kawashima, K.; Uchida, H.; Watanabe, M. Electronic Structures of Pt-Co and Pt-Ru Alloys for CO-tolerant Anode Catalysts in Polymer Electrolyte Fuel Cells Studied by EC-XPS. J. Phys. Chem. B 2006, 110, 23489–23496. [Google Scholar] [CrossRef]
- Zhang, J.; Qu, X.; Han, Y.; Shen, L.; Yin, S.; Li, G.; Jiang, Y.; Sun, S. Engineering PtRu Bimetallic Nanoparticles with Adjustable Alloying Degree for Methanol Electrooxidation: Enhanced Catalytic Performance. Appl. Catal. B Environ. 2020, 263, 118345. [Google Scholar] [CrossRef]
- Chetty, R.; Kundu, S.; Xia, W.; Bron, M.; Schuhmann, W.; Chirila, V.; Brandl, W.; Reinecke, T.; Muhler, M. PtRu Nanoparticles Supported on Nitrogen-doped Multiwalled Carbon Nanotubes as Catalyst for Methanol Electrooxidation. Electrochim. Acta 2009, 54, 4208–4215. [Google Scholar] [CrossRef]
- Carmo, M.; Paganin, V.A.; Rosolen, J.M.; Gonzalez, E.R. Alternative Supports for the Preparation of Catalysts for Low-temperature Fuel Cells: The Use of Carbon Nanotubes. J. Power Sources 2005, 142, 169–176. [Google Scholar] [CrossRef]
- Takasu, Y.; Fujiwara, T.; Murakami, Y.; Sasaki, K.; Oguri, M.; Asaki, T.; Sugimoto, W. Effect of Structure of Carbon-supported PtRu Electrocatalysts on the Electrochemical Oxidation of Methanol. J. Electrochem. Soc. 2000, 147, 4421–4427. [Google Scholar] [CrossRef]
- Harish, S.; Baranton, S.; Coutanceau, C.; Joseph, J. Microwave Assisted Polyol Method for the Preparation of Pt/C, Ru/C and PtRu/C Nanoparticles and its Application in Electrooxidation of Methanol. J. Power Sources 2012, 214, 33–39. [Google Scholar] [CrossRef]
- Neto, A.O.; Dias, R.R.; Tusi, M.M.; Linardi, M.; Spinacé, E.V. Electro-oxidation of Methanol and Ethanol Using PtRu/C, PtSn/C and PtSnRu/C Electrocatalysts Prepared by an Alcohol-Reduction Process. J. Power Sources 2007, 166, 87–91. [Google Scholar] [CrossRef]
- Liu, Z.; Lee, J.Y.; Han, M.; Chen, W.; Gan, L.M. Synthesis and Characterization of PtRu/C Catalysts from Microemulsions and Emulsions. J. Mater. Chem. 2002, 12, 2453–2458. [Google Scholar] [CrossRef]
- Liu, Z.L.; Ling, X.Y.; Su, X.D.; Lee, J.Y. Carbon-supported Pt and PtRu Nanoparticles as Catalysts for a Direct Methanol Fuel Cell. J. Phys. Chem. B 2004, 108, 8234–8240. [Google Scholar] [CrossRef]
- Bock, C.; Paquet, C.; Couillard, M.; Botton, G.A.; MacDougall, B.R. Size-selected Synthesis of PtRu Nano-catalysts: Reaction and Size Control Mechanism. J. Am. Chem. Soc. 2004, 126, 8028–8037. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chan, K.Y. Water-in-oil Microemulsion Synthesis of Platinum-Ruthenium Nanoparticles, Their Characterization and Electrocatalytic Properties. Chem. Mater. 2003, 15, 451–459. [Google Scholar] [CrossRef]
- Okaya, K.; Yano, H.; Uchida, H.; Watanabe, M. Control of Particle Size of Pt and Pt Alloy Electrocatalysts Supported on Carbon Black by the Nanocapsule Method. ACS Appl. Mater. Interfaces 2010, 2, 888–895. [Google Scholar] [CrossRef]
- Bang, J.H.; Han, K.; Skrabalak, S.E.; Kim, H.; Suslick, K.S. Porous Carbon Supports Prepared by Ultrasonic Spray Pyrolysis for Direct Methanol Fuel Cell Electrodes. J. Phys. Chem. C 2007, 111, 10959–10964. [Google Scholar] [CrossRef]
- Xue, X.; Lu, T.; Liu, C.; Xing, W. Simple and Controllable Synthesis of Highly Dispersed Pt–Ru/C Catalysts by a Two-step Spray Pyrolysis Process. Chem. Commun. 2005, 12, 1601–1603. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.C.M.; Ntais, S.; Rajaraman, V.; Teixeira-Neto, É.; Teixeira-Neto, Â.A.; Neto, A.O.; Antoniassi, R.M.; Spinacé, E.V.; Baranova, E.A. The Catalytic Activity of Pt:Ru Nanoparticles for Ethylene Glycol and Ethanol Electrooxidation in a Direct Alcohol Fuel Cell. Electrocatalysis 2019, 10, 203–213. [Google Scholar] [CrossRef]
- Guo, J.W.; Zhao, T.S.; Prabhuram, J.; Chen, R.; Wong, C.W. Preparation and Characterization of a PtRu/C Nanocatalyst for Direct Methanol Fuel Cells. Electrochim. Acta 2005, 51, 754–763. [Google Scholar] [CrossRef]
- Montiel, G.; Fuentes-Quezada, E.; Bruno, M.M.; Corti, H.R.; Viva, F.A. Effect of Bimodal Mesoporous Carbon as PtRu Catalyst Support for Direct Methanol Fuel Cells. RSC Adv. 2020, 10, 30631–30639. [Google Scholar] [CrossRef]
- Lizcano-Valbuena, W.H.; Paganin, V.A.; Gonzalez, E.R. Methanol Electro-oxidation on Gas Diffusion Electrodes Prepared with Pt-Ru/C Catalysts. Electrochim. Acta 2002, 47, 3715–3722. [Google Scholar] [CrossRef]
- Ohno, I.; Wakabayashi, O.; Haruyama, S. Anodic-Oxidation of Reductants in Electroless Plating. J. Electrochem. Soc. 1985, 132, 2323–2330. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Kang, Y.-Y.; Yang, H.; Cai, W.-B. Boron-doped Palladium Nanoparticles on Carbon Black as a Superior Catalyst for Formic Acid Electro-oxidation. J. Phys. Chem. C 2009, 113, 8366–8372. [Google Scholar] [CrossRef]
- Dong, Y.; Zhou, Y.-W.; Wang, M.-Z.; Zheng, S.-L.; Jiang, K.; Cai, W.-B. Facile Aqueous Phase Synthesis of Carbon Supported B-doped Pt3Ni Nanocatalyst for Efficient Oxygen Reduction Reaction. Electrochim. Acta 2017, 246, 242–250. [Google Scholar] [CrossRef]
- Huang, J.; Ding, C.; Yang, Y.; Liu, G.; Cai, W.-B. An Alternate Aqueous Phase Synthesis of the Pt3Co/C Catalyst towards Efficient Oxygen Reduction Reaction. Chin. J. Catal. 2019, 40, 1895–1903. [Google Scholar] [CrossRef]
- Wang, D.; Zhuang, L.; Lu, J. An Alloying-Degree-Controlling Step in the Impregnation Synthesis of PtRu/C Catalysts. J. Phys. Chem. C 2007, 111, 16416–16422. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, S.; Shi, F.; Chen, K.; Nie, Y.; Wang, Y.; Wu, R.; Li, J.; Zhang, Y.; Ding, W.; et al. Structural Evolution of Solid Pt Nanoparticles to a Hollow PtFe Alloy with a Pt-skin Surface Via Space-confined Pyrolysis and the Nanoscale Kirkendall Effect. Adv. Mater. 2016, 28, 10673–10678. [Google Scholar] [CrossRef]
- Gojković, S.L.; Vidaković, T.R.; Đurović, D.R. Kinetic Study of Methanol Oxidation on Carbon-supported PtRu Electrocatalyst. Electrochim. Acta 2003, 48, 3607–3614. [Google Scholar] [CrossRef]
- Antolini, E.; Cardellini, F. Formation of Carbon Supported PtRu Alloys: An XRD Analysis. J. Alloys Compd. 2001, 315, 118–122. [Google Scholar] [CrossRef]
- Tian, M.; Shi, S.; Shen, Y.; Yin, H. PtRu Alloy Nanoparticles Supported on Nanoporous Gold as an Efficient Anode Catalyst for Direct Methanol Fuel Cell. Electrochim. Acta 2019, 293, 390–398. [Google Scholar] [CrossRef]
- Wang, H.S.; Wingender, C.; Baltruschat, H.; Lopez, M.; Reetz, M.T. Methanol Oxidation on Pt, PtRu, and Colloidal Pt Electrocatalysts: A DEMS Study of Product Formation. J. Electroanal. Chem. 2001, 509, 163–169. [Google Scholar] [CrossRef]
- Parsons, R.; Vandernoot, T. The Oxidation of Small Organic-Molecules—A Survey of Recent Fuel-cell Related Research. J. Electroanal. Chem. 1988, 257, 9–45. [Google Scholar] [CrossRef]
- Sato, T.; Okaya, K.; Kunimatsu, K.; Yano, H.; Watanabe, M.; Uchida, H. Effect of Particle Size and Composition on CO-tolerance at Pt–Ru/C Catalysts Analyzed by in situ Attenuated Total Reflection FTIR Spectroscopy. ACS Catal. 2012, 2, 450–455. [Google Scholar] [CrossRef]
- Watanabe, M.; Sato, T.; Kunimatsu, K.; Uchida, H. Temperature Dependence of CO-adsorption of Carbon Monoxide and Water on Highly Dispersed Pt/C and PtRu/C Electrodes Studied by in-situ ATR-FTIRAS. Electrochim. Acta 2008, 53, 6928–6937. [Google Scholar] [CrossRef]
- Manoharan, R.; Goodenough, J.B. Methanol Oxidation in Acid on Ordered NiTi. J. Mater. Chem. 1992, 2, 875–887. [Google Scholar] [CrossRef]
- Kuai, L.; Wang, S.; Geng, B. Gold–Platinum Yolk–shell Structure: A Facile Galvanic Displacement Synthesis and Highly Active Electrocatalytic Properties for Methanol Oxidation with Super CO-tolerance. Chem. Commun. 2011, 47, 6093. [Google Scholar] [CrossRef]
- Chen, X.; Jiang, Y.; Sun, J.; Jin, C.; Zhang, Z. Highly Active Nanoporous Pt-based Alloy as Anode and Cathode Catalyst for Direct Methanol Fuel Cells. J. Power Sources 2014, 267, 212–218. [Google Scholar] [CrossRef]
- Lee, Y.-W.; Lee, J.-Y.; Kwak, D.-H.; Hwang, E.-T.; Sohn, J.I.; Park, K.-W. Pd@Pt Core–shell Nanostructures for Improved Electrocatalytic Activity in Methanol Oxidation Reaction. Appl. Catal. B Environ. 2015, 179, 178–184. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, X.; Schechter, J.M.; Yang, Y. Revisiting the Oxidation Peak in the Cathodic Scan of the Cyclic Voltammogram of Alcohol Oxidation on Noble Metal Electrodes. RSC Adv. 2016, 6, 5384–5390. [Google Scholar] [CrossRef]
- Chung, D.Y.; Lee, K.-J.; Sung, Y.-E. Methanol Electro-oxidation on the Pt Surface: Revisiting the Cyclic Voltammetry Interpretation. J. Phys. Chem. C 2016, 120, 9028–9035. [Google Scholar] [CrossRef]
- Jing, F.; Sun, R.; Wang, S.; Sun, H.; Sun, G. Effect of the Anode Structure on the Stability of a Direct Methanol Fuel Cell. Energy Fuels 2020, 34, 3850–3857. [Google Scholar] [CrossRef]
- Alias, M.S.; Kamarudin, S.K.; Zainoodin, A.M.; Masdar, M.S. Active Direct Methanol Fuel Cell: An Overview. Int. J. Hydrogen Energy 2020, 45, 19620–19641. [Google Scholar] [CrossRef]
- Zhou, Y.-W.; Chen, Y.-F.; Jiang, K.; Liu, Z.; Mao, Z.-J.; Zhang, W.-Y.; Lin, W.-F.; Cai, W.-B. Probing the Enhanced Methanol Electrooxidation Mechanism on Platinum-Metal Oxide Catalyst. Appl. Catal. B Environ. 2021, 280, 119393. [Google Scholar] [CrossRef]
- Iwasita, T.; Nart, F.C.; Vielstich, W. An FTIR Study of the Catalytic Activity of a 85-15 Pt-Ru Alloy for Methanol Oxidation. Ber. Bunsen Ges. Phys. Chem 1990, 94, 1030–1034. [Google Scholar] [CrossRef]
- Chen, Y.X.; Miki, A.; Ye, S.; Sakai, H.; Osawa, M. Formate, an Active Intermediate for Direct Oxidation of Methanol on Pt Electrode. J. Am. Chem. Soc. 2003, 125, 3680–3681. [Google Scholar] [CrossRef] [PubMed]
- Yajima, T.; Uchida, H.; Watanabe, M. In-situ ATR-FTIR Spectroscopic Study of Electro-oxidation of Methanol and Adsorbed CO at Pt-Ru Alloy. J. Phys. Chem. B 2004, 108, 2654–2659. [Google Scholar] [CrossRef]
- Wang, H.; Jusys, Z.; Behm, R.J. Ethanol Electro-oxidation on Carbon-supported Pt, PtRu and Pt3Sn Catalysts: A Quantitative DEMS Study. J. Power Sources 2006, 154, 351–359. [Google Scholar] [CrossRef]
- Jusys, Z.; Kaiser, J.; Behm, R.J. Composition and Activity of High Surface Area PtRu Catalysts towards Adsorbed CO and Methanol Electrooxidation—A DEMS Study. Electrochim. Acta 2002, 47, 3693–3706. [Google Scholar] [CrossRef]
- Wang, J.-Y.; Zhang, H.-X.; Jiang, K.; Cai, W.-B. From HCOOH to CO at Pd Electrodes: A Surface-enhanced Infrared Spectroscopy Study. J. Am. Chem. Soc. 2011, 133, 14876–14879. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-X.; Wang, S.-H.; Jiang, K.; André, T.; Cai, W.-B. In situ Spectroscopic Investigation of CO Accumulation and Poisoning on Pd Black Surfaces in Concentrated HCOOH. J. Power Sources 2012, 199, 165–169. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Adding Mode | Pt (wt.%) | Ru (wt.%) | B (at.%) | Pt/Ru Ratio of Charge | Pt/Ru Atomic Ratio |
|---|---|---|---|---|---|---|
| PtRu/C(DMAB) | reverse addition | 19.0 | 10.2 | 1.27 | 1.0:1 | 1.0:1 |
| PtRu/C(NaBH4) | 19.2 | 9.9 | 0.90 | 1.0:1 | ||
| PtRu/C(DMAB) | regular addition | 18.5 | 7.0 | 1.32 | 1.4:1 | |
| PtRu/C(NaBH4) | 20.0 | 6.8 | 0.88 | 1.5:1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Zhou, Y.-W.; Jin, Z.; Chen, C.; Li, H.; Cai, W.-B. Alternative Aqueous Phase Synthesis of a PtRu/C Electrocatalyst for Direct Methanol Fuel Cells. Catalysts 2021, 11, 925. https://doi.org/10.3390/catal11080925
Wang Q, Zhou Y-W, Jin Z, Chen C, Li H, Cai W-B. Alternative Aqueous Phase Synthesis of a PtRu/C Electrocatalyst for Direct Methanol Fuel Cells. Catalysts. 2021; 11(8):925. https://doi.org/10.3390/catal11080925
Chicago/Turabian StyleWang, Qijun, Ya-Wei Zhou, Zhao Jin, Chunguang Chen, Hong Li, and Wen-Bin Cai. 2021. "Alternative Aqueous Phase Synthesis of a PtRu/C Electrocatalyst for Direct Methanol Fuel Cells" Catalysts 11, no. 8: 925. https://doi.org/10.3390/catal11080925
APA StyleWang, Q., Zhou, Y.-W., Jin, Z., Chen, C., Li, H., & Cai, W.-B. (2021). Alternative Aqueous Phase Synthesis of a PtRu/C Electrocatalyst for Direct Methanol Fuel Cells. Catalysts, 11(8), 925. https://doi.org/10.3390/catal11080925