
Alkoxy-Functionalized Schiff-Base Ligation at Aluminum and Zinc: Synthesis, Structures and ROP Capability


 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
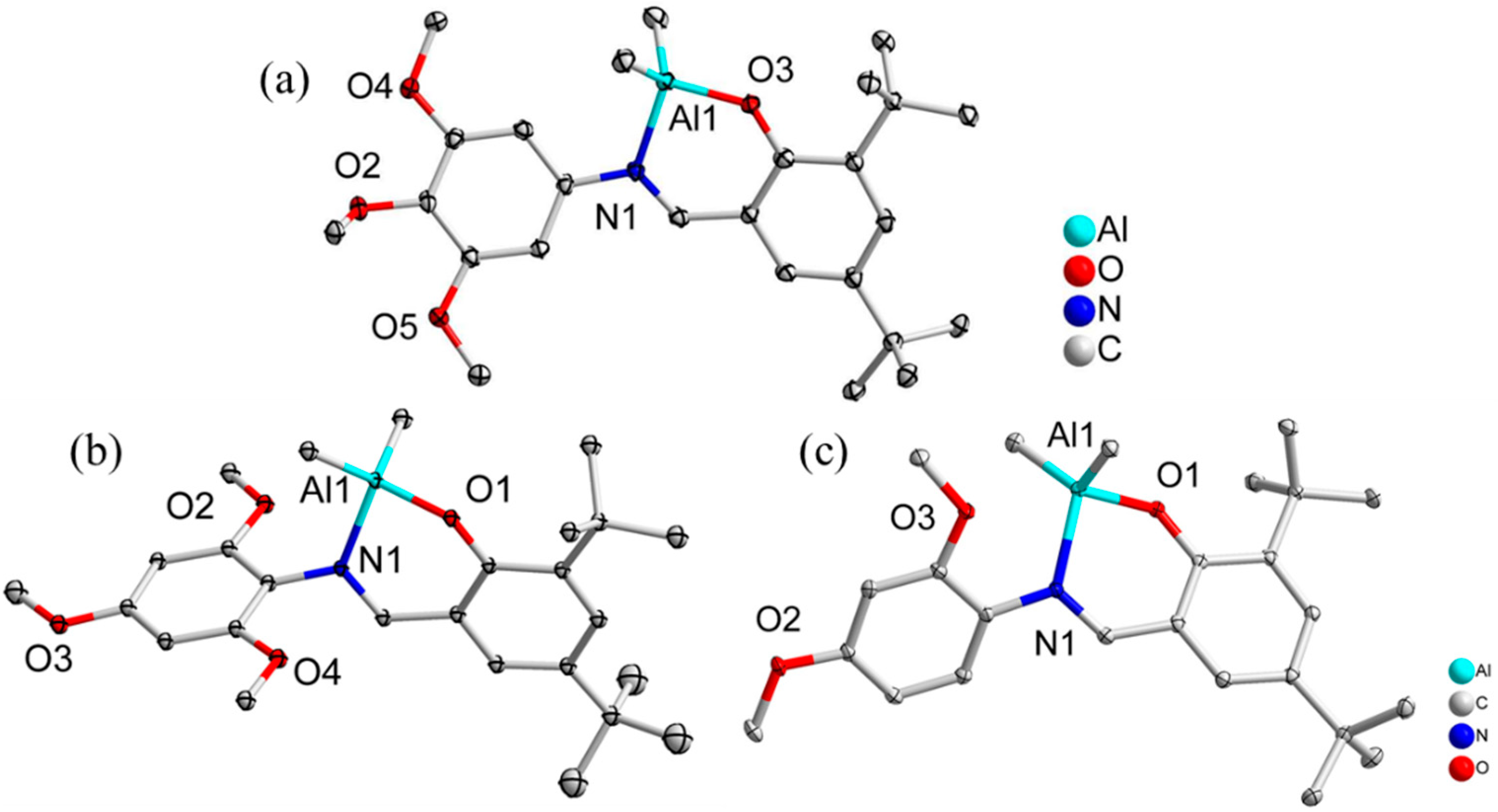
2.1. Synthesis and Characterization of Schiff-Base Aluminum Complexes
2.2. Synthesis and Characterization of Schiff-Base Zinc Complexes
3. Ring Opening Polymerization (ROP)
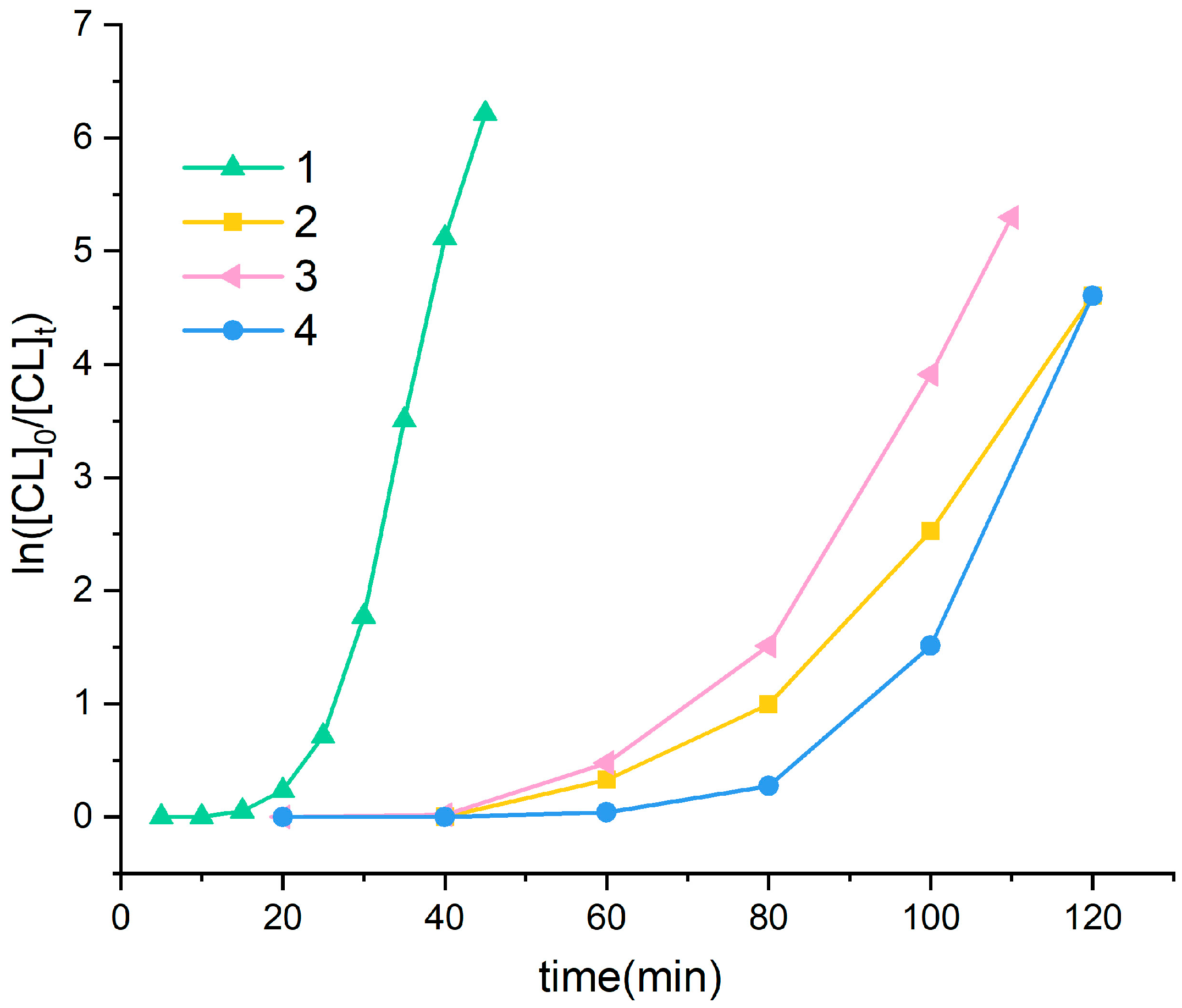
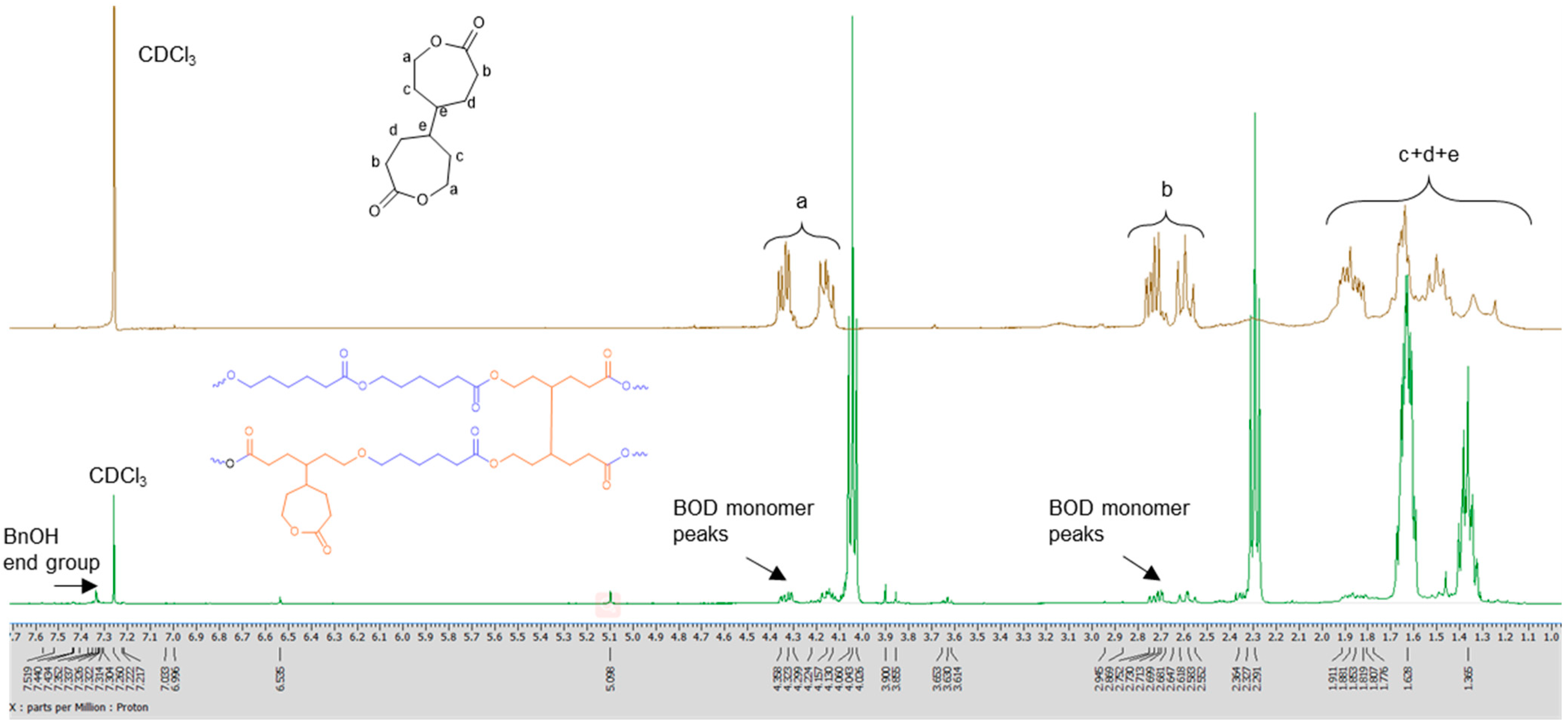
3.1. Ring Opening Polymerization of ε-Caprolactone (ε-CL)
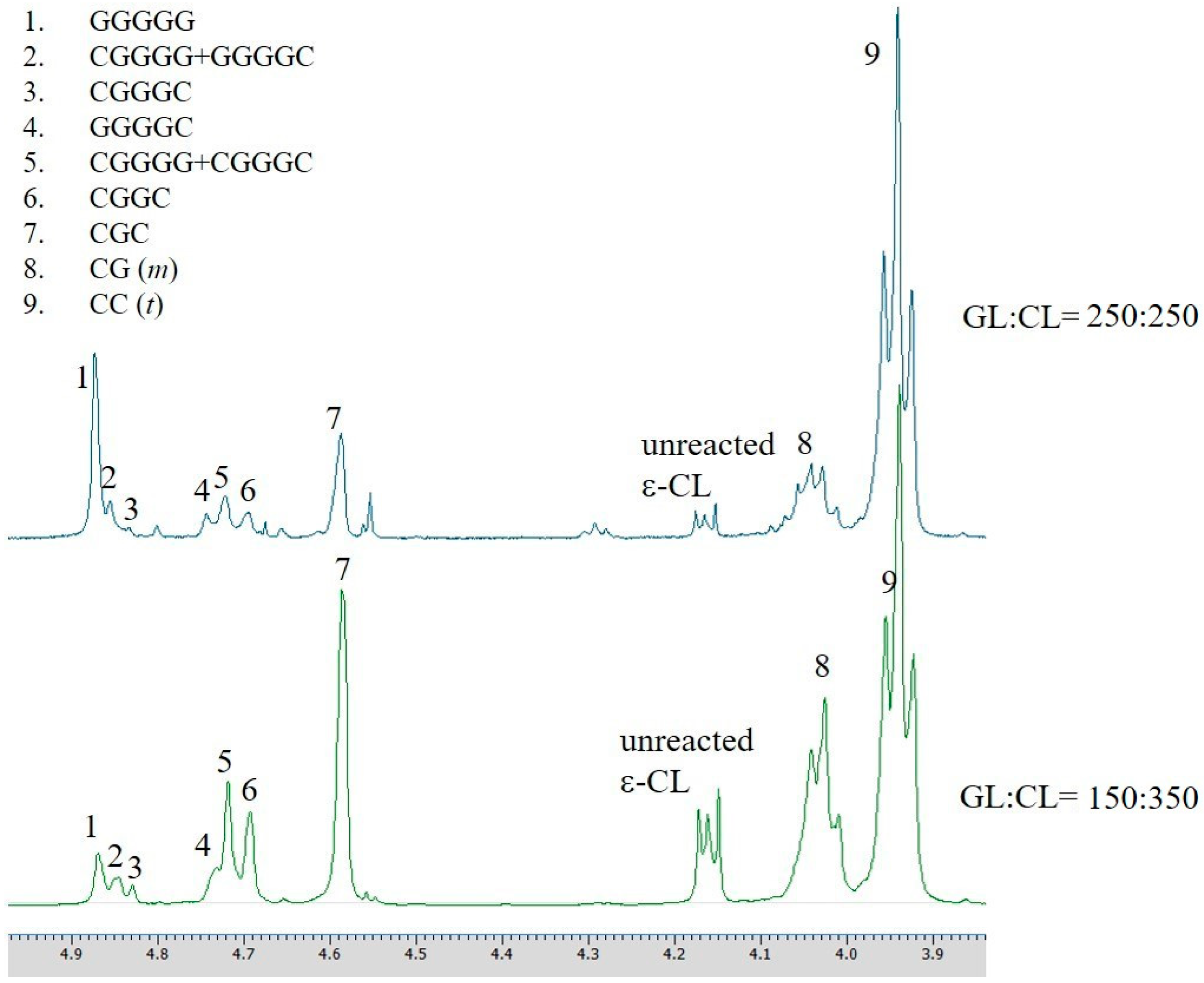
3.2. The ROP of Copolymer P(CL-co-GL) Catalyzed by the Aluminum Complexes
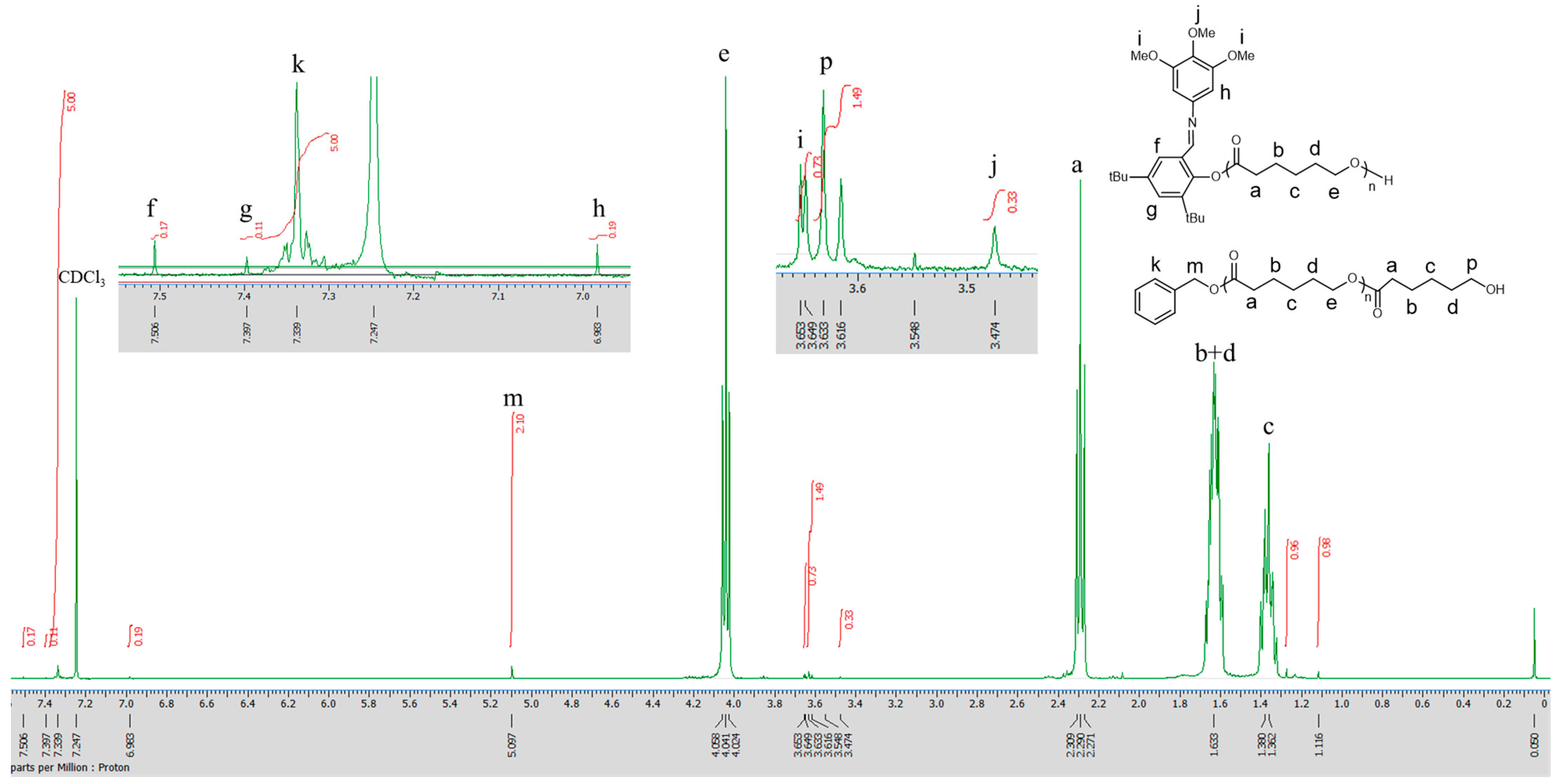
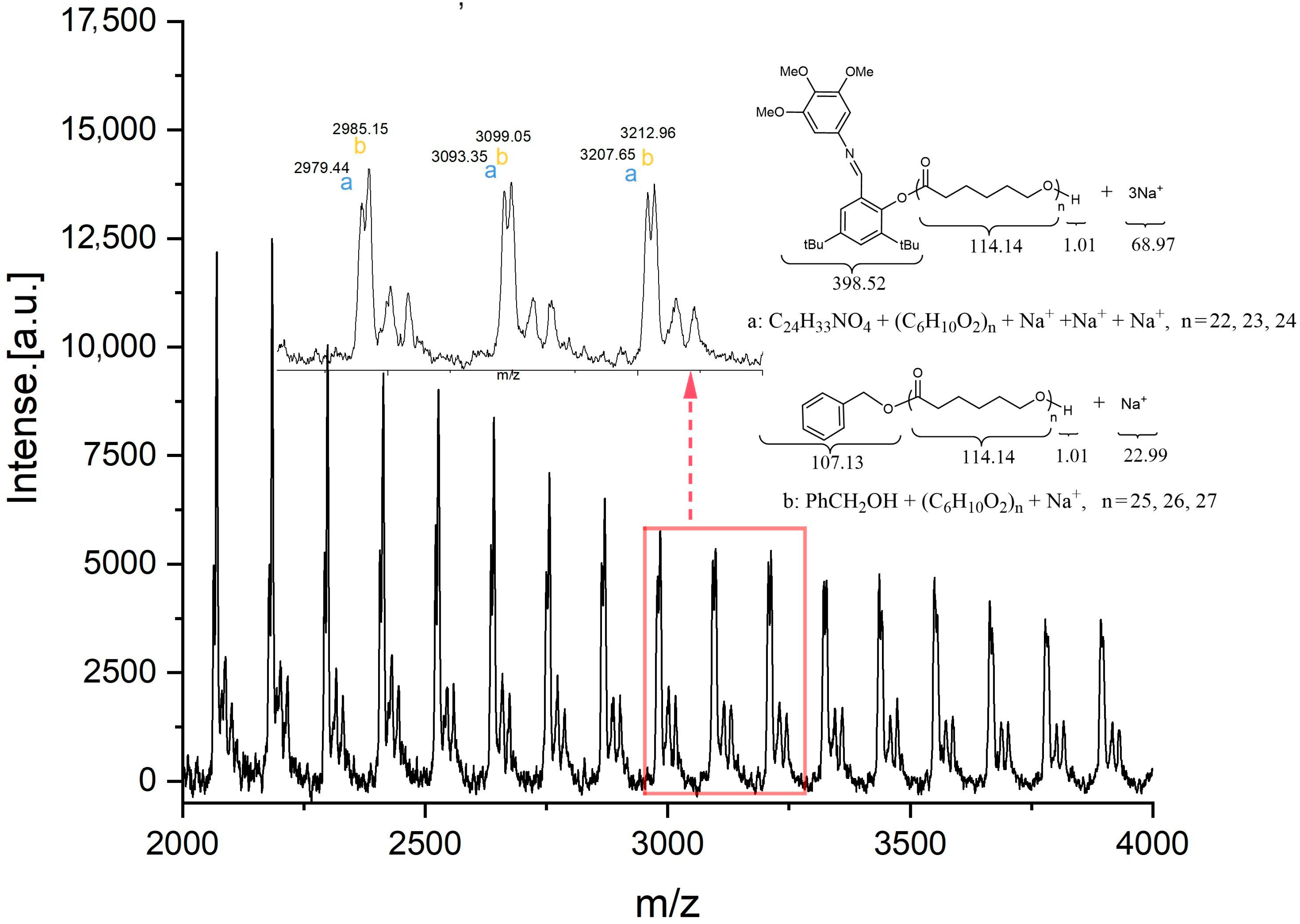
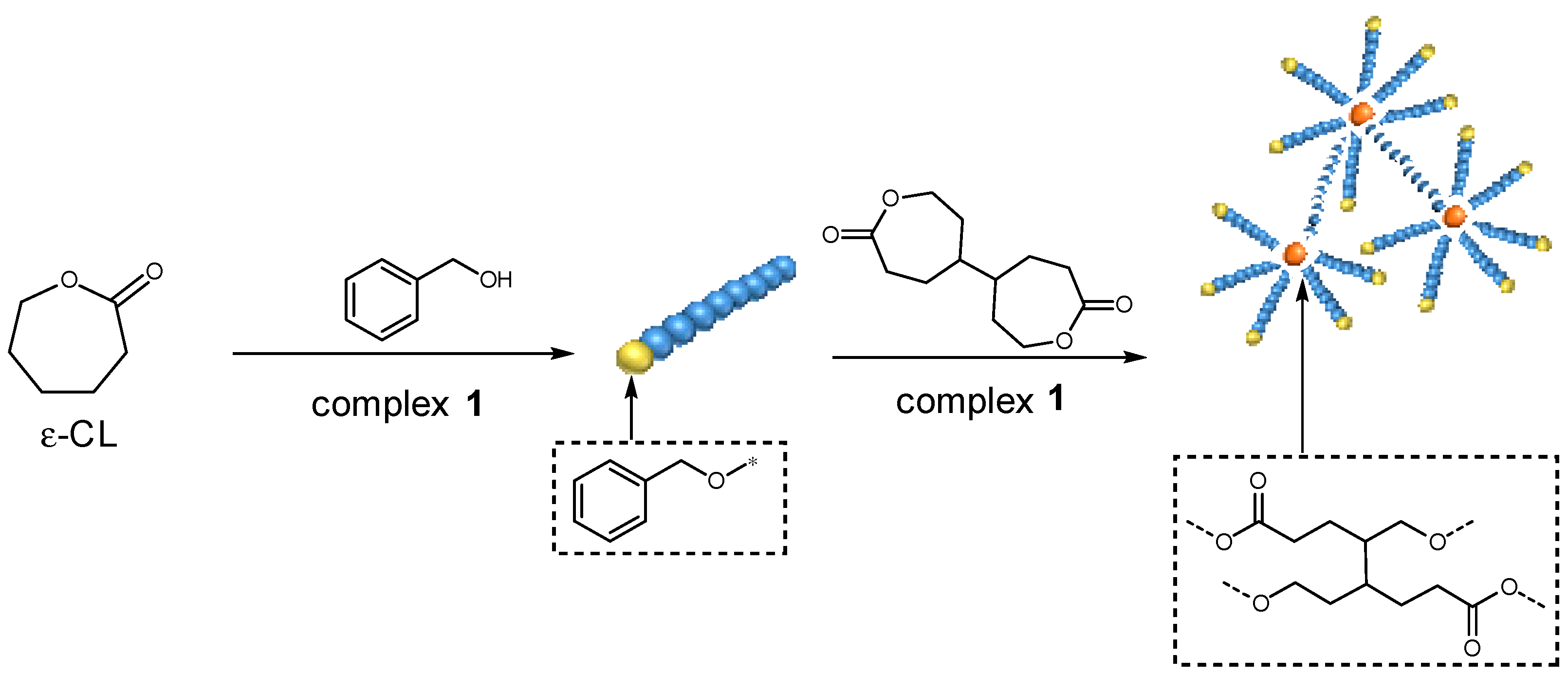
3.3. The ROP of Cross-Linked PCL Catalyzed by the Aluminum Complexes
4. Materials and Methods
4.1. Synthesis of 2,4-Di-Tert-Butyl-6-(((3,4,5-Trimethoxyphenyl)Imino)Methyl)Phenol (L1H)
4.2. Synthesis of 2,4-Di-Tert-Butyl-6-(((2,4,6-Trimethoxyphenyl)Imino)Methyl)Phenol (L2H)
4.3. Synthesis of 2,4-Di-Tert-Butyl-6-(((2,4-Trimethoxyphenyl)Imino)Methyl)Phenol (L3H)
4.4. Synthesis of 2,4-Di-Tert-Butyl-6-((Phenylimino)Methyl)Phenol (L4H)
4.5. Synthesis of [Al(L1)(Me)2] (1)
4.6. Synthesis of [Al(L2)(Me)2] (2)
4.7. Synthesis of [Al(L3)(Me)2] (3)
4.8. Synthesis of [Al(L4)(Me)2] (4)
4.9. Synthesis of [Zn(L1)2]·2CH3CN (5)
4.10. Synthesis of [Zn(L2)2] (6)
4.11. Synthesis of [Zn(L3)2] (7)
4.12. Synthesis of [Zn(L4)2] (8)
4.13. ROP of ε-Caprolactone (ε-CL)
4.14. ROP of Copolymerization of ε-Caprolactone (ε-CL) and Glycolide (GL)
4.15. Polymerization Kinetics
4.16. 4,4′-Bioxepane-7,7′-Dione (BOD) Cross Linker
4.17. X-ray Crystallography
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 1 | 2 | 3 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|
| Empirical formula | C26H38NO4Al | C26H38NO4Al | C25H36NO3Al | C52H70N4O8Zn | C48H64N2O8Zn | C48H60N2O6Zn |
| Formula weight | 455.55 | 455.55 | 425.53 | 944.49 | 862.40 | 802.33 |
| Crystal system | Tetragonal | Triclinic | Monoclinic | Orthorhombic | Monoclinic | Monoclinic |
| Temp (K) | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) | 150(2) |
| Wavelength/Å | 1.54178 | 0.71075 | 1.54178 | 0.71075 | 1.54178 | 0.71073 |
| Space group | P42/n | P-1 | P 21/c | Pbca | I 2/a | P 21/n |
| a/Å | 28.9022(5) | 8.77450(10) | 10.49237(5) | 16.8034(4) | 13.0291(4) | 13.3203(9) |
| b/Å | 28.9022(5) | 11.85330(10) | 20.94049(12) | 20.9651(5) | 27.0249(8) | 10.1701(4) |
| c/Å | 6.0320(2) | 12.71810(10) | 11.23374(5) | 28.4430(7) | 27.2058(8) | 32.869(2) |
| α/° | 90 | 101.2730(10) | 90 | 90 | 90 | 90 |
| β/° | 90 | 94.7670(10) | 94.2834(4) | 90 | 90.472(3) | 92.384(5) |
| γ/° | 90 | 93.6800(10) | 90 | 90 | 90 | 90 |
| V/Å3 | 5038.8(2) | 1288.38(2) | 2461.33(2) | 10020.0(4) | 9579.1(5) | 4448.9(4) |
| Z | 8 | 2 | 4 | 8 | 8 | 4 |
| Dcalc/g cm−3 | 1.201 | 1.174 | 1.148 | 1.252 | 1.196 | 1.198 |
| F (000) | 1968 | 492 | 920 | 4032 | 3680 | 1712 |
| µ/mm−1 | 0.948 | 0.109 | 0.906 | 0.546 | 1.121 | 0.599 |
| θ range | 2.162–64.995 | 2.337–24.998 | 3.896–75.379 | 2.071–25.000 | 2.3040–68.480 | 1.240–26.215 |
| Crystal size/mm | 0.300 × 0.200 × 0.200 | 0.340 × 0.300 × 0.200 | 0.150 × 0.120 × 0.110 | 0.080 × 0.050 × 0.030 | 0.320 × 0.040 × 0.040 | 0.400 × 0.320 × 0.240 |
| Reflns collected | 66,305 | 9350 | 85,197 | 50,096 | 8821 | 20,791 |
| Reflns unique | 4278 | 9350 | 4659 | 8814 | 8821 | 8877 |
| Rint | 0.1663 | 0.0234 | 0.0468 | 0.0650 | 0.1309 | 0.0551 |
| R1;wR2 [I > 2σ(I)] | 0.0691; 0.1785 | 0.0420; 0.1202 | 0.0327; 0.0319 | 0.0434; 0.0925 | 0.0441; 0.1179 | 0.0386; 0.0644 |
| R1; wR2 (all data) | 0.0828; 0.1879 | 0.0445; 0.1226 | 0.0856; 0.0850 | 0.0620; 0.1003 | 0.0510; 0.1212 | 0.0868; 0.0709 |
| Parameters | 300 | 301 | 281 | 606 | 551 | 490 |
| GOF (F2) | 1.050 | 1.030 | 1.036 | 1.061 | 1.058 | 0.765 |
| Largest diff. peak and hole/e.Å−3 | 0.348 and | 0.579 and | 0.268 and | 0.298 and | 1.063 and | 0.396 and |
| −0.367 | −0.339 | −0.267 | −0.412 | −0.566 | −0.358 |
References
- Makio, H.; Terao, H.; Iwashita, A.; Fujita, T. FI catalysts for olefin polymerization--a comprehensive treatment. Chem. Rev. 2011, 111, 2363–2449. [Google Scholar] [CrossRef]
- Redshaw, C.; Tang, Y. Tridentate ligands and beyond in group IV metal α-olefin homo-/co-polymerization catalysis. Chem. Soc. Rev. 2012, 41, 4484–4510. [Google Scholar] [CrossRef]
- Gao, J.; Zhu, D.; Zhang, W.; Solan, G.A.; Ma, Y.; Sun, W.-H. Recent progress in the application of group 1, 2 & 13 metal complexes as catalysts for the ring opening polymerization of cyclic esters. Inorg. Chem. Front. 2019, 6, 2619–2652. [Google Scholar]
- Wei, Y.; Wang, S.; Zhou, S. Aluminum alkyl complexes: Synthesis, structure, and application in ROP of cyclic esters. Dalton Trans. 2016, 45, 4471–4485. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yu, T.-L.; Chen, C.-T.; Lin, C.-C. Recent developments in main group metal complexes catalyzed/initiated polymerization of lactides and related cyclic esters. Coord. Chem. Rev. 2006, 250, 602–626. [Google Scholar] [CrossRef]
- Fuoco, T.; Pappalardo, D. Aluminum Alkyl Complexes Bearing Salicylaldiminato Ligands: Versatile Initiators in the Ring-Opening Polymerization of Cyclic Esters. Catalysts 2017, 7, 64. [Google Scholar] [CrossRef]
- Santoro, O.; Zhang, X.; Redshaw, C. Synthesis of biodegradable polymers: A review on the use of Schiff-base metal complexes as catalysts for the Ring Opening Polymerization (ROP) of cyclic esters. Catalysts 2020, 10, 800. [Google Scholar] [CrossRef]
- Guo, L.-h.; Gao, H.-y.; Zhang, L.; Zhu, F.-m.; Wu, Q. An Unsymmetrical Iron(II) Bis(imino)pyridyl Catalyst for Ethylene Polymerization: Effect of a Bulky Ortho Substituent on the Thermostability and Molecular Weight of Polyethylene. Organometallics 2010, 29, 2118–2125. [Google Scholar] [CrossRef]
- Ghaffari, A.; Behzad, M.; Pooyan, M.; Rudbari, H.A.; Bruno, G. Crystal structures and catalytic performance of three new methoxy substituted salen type nickel (II) Schiff base complexes derived from meso-1, 2-diphenyl-1, 2-ethylenediamine. J. Mol. Struct. 2014, 1063, 1–7. [Google Scholar] [CrossRef]
- Arbaoui, A.; Redshaw, C.; Hughes, D.L. Multinuclear alkylaluminium macrocyclic Schiff base complexes: Influence of procatalyst structure on the ring opening polymerisation of epsilon-caprolactone. Chem. Commun. 2008, 39, 4717–4719. [Google Scholar] [CrossRef]
- Hsu, C.-Y.; Tseng, H.-C.; Vandavasi, J.K.; Lu, W.-Y.; Wang, L.-F.; Chiang, M.Y.; Lai, Y.-C.; Chen, H.-Y.; Chen, H.-Y. Investigation of the dinuclear effect of aluminum complexes in the ring-opening polymerization of ε-caprolactone. RSC Adv. 2017, 7, 18851–18860. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Iwasa, N.; Nomura, K. Synthesis of Al complexes containing phenoxy-imine ligands and their use as the catalyst precursors for efficient living ring-opening polymerisation of epsilon-caprolactone. Dalton Trans. 2008, 3978–3988. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Y.; Sun, W.-H.; Wang, L.; Redshaw, C. Dimethylaluminium aldiminophenolates: Synthesis, characterization and ring-opening polymerization behavior towards lactides. Dalton Trans. 2012, 41, 11587–11596. [Google Scholar] [CrossRef]
- Aeilts, S.L.; Coles, M.P.; Swenson, D.C.; Jordan, R.F.; Young, V.G. Aluminum Alkyl Complexes Containing Guanidinate Ligands. Organometallics 1998, 17, 3265–3270. [Google Scholar] [CrossRef]
- Sarma, B.D.; Bailar, J.C., Jr. The stereochemistry of metal chelates with polydentate ligands. Part, I. J. Am. Chem. Soc. 1955, 77, 5476–5480. [Google Scholar] [CrossRef]
- Singh, B.K.; Prakash, A.; Rajour, H.K.; Bhojak, N.; Adhikari, D. Spectroscopic characterization and biological activity of Zn(II), Cd(II), Sn(II) and Pb(II) complexes with Schiff base derived from pyrrole-2-carboxaldehyde and 2-amino phenol. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 76, 376–383. [Google Scholar] [CrossRef]
- Jain, A.K.; Gupta, A.; Bohra, R.; Lorenz, I.-P.; Mayer, P. Synthesis and structural elucidation of some novel aluminium(III) complexes with Schiff bases: Crystal and molecular structure of [Al{O(C6H4)CHNC6H5}2{HO(C6H4)CHNC6H5}2]Br. Polyhedron 2006, 25, 654–662. [Google Scholar] [CrossRef]
- Niu, M.J.; Li, Z.; Chang, G.L.; Kong, X.J.; Hong, M.; Zhang, Q.F. Crystal Structure, Cytotoxicity and Interaction with DNA of Zinc (II) Complexes with o-Vanillin Schiff Base Ligands. PLoS ONE 2015, 10, e0130922. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, Y.; Cui, Y.; Zhang, X.; Zhu, H.-L.; Zeng, Q. Synthesis, molecular docking and biological evaluation of Schiff base transition metal complexes as potential urease inhibitors. Eur. J. Med. Chem. 2010, 45, 4473–4478. [Google Scholar] [CrossRef]
- Arbaoui, A.; Redshaw, C.; Sanchez-Ballester, N.M.; Elsegood, M.R.J.; Hughes, D.L. Bimetallic copper(II) and zinc(II) complexes of acyclic Schiff base ligands derived from amino acids. Inorg. Chim. Acta 2011, 365, 96–102. [Google Scholar] [CrossRef]
- Chakraborty, B.; Banerjee, S. Synthesis, characterization, and crystal structures of cobalt(II), copper(II), and zinc(II) complexes of a bidentate iminophenol. J. Coord. Chem. 2013, 66, 3619–3628. [Google Scholar] [CrossRef]
- Oliveira, A.; Ferreira, L.; Dias, M.; Bitzer, R.; Nascimento, M. Ring Opening Polymerization of L-Lactide with Two Different Zinc(Ii) Phenoxy-Imine Complexes as Initiators. Química Nova 2019, 42, 505–512. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, W.; Liu, D.; Li, S.; Liu, X.; Cui, D.; Chen, X. Magnesium and Zinc Complexes Supported by N,O-Bidentate Pyridyl Functionalized Alkoxy Ligands: Synthesis and Immortal ROP of ε-CL andl-LA. Organometallics 2012, 31, 4182–4190. [Google Scholar] [CrossRef]
- Nassar, A.M.; Hassan, A.M.; Shoeib, M.A.; El kmash, A.N. Synthesis, Characterization and Anticorrosion Studies of New Homobimetallic Co(II), Ni(II), Cu(II), and Zn(II) Schiff Base Complexes. J. Bio-Tribo-Corros. 2015, 1, 19. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.-L.; Lin, Y.-F.; Jiang, M.-T.; Lu, W.-Y.; Vandavasi, J.K.; Wang, L.-F.; Lai, Y.-C.; Chiang, M.Y.; Chen, H.-Y. Improvement in Aluminum Complexes Bearing Schiff Bases in Ring-Opening Polymerization of ε-Caprolactone: A Five-Membered-Ring System. Organometallics 2017, 36, 1936–1945. [Google Scholar] [CrossRef]
- Wei, Y.; Song, L.; Jiang, L.; Huang, Z.; Wang, S.; Yuan, Q.; Mu, X.; Zhu, X.; Zhou, S. Aluminum complexes with Schiff base bridged bis(indolyl) ligands: Synthesis, structure, and catalytic activity for polymerization of rac-lactide. Dalton Trans. 2019, 48, 15290–15299. [Google Scholar] [CrossRef]
- Ajellal, N.; Carpentier, J.-F.; Guillaume, C.; Guillaume, S.M.; Helou, M.; Poirier, V.; Sarazin, Y.; Trifonov, A. Metal-catalyzed immortal ring-opening polymerization of lactones, lactides and cyclic carbonates. Dalton Trans. 2010, 39, 8363–8376. [Google Scholar] [CrossRef]
- Qin, L.; Zhang, Y.; Chao, J.; Cheng, J.; Chen, X. Four-and five-coordinate aluminum complexes supported by N, O-bidentate β-pyrazylenolate ligands: Synthesis, structure and application in ROP of ε-caprolactone and lactide. Dalton Trans. 2019, 48, 12315–12325. [Google Scholar] [CrossRef]
- Kong, W.L.; Chai, Z.Y.; Wang, Z.X. Synthesis of N,N,O-chelate zinc and aluminum complexes and their catalysis in the ring-opening polymerization of epsilon-caprolactone and rac-lactide. Dalton Trans. 2014, 43, 14470–14480. [Google Scholar] [CrossRef]
- Munzeiwa, W.A.; Nyamori, V.O.; Omondi, B. N,O-Amino-phenolate Mg(II) and Zn(II) Schiff base complexes: Synthesis and application in ring-opening polymerization of ε-caprolactone and lactides. Inorg. Chim. Acta 2019, 487, 264–274. [Google Scholar] [CrossRef]
- Mata-Mata, J.L.; Gutiérrez, J.A.; Paz-Sandoval, M.A.; Madrigal, A.R.; Martínez-Richa, A. Ring-opening polymerization of ϵ-caprolactone initiated with different ruthenium derivatives: Kinetics and mechanism studies. J. Polym. Sci. Part A Polym. Chem. 2006, 44, 6926–6942. [Google Scholar] [CrossRef]
- Hammett, L.P. The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives. J. Am. Chem. Soc. 1937, 59, 96–103. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, Q.; Cui, Y.; He, J.; Zhang, Y. Living/controlled ring-opening (co)polymerization of lactones by Al-based catalysts with different sidearms. Dalton Trans. 2019, 48, 7167–7178. [Google Scholar] [CrossRef] [PubMed]
- Rae, A.; Gaston, A.J.; Greindl, Z.; Garden, J.A. Electron rich (salen)AlCl catalysts for lactide polymerisation: Investigation of the influence of regioisomers on the rate and initiation efficiency. Eur. Polym. J. 2020, 138, 109917. [Google Scholar] [CrossRef]
- Hung, W.-C.; Huang, Y.; Lin, C.-C. Efficient initiators for the ring-opening polymerization of L-lactide: Synthesis and characterization of NNO-tridentate Schiff-base zinc complexes. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 6466–6476. [Google Scholar] [CrossRef]
- Wang, J.; Wang, L.; Zhou, Z.; Lai, H.; Xu, P.; Liao, L.; Wei, J. Biodegradable polymer membranes applied in guided bone/tissue regeneration: A review. Polymers 2016, 8, 115. [Google Scholar] [CrossRef]
- Van den Vreken, N.M.; Dubruel, P.; Verbeeck, R.M. The effect of a photopolymerizable poly (ε-caprolactone-co-glycolide) matrix on the cement reactions of tetracalcium phosphate and tetracalcium phosphate–monocalcium phosphate monohydrate mixtures. J. Mater. Chem. B 2013, 1, 1584–1594. [Google Scholar] [CrossRef]
- Cooper, K.; Nathan, A.; Vyakarnam, M. Poly (ε-Caprolactone-co-Glycolide): Biomedical Applications of a Unique Elastomer. Biodegrad. Polym. Clin. Use Clin. Dev. 2011, 401–415. [Google Scholar]
- Kasperczyk, J. Copolymerization of glycolide and ϵ-caprolactone, 1. Analysis of the copolymer microstructure by means of 1H and 13C NMR spectroscopy. Macromol. Chem. Phys. 1999, 200, 903–910. [Google Scholar] [CrossRef]
- Meduri, A.; Fuoco, T.; Lamberti, M.; Pellecchia, C.; Pappalardo, D. Versatile Copolymerization of Glycolide and rac-Lactide by Dimethyl(salicylaldiminato)aluminum Compounds. Macromolecules 2014, 47, 534–543. [Google Scholar] [CrossRef]
- Li, S. Structure-Property Relationships of Copolymers Obtained by Ring-Opening Polymerization of Glycolide and E-Caprolactone. Part 1. Synthesis and Characterization. Biomacromolecules 2005, 6, 483–488. [Google Scholar] [CrossRef]
- Zheng, Y.; Turner, W.; Zong, M.; Irvine, D.J.; Howdle, S.M.; Thurecht, K.J. Biodegradable Core−Shell Materials via RAFT and ROP: Characterization and Comparison of Hyperbranched and Microgel Particles. Macromolecules 2011, 44, 1347–1354. [Google Scholar] [CrossRef]
- Yang, W.; Zhao, K.Q.; Wang, B.Q.; Redshaw, C.; Elsegood, M.R.; Zhao, J.L.; Yamato, T. Manganese coordination chemistry of bis(imino)phenoxide derived [2 + 2] Schiff-base macrocyclic ligands. Dalton Trans. 2016, 45, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Wiltshire, J.T.; Qiao, G.G. Degradable core cross-linked star polymers via ring-opening polymerization. Macromolecules 2006, 39, 4282–4285. [Google Scholar] [CrossRef]
- Sheldrick, G. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
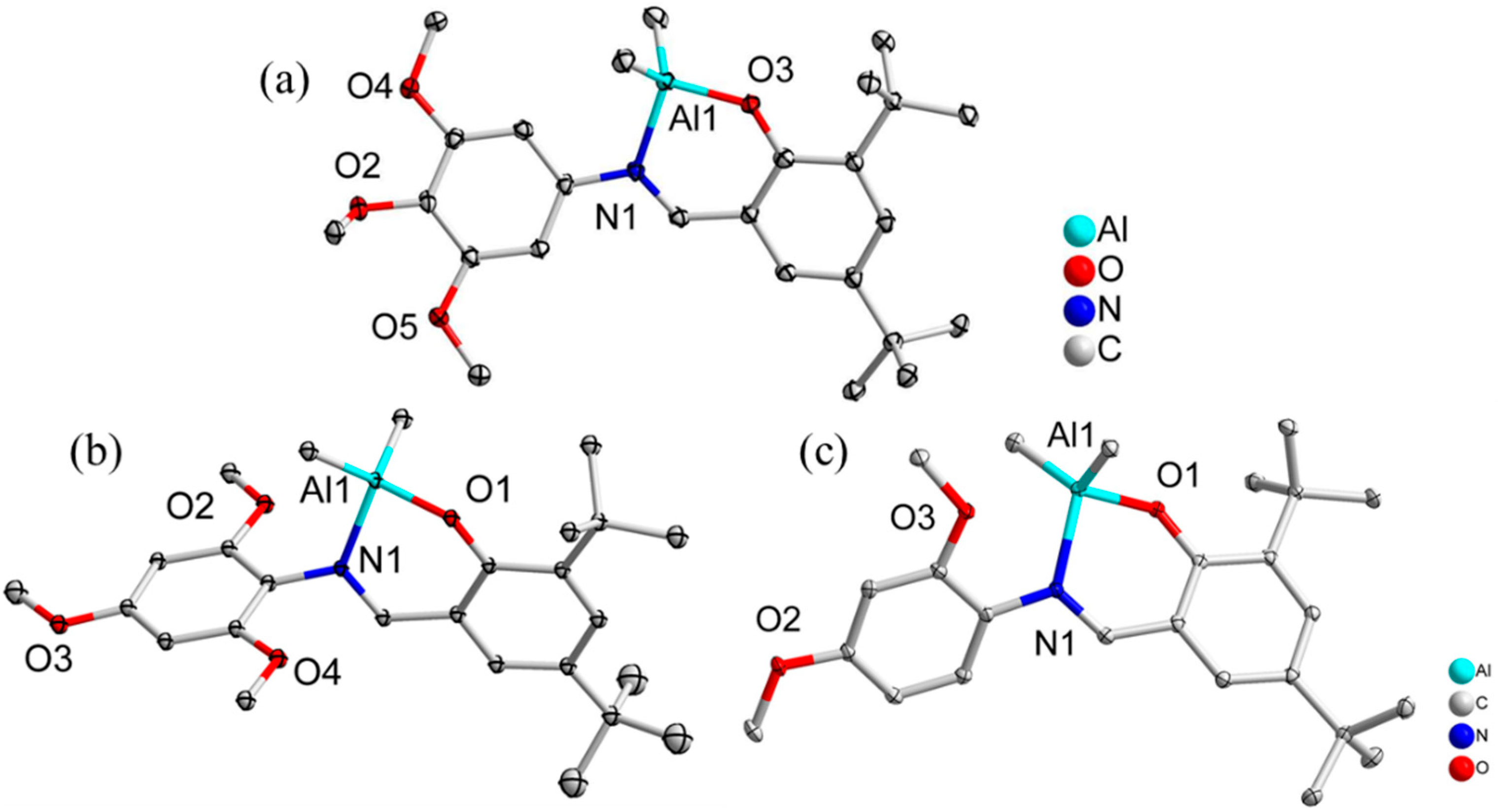


 ), 2 (
), 2 (  ), 3 (
), 3 (  ), 4 (
), 4 (  ); reaction conditions: [CL]:[catalyst]:[BnOH] = 250:1:1, [CL] = 16.44 mmol, [BnOH] = 0.01 M, reaction temperature: 100 °C.
), 2 ( ), 3 ( ), 4 ( ); reaction conditions: [CL]:[catalyst]:[BnOH] = 250:1:1, [CL] = 16.44 mmol, [BnOH] = 0.01 M, reaction temperature: 100 °C.
); reaction conditions: [CL]:[catalyst]:[BnOH] = 250:1:1, [CL] = 16.44 mmol, [BnOH] = 0.01 M, reaction temperature: 100 °C.
), 2 ( ), 3 ( ), 4 ( ); reaction conditions: [CL]:[catalyst]:[BnOH] = 250:1:1, [CL] = 16.44 mmol, [BnOH] = 0.01 M, reaction temperature: 100 °C.
 ), 6 (
), 6 (  ), 7 (
), 7 (  ), 8 (
), 8 (  ); Reaction conditions: [CL]:[catalyst]:[BnOH] = 250:1:1, [CL] = 16.44 mmol, [BnOH] = 0.01 M, reaction temperature: 100 °C.
), 6 ( ), 7 ( ), 8 ( ); Reaction conditions: [CL]:[catalyst]:[BnOH] = 250:1:1, [CL] = 16.44 mmol, [BnOH] = 0.01 M, reaction temperature: 100 °C.
); Reaction conditions: [CL]:[catalyst]:[BnOH] = 250:1:1, [CL] = 16.44 mmol, [BnOH] = 0.01 M, reaction temperature: 100 °C.
), 6 ( ), 7 ( ), 8 ( ); Reaction conditions: [CL]:[catalyst]:[BnOH] = 250:1:1, [CL] = 16.44 mmol, [BnOH] = 0.01 M, reaction temperature: 100 °C.





| - | 1 | 2 | 3 | - | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Bond length(Å) | - | - | - | - | - | - | - |
| Al(1)–O(1) | 1.7650(2) | 1.7895(11) | 1.8038(8) | Zn(1)–O(1) | 1.9077(15) | 1.9335(19) | 1.9583(17) |
| Al(1)–C(22) | 1.9550(4) | 1.9592(17) | 1.9626(12) | Zn(1)–O(2) | 1.9088(15) | 1.9338(18) | 1.9573(18) |
| Al(1)–C(23) | 1.9610(3) | 1.9614(17) | 1.9652(12) | Zn(1)–N(1) | 1.9998(18) | 2.026(2) | 2.027(3) |
| Al(1)–N(1) | 1.9800(3) | 1.9570(14) | 1.9923(10) | Zn(1)–N(2) | 2.0089(18) | 2.032(2) | 2.026(2) |
| Bond angles(°) | - | - | - | - | - | - | - |
| O(1)–Al(1)–C(22) | 109.08(14) | 109.00(6) | 107.68(5) | O(1)–Zn(1)–O(2) | 122.14(7) | 97.45(8) | 117.54(8) |
| O(1)–Al(1)–C(23) | 113.48(13) | 112.77(6) | 105.41(4) | O(1)–Zn(1)–N(1) | 96.84(7) | 93.54(9) | 101.30(8) |
| C(22)–Al(1)–C(23) | 119.12(16) | 121.35(7) | 123.31(5) | O(2)–Zn(1)–N(1) | 111.25(7) | 146.25(9) | 91.96(9) |
| O(1)–Al(1)–N(1) | 94.59(10) | 93.74(5) | 91.19(4) | O(1)–Zn(1)–N(2) | 111.20(7) | 138.49(8) | 92.29(8) |
| C(22)–Al(1)–N(1) | 108.91(13) | 110.62(7) | 111.29(5) | O(2)–Zn(1)–N(2) | 95.76(7) | 92.70(8) | 105.58(9) |
| C(23)–Al(1)–N(1) | 108.81(13) | 105.78(7) | 112.63(5) | N(1)–Zn(1)–N(2) | 121.63(7) | 99.93(8) | 149.77(9) |
| - | ν (C=N) | ν (C–O) | ν (M–O) | ν (M–N) | ν (Al–C) |
|---|---|---|---|---|---|
| 1 | 1613 | 1237 | 708 | 587 | 609 |
| 2 | 1614 | 1230 | 705 | 575 | 604 |
| 3 | 1615 | 1240 | 755 | 579 | 676 |
| 5 | 1612 | 1237 | 661 | 598 | - |
| 6 | 1614 | 1227 | 687 | 571 | - |
| 7 | 1616 | 1258 | - | - | - |
| L1H | 1614 | 1249 | - | - | - |
| L2H | 1619 | 1230 | - | - | - |
| L3H | 1616 | 1250 | - | - | - |
| Entry | Complex | [CL]:[Cat] | T | Time | Conversion | Mn,calc | Mn,GPC | Mw,GPC | Mw/Mne |
|---|---|---|---|---|---|---|---|---|---|
| :[BnOH] | (°C) | (min) | (%) b | (Da) c | (Da) d | (Da) d | |||
| 1 | 1 | 250:1:1 | 100 | 40 | 99 | 28,358 | 17,000 | 28,000 | 1.74 |
| 2 | 2 | 250:1:1 | 100 | 120 | 99 | 28,358 | 16,000 | 30,000 | 1.87 |
| 3 | 3 | 250:1:1 | 100 | 100 | 98 | 27,183 | 16,000 | 30,200 | 1.90 |
| 4 | 4 | 250:1:1 | 100 | 120 | 99 | 28,358 | 12,000 | 28,100 | 2.41 |
| 5 | 5 | 250:1:1 | 100 | 460 | 95 | 27,183 | 14,500 | 15,000 | 1.03 |
| 6 | 6 | 250:1:1 | 100 | 720 | 99 | 28,358 | 6700 | 7300 | 1.10 |
| 7 | 7 | 250:1:1 | 100 | 470 | 98 | 28,072 | 7100 | 8300 | 1.18 |
| 8 | 8 | 250:1:1 | 100 | 1560 | 95 | 27,183 | 7800 | 9100 | 1.18 |
| 9 | 1 | 250:1:1 | 25 | 720 | 4 | - | - | - | - |
| 10 | 1 | 250:1:1 | 50 | 1080 | 99 | 28,358 | 22,100 | 33,000 | 1.46 |
| 11 | 2 | 250:1:1 | 50 | 1080 | 90 | 25,790 | 20,000 | 39,000 | 1.96 |
| 12 | 3 | 250:1:1 | 50 | 1080 | 95 | 27,183 | 21,000 | 34,000 | 1.62 |
| 13 | 4 | 250:1:1 | 50 | 1080 | 85 | 24,363 | 20,237 | 39,000 | 1.93 |
| 14 | 5 | 250:1:1 | 50 | 1080 | 30 | 8669 | 5900 | 7000 | 1.20 |
| 15 | 6 | 250:1:1 | 50 | 1080 | 20 | 5821 | 3400 | 4400 | 1.28 |
| 16 | 7 | 250:1:1 | 50 | 1080 | 19 | 5530 | 3300 | 4300 | 1.29 |
| 17 | 8 | 250:1:1 | 50 | 1080 | 18 | 5821 | 5700 | 6800 | 1.20 |
| 18 f | 1 | 250:1:1f | 100 | 720 | 99 | 28,358 | 9200 | 16,000 | 1.75 |
| 19 f | 5 | 250:1:1f | 100 | 720 | 7 | - | - | - | - |
| 20 | 1 | 125:1:1 | 100 | 120 | 99 | 14,233 | 7300 | 11,300 | 1.56 |
| 21 | 1 | 500:1:1 | 100 | 120 | 99 | 56,716 | 24,000 | 49,400 | 2.04 |
| 22 | 1 | 250:1:0 | 100 | 35 | 99 | 28,250 | 76,000 | 210,000 | 2.75 |
| 23 | 1 | 250:1:1 | 100 | 40 | 99 | 28,358 | 17,000 | 28,000 | 1.74 |
| 24 | 1 | 250:1:2 | 100 | 115 | 99 | 14,233 | 6500 | 14,600 | 2.25 |
| 25 | 1 | 250:1:4 | 100 | 120 | 99 | 7200 | 3900 | 6200 | 1.60 |
| 26 | 5 | 250:1:0 | 100 | 120 | 40 | 28,250 | 25,500 | 62,500 | 2.45 |
| 27 | 5 | 250:1:1 | 100 | 120 | 51 | 27,183 | 14,500 | 15,000 | 1.03 |
| 28 | 5 | 250:1:2 | 100 | 120 | 26 | 8125 | 7800 | 10,500 | 1.34 |
| 29 | 5 | 250:1:4 | 100 | 120 | 13 | 4959 | 4000 | 5300 | 1.33 |
| Entry. | [GL]:[Cl] | Conversion | Conversion | l eGLc | l eCLc | R d | Mn,GPC | MW,GPC | Mw/Mnf |
|---|---|---|---|---|---|---|---|---|---|
| (%) | (%) of GL b | (%) of CL b | (Da) e | (Da) e | |||||
| 1 | 150:350 | 97 | 86 | 0.79 | 2.97 | 0.96 | 19,900 | 59,400 | 2.99 |
| 2 | 250:250 | 98 | 97 | 1.31 | 5.73 | 0.58 | 8400 | 24,500 | 2.88 |
| Entry. | [CL]:[BOD] | Conversion (%) of CL b | Conversion (%) of BOD b | Mn,GPC (Da) c | Mw,GPC (Da) c | Mw/Mnd |
|---|---|---|---|---|---|---|
| 1 | 250:250 | 99 | 51 | 308,000 | 526,000 | 1.71 |
| 2 | 250:0 | 99 | - | 17,000 | 28,000 | 1.74 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Chen, K.; Chicoma, M.; Goins, K.; Prior, T.J.; Nile, T.A.; Redshaw, C. Alkoxy-Functionalized Schiff-Base Ligation at Aluminum and Zinc: Synthesis, Structures and ROP Capability. Catalysts 2021, 11, 1090. https://doi.org/10.3390/catal11091090
Zhang X, Chen K, Chicoma M, Goins K, Prior TJ, Nile TA, Redshaw C. Alkoxy-Functionalized Schiff-Base Ligation at Aluminum and Zinc: Synthesis, Structures and ROP Capability. Catalysts. 2021; 11(9):1090. https://doi.org/10.3390/catal11091090
Chicago/Turabian StyleZhang, Xin, Kai Chen, Melissa Chicoma, Kimberly Goins, Timothy J. Prior, Terence A. Nile, and Carl Redshaw. 2021. "Alkoxy-Functionalized Schiff-Base Ligation at Aluminum and Zinc: Synthesis, Structures and ROP Capability" Catalysts 11, no. 9: 1090. https://doi.org/10.3390/catal11091090
APA StyleZhang, X., Chen, K., Chicoma, M., Goins, K., Prior, T. J., Nile, T. A., & Redshaw, C. (2021). Alkoxy-Functionalized Schiff-Base Ligation at Aluminum and Zinc: Synthesis, Structures and ROP Capability. Catalysts, 11(9), 1090. https://doi.org/10.3390/catal11091090