
Synthesis of Indoles by Palladium-Catalyzed Reductive Cyclization of β-Nitrostyrenes with Phenyl Formate as a CO Surrogate




Abstract
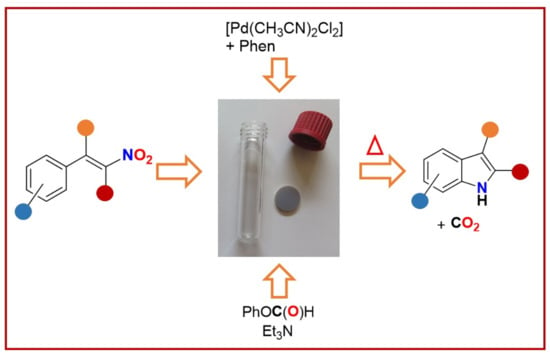
:
1. Introduction
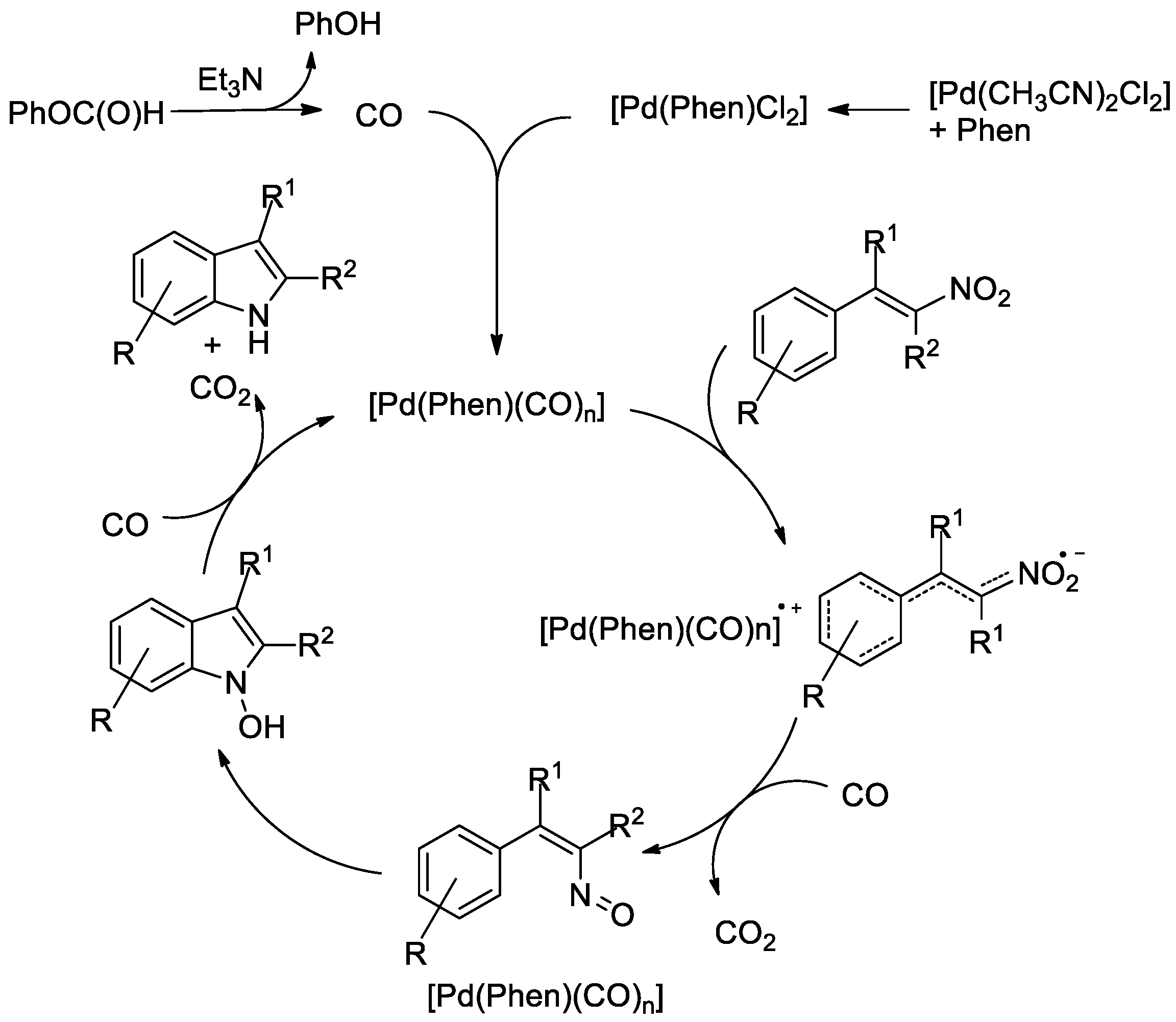
2. Results and Discussion
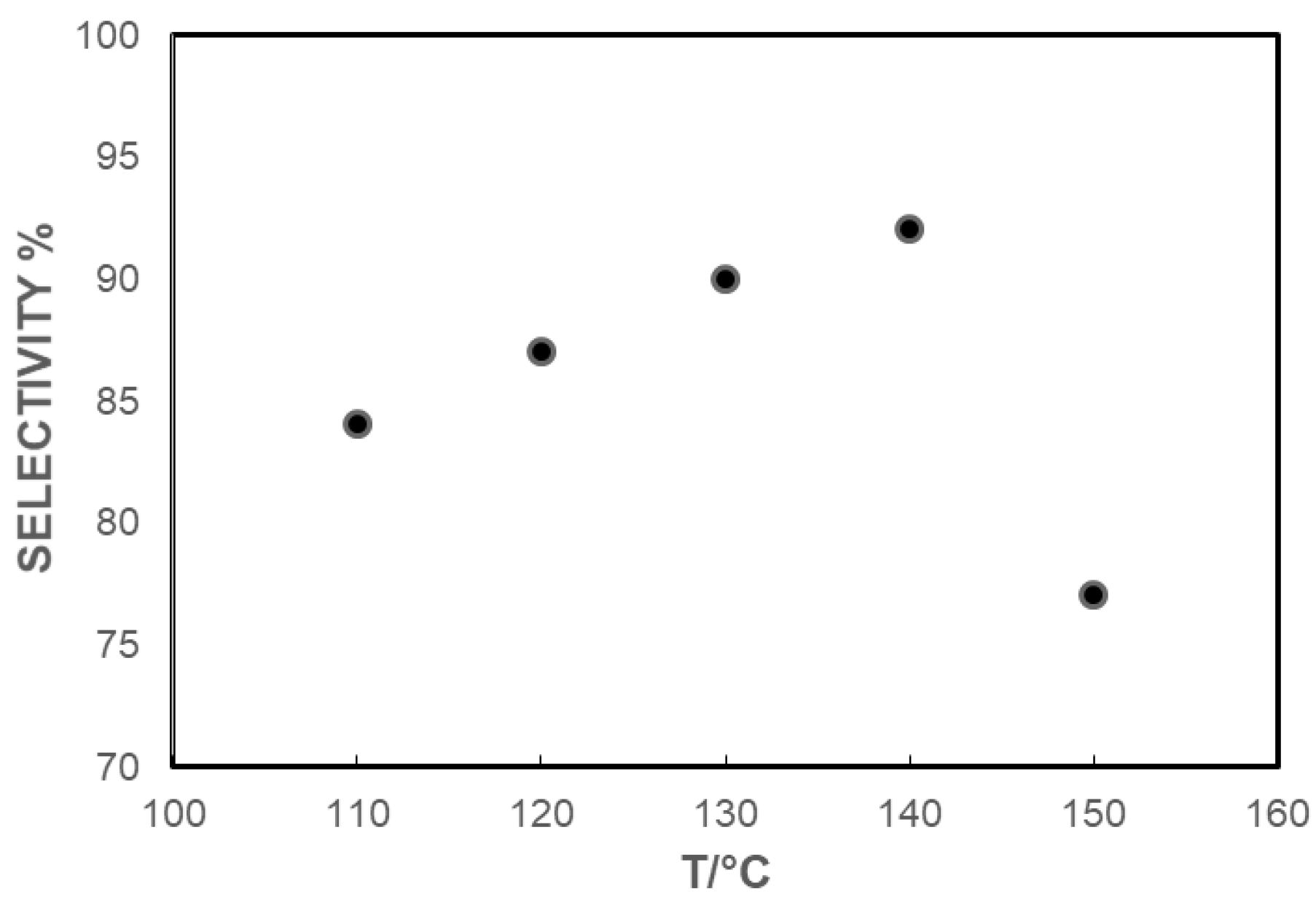
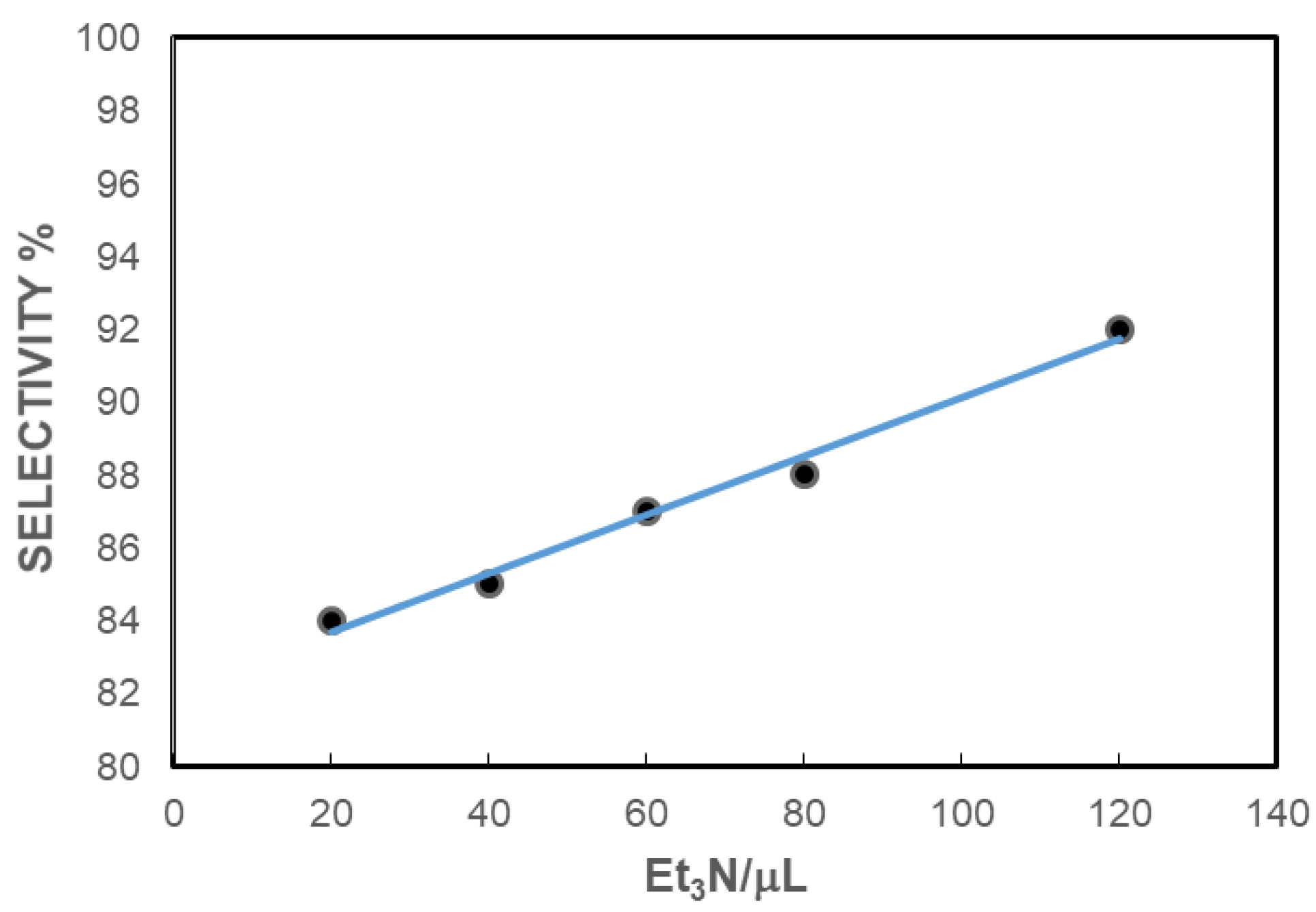
Optimization of the Reaction Conditions
3. Materials and Methods
3.1. General Procedures
3.2. Apparatus
3.3. General Methods for Catalytic Reactions
3.4. Characterization Data for Indoles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bandini, M.; Eichholzer, A. Catalytic Functionalization of Indoles in a New Dimension. Angew. Chem. Int. Ed. 2009, 48, 9608–9644. [Google Scholar] [CrossRef]
- Mathada, B.S.; Yernale, N.G.; Basha, J.N.; Badiger, J. An insight into the advanced synthetic recipes to access ubiquitous indole heterocycles. Tetrahedron Lett. 2021, 85, 153458. [Google Scholar] [CrossRef]
- Ayesha; Bilal, M.; Rasool, N.; Khan, S.G.; Rashid, U.; Altaf, H.; Ali, I. Synthesis of Indoles via Intermolecular and Intramolecular Cyclization by Using Palladium-Based Catalysts. Catalysts 2021, 11, 1018. [Google Scholar] [CrossRef]
- Bartoli, G.; Dalpozzo, R.; Nardi, M. Applications of Bartoli indole synthesis. Chem. Soc. Rev. 2014, 43, 4728–4750. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, G.R.; Kuethe, J.T. Practical Methodologies for the Synthesis of Indoles. Chem. Rev. 2006, 106, 2875–2911. [Google Scholar] [CrossRef]
- Cenini, S.; Ragaini, F. Catalytic Reductive Carbonylation of Organic Nitro Compounds; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1996. [Google Scholar]
- Soderberg, B.C.G. Synthesis of heterocycles via intramolecular annulation of nitrene intermediates. Curr. Org. Chem. 2000, 4, 727–764. [Google Scholar] [CrossRef]
- Ragaini, F.; Cenini, S.; Gallo, E.; Caselli, A.; Fantauzzi, S. Fine chemicals by reductive carbonylation of nitroarenes, catalyzed by transition metal complexes. Curr. Org. Chem. 2006, 10, 1479–1510. [Google Scholar] [CrossRef]
- Ferretti, F.; Formenti, D.; Ragaini, F. The reduction of organic nitro compounds by carbon monoxide as an effective strategy for the synthesis of N-heterocyclic compounds: A personal account. Rend. Lincei 2017, 28, 97–115. [Google Scholar] [CrossRef]
- Ferretti, F.; Ramadan, D.R.; Ragaini, F. Transition Metal Catalyzed Reductive Cyclization Reactions of Nitroarenes and Nitroalkenes. ChemCatChem 2019, 11, 4450–4488. [Google Scholar] [CrossRef]
- Afanasyev, O.I.; Chusov, D. Carbon Monoxide Related Reductions. Ineos Open 2020, 3, 133–139. [Google Scholar] [CrossRef]
- Tsygankov, A.A.; Makarova, M.; Chusov, D. Carbon monoxide as a selective reducing agent in organic chemistry. Mendeleev Commun. 2018, 28, 113–122. [Google Scholar] [CrossRef]
- Peng, J.B.; Qi, X.X.; Wu, X.F. Recent Achievements in Carbonylation Reactions: A Personal Account. Synlett 2017, 28, 175–194. [Google Scholar]
- Wu, L.; Liu, Q.; Jackstell, R.; Beller, M. Carbonylations of Alkenes with CO Surrogates. Angew. Chem. Int. Ed. 2014, 53, 6310–6320. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Manabe, K. Formic Acid Derivatives as Practical Carbon Monoxide Surrogates for Metal-Catalyzed Carbonylation Reactions. Synlett 2014, 25, 1971–1986. [Google Scholar] [CrossRef]
- Gautam, P.; Bhanage, B.M. Recent advances in the transition metal catalyzed carbonylation of alkynes, arenes and aryl halides using CO surrogates. Catal. Sci. Technol. 2015, 5, 4663–4702. [Google Scholar] [CrossRef]
- Friis, S.D.; Lindhardt, A.T.; Skrydstrup, T. The Development and Application of Two-Chamber Reactors and Carbon Monoxide Precursors for Safe Carbonylation Reactions. Acc. Chem. Res. 2016, 49, 594–605. [Google Scholar] [CrossRef] [Green Version]
- Fouad, M.A.; Ferretti, F.; Formenti, D.; Milani, F.; Ragaini, F. Synthesis of Indoles by Reductive Cyclization of Nitro Compounds Using Formate Esters as CO Surrogates. Eur. J. Org. Chem. 2021, 2021, 4876–4894. [Google Scholar] [CrossRef]
- Formenti, D.; Ferretti, F.; Ragaini, F. Synthesis of N-Heterocycles by Reductive Cyclization of Nitro Compounds using Formate Esters as Carbon Monoxide Surrogates. ChemCatChem 2018, 10, 148–152. [Google Scholar] [CrossRef]
- EL-Atawy, M.A.; Formenti, D.; Ferretti, F.; Ragaini, F. Synthesis of 3,6-Dihydro-2H-[1,2]-Oxazines from Nitroarenes and Conjugated Dienes, Catalyzed by Palladium/Phenanthroline Complexes and Employing Phenyl Formate as a CO Surrogate. ChemCatChem 2018, 10, 4707–4717. [Google Scholar] [CrossRef] [Green Version]
- Ramadan, D.R.; Ferretti, F.; Ragaini, F. Synthesis of Carbazoles: Use of Formate Esters as CO Surrogates and Pd/Phen Complexes as Catalysts for Reductive Cyclization of 2-Nitrobiphenyls. In Proceedings of the Cutting-Edge Homogeneous Catalysis (CEHC-1), Toulouse, France, 4–6 May 2021; p. 26. [Google Scholar]
- Qi, X.; Zhou, R.; Peng, J.-B.; Ying, J.; Wu, X.-F. Selenium-Catalyzed Carbonylative Synthesis of 2-Benzimidazolones from 2-Nitroanilines with TFBen as the CO Source. Eur. J. Org. Chem. 2019, 2019, 5161–5164. [Google Scholar] [CrossRef]
- Zhou, R.; Qi, X.; Wu, X.-F. Selenium-Catalyzed Carbonylative Synthesis of 3,4-Dihydroquinazolin-2(1H)-one Derivatives with TFBen as the CO Source. ACS Comb. Sci. 2019, 21, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Tollari, S.; Cenini, S.; Crotti, C.; Gianella, E. Synthesis of heterocycles via palladium-catalyzed carbonylation of ortho-substituted organic nitro compounds in relatively mild conditions. J. Mol. Catal. 1994, 87, 203–214. [Google Scholar] [CrossRef]
- Tollari, S.; Cenini, S.; Ragaini, F.; Cassar, L. Intramolecular Amination of Olefins-Synthesis of 2-Substituted-4-Quinolones from 2-Nitrochalcones Catalyzed by Ruthenium. J. Chem. Soc. Chem. Comm. 1994, 1994, 1741–1742. [Google Scholar] [CrossRef]
- Rahman, S.M.A.; Söderberg, B.C.G. A palladium-catalyzed Barluenga cross-coupling–Reductive cyclization sequence to substituted indoles. Tetrahedron 2021, 94, 132331. [Google Scholar] [CrossRef] [PubMed]
- Ansari, N.H.; Dacko, C.A.; Akhmedov, N.G.; Söderberg, B.C.G. Double Palladium Catalyzed Reductive Cyclizations. Synthesis of 2,2′-, 2,3′-, and 3,3′-Bi-1H-indoles, Indolo[3,2-b]indoles, and Indolo[2,3-b]indoles. J. Org. Chem. 2016, 81, 9337–9349. [Google Scholar] [CrossRef]
- Hubbard, J.W.; Piegols, A.M.; Soderberg, B.C.G. Palladium-catalyzed N-heteroannulation of N-allyl- or N-benzyl-2-nitrobenzenamines: Synthesis of 2-substituted benzimidazoles. Tetrahedron 2007, 63, 7077–7085. [Google Scholar] [CrossRef]
- Soderberg, B.C.; Shriver, J.A. Palladium-catalyzed synthesis of indoles by reductive N-heteroannulation of 2-nitrostyrenes. J. Org. Chem. 1997, 62, 5838–5845. [Google Scholar] [CrossRef]
- Okuro, K.; Gurnham, J.; Alper, H. Ionic Diamine Rhodium Complex Catalyzed Reductive N-Heterocyclization of 2-Nitrovinylarenes. J. Org. Chem. 2011, 76, 4715–4720. [Google Scholar] [CrossRef]
- O’Dell, D.K.; Nicholas, K.M. Synthesis of 3-substituted quinolines via transition-metal-catalyzed reductive cyclization of o-nitro Baylis-Hillman acetates. J. Org. Chem. 2003, 68, 6427–6430. [Google Scholar] [CrossRef]
- Ragaini, F.; Cenini, S.; Borsani, E.; Dompe, M.; Gallo, E.; Moret, M. Synthesis of N-arylpyrroles, hetero-Diels-Alder adducts, and allylic amines by reaction of unfunctionalized dienes with nitroarenes and carbon monoxide, catalyzed by Ru(CO)3(Ar-BIAN). Organometallics 2001, 20, 3390–3398. [Google Scholar] [CrossRef]
- Ragaini, F.; Ventriglia, F.; Hagar, M.; Fantauzzi, S.; Cenini, S. Synthesis of Indoles by Intermolecular Cyclization of Unfunctionalized Nitroarenes and Alkynes: One-Step Synthesis of the Skeleton of Fluvastatin. Eur. J. Org. Chem. 2009, 2009, 2185–2189. [Google Scholar] [CrossRef]
- Penoni, A.; Nicholas, K.M. A novel and direct synthesis of indoles via catalytic reductive annulation of nitroaromatics with alkynes. Chem. Commun. 2002, 2002, 484–485. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, T.H.H.; Dong, V.M. Indole synthesis: Palladium-catalyzed C-H bond amination via reduction of nitroalkenes with carbon monoxide. Tetrahedron 2009, 65, 3062–3068. [Google Scholar] [CrossRef]
- Ferretti, F.; El-Atawy, M.A.; Muto, S.; Hagar, M.; Gallo, E.; Ragaini, F. Synthesis of Indoles by Palladium-Catalyzed Reductive Cyclization of β-Nitrostyrenes with Carbon Monoxide as the Reductant. Eur. J. Org. Chem. 2015, 2015, 5712–5715. [Google Scholar] [CrossRef]
- El-Atawy, M.A.; Ferretti, F.; Ragaini, F. Palladium-Catalyzed Intramolecular Cyclization of Nitroalkenes: Synthesis of Thienopyrroles. Eur. J. Org. Chem. 2017, 2017, 1902–1910. [Google Scholar] [CrossRef]
- EL-Atawy, M.A.; Ferretti, F.; Ragaini, F. A Synthetic Methodology for Pyrroles from Nitrodienes. Eur. J. Org. Chem. 2018, 2018, 4818–4825. [Google Scholar] [CrossRef]
- Su, Z.; Liu, B.; Liao, H.; Lin, H.-W. Synthesis of N-Heterocycles by Reductive Cyclization of Nitroalkenes Using Molybdenum Hexacarbonyl as Carbon Monoxide Surrogate. Eur. J. Org. Chem. 2020, 2020, 4059–4066. [Google Scholar] [CrossRef]
- Guan, X.; Zhu, H.; Zhao, Y.; Driver, T.G. Pd-Catalyzed Reductive Cyclization of Nitroarenes with CO2 as the CO Source. Eur. J. Org. Chem. 2020, 2020, 57–60. [Google Scholar] [CrossRef] [Green Version]
- Ragaini, F.; Cenini, S.; Brignoli, D.; Gasperini, M.; Gallo, E. Synthesis of oxazines and N-arylpyrroles by reaction of unfunctionalized dienes with nitroarenes and carbon monoxide, catalyzed by palladium-phenanthroline complexes. J. Org. Chem. 2003, 68, 460–466. [Google Scholar] [CrossRef]
- Smitrovich, J.H.; Davies, I.W. Catalytic C-H functionalization driven by CO as a stoichiometric reductant: Application to carbazole synthesis. Org. Lett. 2004, 6, 533–535. [Google Scholar] [CrossRef]
- Davies, I.W.; Smitrovich, J.H.; Sidler, R.; Qu, C.; Gresham, V.; Bazaral, C. A highly active catalyst for the reductive cyclization of ortho-nitrostyrenes under mild conditions. Tetrahedron 2005, 61, 6425–6437. [Google Scholar] [CrossRef]
- Clawson, R.W.; Dacko, C.A.; Deavers, R.E.; Akhmedov, N.G.; Soderberg, B.C.G. Attempted synthesis of 3-hydroxy-2-octadecylindole. Proposed structural revision of previously prepared 3-hydroxy-2-octadecylindole and a proposed structure of fistulosin. Tetrahedron 2009, 65, 8786–8793. [Google Scholar] [CrossRef]
- Alessio, E.; Mestroni, G. Catalytic synthesis of aromatic urethanes from nitroaromatic compounds and carbon monoxide, using palladium + 1,10-phenanthroline derivatives as catalyst precursors. J. Mol. Catal. 1984, 26, 337–340. [Google Scholar] [CrossRef]
- Bontempi, A.; Alessio, E.; Chanos, G.; Mestroni, G. Reductive carbonylation of nitroaromatic compounds to urethanes catalyzed by (di-1,10-phenanthroline)palladium bis(hexafluorophosphate) and related complexes. J. Mol. Catal. 1987, 42, 67–80. [Google Scholar] [CrossRef]
- Wehman, P.; Kaasjager, V.E.; Delange, W.G.J.; Hartl, F.; Kamer, P.C.J.; van Leeuwen, P.W.N.M.; Fraanje, J.; Goubitz, K. Subtle Balance between Various Phenanthroline Ligands and Anions in the Palladium-Catalyzed Reductive Carbonylation of Nitrobenzene. Organometallics 1995, 14, 3751–3761. [Google Scholar] [CrossRef] [Green Version]
- Gasperini, M.; Ragaini, F.; Cazzaniga, C.; Cenini, S. Carbonylation of dinitrotoluene to dimethyl toluenedicarbamate; high efficiency of phosphorus acids as promoters for the palladium-phenanthroline catalytic system. Adv. Synth. Catal. 2005, 347, 105–120. [Google Scholar] [CrossRef]
- Gasperini, M.; Ragaini, F.; Remondini, C.; Caselli, A.; Cenini, S. The palladium-phenanthroline catalyzed carbonylation of nitroarenes to diarylureas: Effect of chloride and diphenylphosphinic acid. J. Organomet. Chem. 2005, 690, 4517–4529. [Google Scholar] [CrossRef]
- Ferretti, F.; Ragaini, F.; Lariccia, R.; Gallo, E.; Cenini, S. New Nonsymmetric Phenanthrolines as Very Effective Ligands in the Palladium-Catalyzed Carbonylation of Nitrobenzene. Organometallics 2010, 29, 1465–1471. [Google Scholar] [CrossRef]
- Ferretti, F.; Gallo, E.; Ragaini, F. Nitrogen ligands effects in the palladium-catalyzed carbonylation reaction of nitrobenzene to give N-methyl phenylcarbamate. J. Organomet. Chem. 2014, 771, 59–67. [Google Scholar] [CrossRef]
- Ferretti, F.; Gallo, E.; Ragaini, F. Mineral Oil/Methanol: A Cheap Biphasic Reaction Medium with Thermomorphic Properties and Its Application to the Palladium-Catalyzed Carbonylation of Nitrobenzene to Methyl Phenylcarbamate. ChemCatChem 2015, 7, 2241–2247. [Google Scholar] [CrossRef]
- Carter, M.E.; Nash Jr, J.L.; Drueke Jr, J.W.; Schwietert, J.W.; Butler, G.B. Anionic-Initiated Polymerization of beta -Nitrostyrenes. J. Polym. Sci. Polym. Chem. Ed. 1978, 16, 937–959. [Google Scholar] [CrossRef]
- Berman, R.S.; Kochi, J.K. Kinetics and mechanism of oxygen atom transfer from nitro compounds mediated by nickel(0) complexes. Inorg. Chem. 1980, 19, 248–254. [Google Scholar] [CrossRef]
- Skoog, S.J.; Gladfelter, W.L. Activation of Nitroarenes in the Homogenous Catalytic Carbonylation of Nitroaromatics via an Oxygen-Atom-Transfer Mechanism Induced by Inner Sphere Electron Transfer. J. Am. Chem. Soc. 1997, 119, 11049–11060. [Google Scholar] [CrossRef]
- Kunin, A.J.; Noirot, M.D.; Gladfelter, W.L. In situ FTIR spectroscopy at elevated carbon monoxide pressure. Evidence for single electron transfer in the catalytic carbonylation of nitroaromatics. J. Am. Chem. Soc. 1989, 111, 2739–2741. [Google Scholar] [CrossRef]
- Belousov, Y.A. Radical chemistry of iron carbonyls. Russ. Chem. Rev. 2007, 76, 41–58. [Google Scholar] [CrossRef]
- Belousov, Y.A.; Kolosova, T.A. Electron-spin resonance study of the reaction of iron carbonyls with nitro and nitrosoparaffines—A mechanism of the reductive carbonylation of nitrocompounds. Polyhedron 1987, 6, 1959–1970. [Google Scholar] [CrossRef]
- Ragaini, F.; Song, J.S.; Ramage, D.L.; Geoffroy, G.L.; Yap, G.A.P.; Rheingold, A.L. Radical Processes in the Reduction of Nitrobenzene Promoted by Iron Carbonyl Clusters. X-Ray Crystal-Structures of [Fe3(CO)9(μ3-NPh)]2−, [HFe3(CO)9(μ3-NPh)]−, and the Radical-Anion [Fe3(CO)11]−•. Organometallics 1995, 14, 387–400. [Google Scholar] [CrossRef]
- Ragaini, F. Mechanistic study of the phase-transfer-catalyzed reduction of nitrobenzene to aniline by iron carbonyl complexes. Role of the radical anion [Fe3(CO)11)]•−. Organometallics 1996, 15, 3572–3578. [Google Scholar] [CrossRef]
- Liu, P.H.; Liao, H.Y.; Cheng, C.H. Electro-transfer from tetracarbonylrhodate(1-) to nitro aromatics-novel interaction of nitro radical-anions with a rhodium(I) center. J. Chem. Soc. Chem. Comm. 1995, 23, 2441–2442. [Google Scholar] [CrossRef] [Green Version]
- Ragaini, F.; Cenini, S.; Demartin, F. Mechanistic Studies of the Carbonylation of Nitrobenzene Catalyzed by the [Rh(CO)4]-/Bipy System. X-Ray Structure of [PPN][Rh(CO)2ON[C6H3Cl2)C(O)O] [PPN+ = (PPh3)2N+; Bipy = 2,2′-Bipyridyl]. J. Chem. Soc. Chem. Comm. 1992, 19, 1467–1468. [Google Scholar] [CrossRef]
- Ragaini, F.; Cenini, S.; Demartin, F. Mechanistic Study of the Carbonylation of Nitrobenzene Catalyzed by the [Rh(CO)4]− Nitrogen Base System. X-Ray Structure of [PPN][Rh(CO)2ON(C6H3Cl2)C(O)O]. Organometallics 1994, 13, 1178–1189. [Google Scholar] [CrossRef]
- Gallo, E.; Ragaini, F.; Cenini, S.; Demartin, F. Investigation of the reactivity of palladium(0) complexes with nitroso compounds: Relevance to the palladium/phenanthroline-catalysed carbonylation reactions of nitroarenes. J. Organomet. Chem. 1999, 586, 190–195. [Google Scholar] [CrossRef]
- Tomson, N.C.; Labios, L.A.; Weyhermüller, T.; Figueroa, J.S.; Wieghardt, K. Redox Noninnocence of Nitrosoarene Ligands in Transition Metal Complexes. Inorg. Chem. 2011, 50, 5763–5776. [Google Scholar] [CrossRef]
- Popp, B.V.; Thorman, J.L.; Morales, C.M.; Landis, C.R.; Stahl, S.S. Inverse-Electron-Demand Ligand Substitution: Experimental and Computational Insights into Olefin Exchange at Palladium(0). J. Am. Chem. Soc. 2004, 126, 14832–14842. [Google Scholar] [CrossRef]
- Todres, Z.V. Cia/Trans Conversion of Potassium Derivatives of 2-Nitrostilbenes and 4-Nitrostilbenes. J. Organomet. Chem. 1992, 441, 349–354. [Google Scholar] [CrossRef]
- Álvarez-Calero, J.M.; Jorge, Z.D.; Massanet, G.M. TiCl4/Et3N-Mediated Condensation of Acetate and Formate Esters: Direct Access to β-Alkoxy- and β-Aryloxyacrylates. Org. Lett. 2016, 18, 6344–6347. [Google Scholar] [CrossRef] [PubMed]
- Rimoldi, M.; Ragaini, F.; Gallo, E.; Ferretti, F.; Macchi, P.; Casati, N. Unexpected isomerism in [Pd(2,9-dimethylphenanthroline)X2] (X = Cl, Br, I) complexes: A neutral and an ionic form exist. Dalton Trans. 2012, 41, 3648–3658. [Google Scholar] [CrossRef]
- Lu, B.-L.; Shi, M. Gold(I)-Catalyzed Tandem Oxidative Ring-Opening/C—C Bond Cleavage Reactions of Vinylidenecyclopropanes with Secondary Amines Under an Oxygen Atmosphere. Chem. Eur. J. 2011, 17, 9070–9075. [Google Scholar] [CrossRef]
- Maity, S.; Naveen, T.; Sharma, U.; Maiti, D. Stereoselective Nitration of Olefins with tBuONO and TEMPO: Direct Access to Nitroolefins under Metal-free Conditions. Org. Lett. 2013, 15, 3384–3387. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Phen (mol%) | T (°C) | Base | Base Amount (mmol) | Solvent | Conv.% 2 | Select.% 3 |
|---|---|---|---|---|---|---|---|
| 1 | 2.5 | 140 | Et3N | 0.29 | CH3CN | 80 | 39 |
| 2 | 10 | 140 | Et3N | 2.30 | CH3CN | >99 | 40 |
| 3 | 10 | 140 | Et3N | 2.30 | THF | 85 | 30 |
| 4 | 10 | 140 | Et3N | 2.30 | DMF | >99 | 41 |
| 5 | 10 | 140 | Et3N | 2.30 | C6H5CH3 | 52 | 12 |
| 6 | 10 | 140 | Et3N | 2.30 | CH3OH | 85 | 9 |
| 7 4 | 10 | 140 | - | - | CH3CN | ~0 | 0 |
| 8 5 | 10 | 140 | Et3N | 2.30 | CH3CN | 61 | 0 |
| 9 | 10 | 120 | Et3N | 0.29 | CH3CN | 32 | 31 |
| 10 | 10 | 120 | Py | 0.29 | CH3CN | <1 | <1 |
| 11 | 10 | 150 | Py | 0.29 | CH3CN | 17 | 18 |
| 12 | 10 | 120 | NaOAc | 0.29 | CH3CN | 62 | 17 |
| 13 | 10 | 120 | Na3PO4 | 0.29 | CH3CN | 78 | 7 |
| 14 6 | 10 | 140 | Et3N | 2.30 | CH3CN | >99 | 26 |
| 15 6 | 10 | 140 | Et3N | 0.29 | CH3CN | 95 | 28 |
| 16 7 | 10 | 140 | Et3N | 2.30 | CH3CN | >99 | 47 |
| Entry | Pd (mol%) | Phen (mol%) | T (°C) | t (h) | Et3N (μL) | PhOC(O)H (μL) | Conv.% 2 | Select.% 3 |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 5 | 110 | 3 | 120 | 260 | >99 | 84 |
| 2 | 1 | 5 | 120 | 3 | 120 | 260 | >99 | 87 |
| 3 | 1 | 5 | 130 | 3 | 120 | 260 | >99 | 90 |
| 4 | 1 | 5 | 140 | 3 | 120 | 260 | >99 | 92 |
| 5 | 1 | 5 | 150 | 3 | 120 | 260 | >99 | 77 |
| 6 | 1 | 5 | 140 | 3 | 20 | 260 | >99 | 84 |
| 7 | 1 | 5 | 140 | 3 | 40 | 260 | >99 | 85 |
| 8 | 1 | 5 | 140 | 3 | 60 | 260 | >99 | 87 |
| 9 | 1 | 5 | 140 | 3 | 80 | 260 | >99 | 88 |
| 10 | 1 | 5 | 140 | 3 | 120 | 200 | 97 | 81 |
| 11 | 1 | 5 | 140 | 3 | 120 | 350 | >99 | 91 |
| 12 | 1 | 5 | 140 | 3 | 200 | 350 | >99 | 90 |
| 13 | 1 | 2.5 | 140 | 3 | 120 | 260 | 94 | 85 |
| 14 | 1 | 2.5 | 140 | 3 | 80 | 260 | >99 | 83 |
| 15 | 1 | 2.5 | 140 | 7 | 40 | 260 | >99 | 87 |
| 16 | 1 | 2.5 | 140 | 16 | 40 | 260 | >99 | 87 |
| 17 | 1 | 10 | 140 | 3 | 120 | 260 | >99 | 90 |
| 18 | 0.5 | 5 | 140 | 3 | 120 | 260 | >99 | 78 |
| 19 | 0.5 | 5 | 140 | 6 | 120 | 260 | >99 | 79 |
| 20 4 | 2 | 5 | 140 | 3 | 120 | 260 | >99 | 90 |
| 21 4 | 2 | 5 | 140 | 1.5 | 120 | 260 | >99 | 90 |
| 22 5 | 1 | 5 | 140 | 3 | 120 | 260 | >99 | 88 |
| 23 6 | 1 | 5 | 140 | 3 | 120 | 260 | >99 | 85 |
| Entry | Substrate | Product | Yield 2 |
|---|---|---|---|
| 1 3 |  |  | 43(47) 4 |
| 2 |  |  | 89(92) 4 |
| 3 |  |  | 66 |
| 4 |  |  | |
| 5 |  |  | 49 |
| 6 |  |  | _ 5 |
| 7 |  |  | 67(1:2.5) |
| 8 |  |  | 70 |
| 9 |  |  | 71(1.3:1) |
| 10 |  |  | 47 |
| 11 |  |  | (34) 4 |
| 12 |  |  | _ 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferretti, F.; Fouad, M.A.; Ragaini, F. Synthesis of Indoles by Palladium-Catalyzed Reductive Cyclization of β-Nitrostyrenes with Phenyl Formate as a CO Surrogate. Catalysts 2022, 12, 106. https://doi.org/10.3390/catal12010106
Ferretti F, Fouad MA, Ragaini F. Synthesis of Indoles by Palladium-Catalyzed Reductive Cyclization of β-Nitrostyrenes with Phenyl Formate as a CO Surrogate. Catalysts. 2022; 12(1):106. https://doi.org/10.3390/catal12010106
Chicago/Turabian StyleFerretti, Francesco, Manar Ahmed Fouad, and Fabio Ragaini. 2022. "Synthesis of Indoles by Palladium-Catalyzed Reductive Cyclization of β-Nitrostyrenes with Phenyl Formate as a CO Surrogate" Catalysts 12, no. 1: 106. https://doi.org/10.3390/catal12010106
APA StyleFerretti, F., Fouad, M. A., & Ragaini, F. (2022). Synthesis of Indoles by Palladium-Catalyzed Reductive Cyclization of β-Nitrostyrenes with Phenyl Formate as a CO Surrogate. Catalysts, 12(1), 106. https://doi.org/10.3390/catal12010106