2.1. Impacts of CuO Contents
Figure 1 shows the XRD patterns of calcined and reduced 25Cu/ASA-4 and xCu–ASA-4 catalysts, and the particle sizes calculated based on Scherrer’s formula and (111) diffractions are given in
Table 1. For all of the calcined catalysts (
Figure 1a), the broad peak centered at 2
θ of about 23° was clearly observed, which is ascribed to amorphous silica. Noteworthy, no diffractions assigned to the crystalline phases of Al
2O
3, SiO
2, and the derived structures including the spinel were observed for all of the catalysts, which is different from those of the mesoporous Cu-Al
2O
3 and Cu-SiO
2-Al
2O
3 reported in the reference [
20]. This may be reasonably explained as the relatively lower calcination temperature of 550 °C, leading to the amorphous nature of ASA. In the case of Cu species, the characteristic diffractions of CuO at 2
θ of 35.5, 38.7, and 48.8° were clearly observed for the samples with CuO contents higher than 20 wt.%. Moreover, the intensity of the CuO peaks was apparently increased when the CuO content was increased from 25 to 35 wt.%. This was directly reflected in the accordingly increased particle size from 15 to 21 nm (
Table 1), which was calculated based on the diffraction of CuO (111) at 2
θ of 35.5°. Thus, the absent diffractions of Cu species for the 10Cu–ASA-4 and 20Cu–ASA-4 catalysts were reasonably attributed to their smaller sizes insensitive to XRD, indicating the highly dispersed states. If the reference catalyst prepared by the impregnation method was compared, the main diffractions of CuO at 2
θ of 35.5, 38.7, 48.8, 58.2, 61.6, 66.1, and 67.9° were fully developed, and the particle size of CuO (28 nm) was much larger than that of 25Cu–ASA-4 (15 nm,
Table 1). These results indicate the superiority of the single-step complex decomposition method for preparing highly dispersed Cu catalysts.
For the reduced
xCu–ASA-4 catalysts (
Figure 1b), ASA was still amorphous as reflected in the broad XRD peak. In contrast, where the Cu species were concerned, new diffractions emerged irrespective of the CuO contents, which can be assigned to Cu
2O (2
θ = 36.6, 42.4, and 61.4°) and Cu
0 (2
θ = 43.4, 50.5, and 74.1°) according to the references [
21,
22]. Moreover, the intensity for both the Cu
0 (111) peak at 2
θ of 43.4° and Cu
2O (111) diffraction at 2
θ of 36.6° was gradually increased with increasing CuO content of xCu–ASA-4, revealing the increased particle sizes of Cu
0 and Cu
2O. This was directly found from the calculated Cu
0 size (
Table 1), i.e., from 12 nm (10Cu–ASA-4) to 33 nm (35Cu–ASA-4). Noteworthy, no Cu species were detected by XRD for the calcined 10Cu–ASA-4 and 20Cu–ASA-4 catalysts while clear XRD diffractions of Cu
0 and Cu
2O appeared for the reduced catalyst even with the lowest CuO content of 10 wt.%. This was consistent with the easy sintering of Cu species during the reduction in hydrogen although it was performed at a temperature as low as 300 °C. If the Cu
0 particle size of the reduced 25Cu/ASA-4 catalyst was examined (35 nm,
Table 1), it was much larger than that of the reduced 25Cu–ASA-4 catalyst (21 nm).
With the use of 25Cu–ASA-4 as a representative, TEM was performed to directly observe the Cu species of the catalysts. For the calcined 25Cu–ASA-4 catalyst (
Figure 2a), the CuO particles identified by the lattice fringe of 0.234 nm were clearly seen and were assigned to CuO (111). Moreover, the size distribution of the CuO particles was relatively narrower, and the average size was 5.3 ± 1.0 nm. When 25Cu–ASA-4 was reduced (
Figure 2b), Cu (111) lattice fringes with a d-spacing of 0.212 nm were ascertained. In comparison with the calcined catalyst, the size distribution of the Cu
0 particles was broader and the average Cu
0 particle size (5.6 ± 1.5 nm) was slightly larger. Although the change trends of the average particle sizes was consistent with that of the XRD results, they were much smaller than those calculated from the XRD diffractions.
The nitrogen adsorption–desorption isotherms of the ASA-4 and
xCu–ASA-4 catalysts are shown in
Figure 3a. Irrespective of the samples, a type IV isotherm with a clear H3 hysteresis loop based on the IUPAC classification [
23] was observed, indicating the irregular porous structure with micro-, meso-, and macropores. If the isotherms in the range of P/P
0 less than 0.05 were examined, the addition of CuO and increasing its content up to 35 wt.% led to very limited changes in the amount of adsorbed N
2, indicating very similar micropores across all of the samples. This observation was consistent with the amorphous properties of ASA. As indicated from the calculated data (
Table 1), the BET specific surface area of ASA-4 was as high as 356.3 m
2·g
−1, characterizing the superiority of the complex decomposition method in the preparation of the amorphous oxides. Moreover, even a slightly higher BET specific surface area of 357.7 m
2·g
−1 was obtained for 10Cu–ASA-4, which agrees with the high dispersion of CuO in ASA revealed in the XRD characterizations. In the cases of Cu–ASA-4 catalysts with CuO contents from 20 to 35 wt.%, the BET specific surface area, the total pore volume, and the average pore diameter were very similar and in agreement with the high dispersion of CuO in the amorphous silica–alumina composites.
Figure 4 shows the reducibility of the
xCu–ASA-4 and 25Cu/ASA-4 catalysts. For all of the samples, only one asymmetric H
2-consumption peak centered below 300 °C was observed throughout the H
2-TPR process until 600 °C. This observation clearly indicates that the difficult reducible Cu species such as the CuAl
2O
4 spinel were not formed over all of the catalysts, which is supported by the XRD results (
Figure 1a). Considering the irreducible nature of ASA, the H
2 consumption exclusively originated from the reduction of CuO, which was detected by XRD. This was further supported by the continuously increased intensity of the H
2-TPR peak with the increasing CuO content of
xCu–ASA-4. If the temperature at the peak maximum was examined, it continuously decreased from about 272 to ~258 °C with increasing CuO content in
xCu–ASA-4 from 10 to 30 wt.%. By correlating this observation with the calculated CuO particle sizes (
Table 1) and XRD patterns, it can be explained as the increased difficulty for the reduction of the highly dispersed CuO with smaller sizes, which is consistent with the reference results [
20,
24]. This explanation was further supported by the following facts, i.e., almost the same peak temperatures (~258 °C) for 30Cu–ASA-4 and 35Cu–ASA-4 with comparable CuO sizes of 20–21 nm, and the lowest peak temperature of ~250 °C for 25Cu/ASA-4 with the largest CuO particle size (35 nm). As reported in the literature, one of the possible routes for the reduction of CuO in H
2 is the two-step process of CuO to Cu
2O and the subsequent Cu
2O conversion to Cu
0 [
25]. Thus, the asymmetry of the H
2-TPR peak may be due to the varied Cu
0/Cu
+ ratios of different catalysts as revealed from the XRD patterns of the reduced catalysts (
Figure 1b). Alternatively, the asymmetric peak may also originate from the varied reducibility of CuO particles with different sizes as indicated from the broad size distribution over the catalyst (
Figure 2).
The catalytic results of xCu–ASA-4 for SRD are given in
Figure 5. As indicated from the time-on-stream (TOS) data, a volcano shape was ascertained for the changing profile of the initial DME conversion versus the CuO content over the catalysts (
Figure 5a), i.e., 25Cu–ASA-4 (~90%) > 30Cu–ASA-4 (~85%) > 20Cu–ASA-4 (~82%) ≈ 35Cu–ASA-4 (~81%) >> 10Cu–ASA-4 (~63%). Moreover, this observation with the same changing order was still applicable for the initial H
2 yields (
Figure 5b). With increasing TOS from 1 to 6 h, a minor decrease in the DME conversion was observed for the
xCu–ASA-4 catalysts, and the decreased trend was slightly more significant for the TOS H
2 yields except 10Cu–ASA-4 with a sharp decrease. To better under these phenomena, the selectivity of the carbon-containing products at a TOS of 6 h was calculated. As shown in
Figure 5c, the selectivity of CH
4, C
2-C
4 hydrocarbons and CO was very low (<1%) irrespective of the catalysts used. In the case of 10Cu–ASA-4, methanol with a selectivity of about 92% was the main carbon-containing product while the selectivity of CO
2 was only about 7%. This indicates that much less methanol formed via DME hydrolysis was converted during the second-step SRM reaction, leading to the much low efficiency of 10Cu–ASA-4 for the SRD reaction. As a result, the DME conversion of ~60% was still preserved at a TOS of 6 h but the H
2 yield was drastically decreased to be less than 5% (
Figure 5a,b). In contrast, methanol was the minor carbon-containing product, and its selectivity at a TOS of 6 h was decreased in the order of 20Cu–ASA-4 (~21%) >> 25Cu–ASA-4 (~8%) ≥ 30Cu–ASA-4 (~7%) ≈ 35Cu–ASA-4 (~7%), indicating that the extent for consuming the methanol intermediate via the SRM reaction was in a reverse order. As (1) the Si/Al molar ratio of ASA for the hydrolysis of DME over the
xCu–ASA-4 catalysts was maintained at 4 and (2) Cu
0 is the active site for the SRM reaction, these SRD results can be reasonably explained if the Cu content and Cu
0 size are taken into account. This was also supported if the catalytic and characterization results of 25Cu–ASA-4 and 25Cu/ASA-4 were compared, i.e., the clearly higher SRD efficiency and obviously smaller Cu
0 particles of 25Cu–ASA-4 than those of 25Cu/ASA-4 (~90% and ~40% of the initial H
2 yields vs. 21 and 35 nm of Cu
0 particles,
Figure 5b and
Table 1). Moreover, the examination of the initial DME conversion and its TOS changing pattern over
xCu–ASA-4 indicated the reciprocal effect between Cu
0 for SRM and ASA for the DME hydrolysis, i.e., the presence of synergetic effects between the two catalytic functions during SRD. This will be further discussed in detail in
Section 2.3.
2.2. Effect of Si/Al Molar Ratios
To a certain extent, the acidity of ASA can be commonly regulated by changing the Si/Al ratios provided that the amorphous structure is consistently preserved [
16,
26]. However, the precursors of Al and Si, the preparation method, and the parameters are important factors in determining the textural and acidic properties of ASA, which essentially originate from the complex arrangements of Si, Al, and O atoms in ASA [
11,
16,
26,
27,
28,
29]. As indicated in
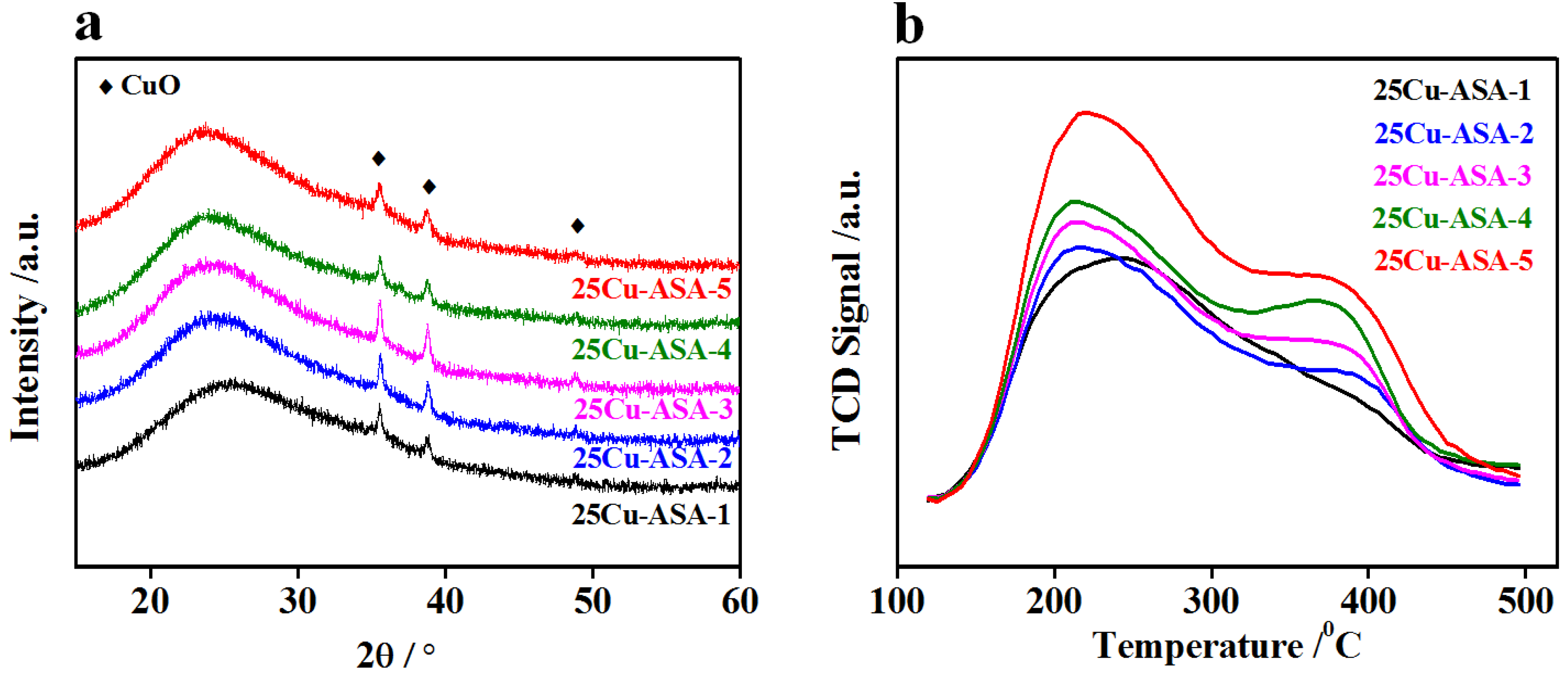
Figure 6a, almost the same XRD profiles characterizing the amorphous silica and crystalline CuO were still observed irrespective of the Si/Al ratios of the 25Cu–ASA-
y catalysts. Moreover, the calculated particle sizes based on the CuO (111) diffraction at 2
θ of 35.5° were only slightly dependent on the Si/Al ratio of ASA, i.e., 15–21 nm (
Table 1). This was well understandable if the textural properties of the catalysts were examined. As shown in
Figure 3b, a typical H4 hysteresis loop for the 25Cu–ASA-
y catalysts (
y = 1, 2, and 3) was transferred to a clear H3 hysteresis loop for 25Cu–ASA-4 and 25Cu–ASA-5 although a type IV isotherm was still observed for all of these catalysts. This indicates that the pore structures of the 25Cu–ASA-
y catalysts (
y = 1, 2 and 3) varied slightly from those of 25Cu–ASA-4 and 25Cu–ASA-5. As a result, with an increase the Si/Al ratio from 1 to 5, the BET surface area of 25Cu–ASA-
y continuously increased from 252.7 to 331.3 m
2·g
−1 but the total pore volume and average pore size varied only slightly. Since the CuO content of the 25Cu–ASA-
y catalysts was kept the same, the slightly smaller CuO size over 25Cu–ASA-4 and 25Cu–ASA-5 can reasonably be ascribed to the higher surface area, which is favorable for dispersing CuO.
To estimate the acidity of the 25Cu–ASA-
y catalysts, NH
3-TPD experiments were performed, and the results are shown in
Figure 6b. Irrespective of the Si/Al ratios of the 25Cu–ASA-
y samples, the pre-adsorbed NH
3 molecules were overwhelmingly desorbed at temperatures ranging from about 150 to 450 °C. In the case of 25Cu–ASA-1, a highly asymmetric and broad peak with a crest temperature of about 240 °C was observed. However, a shoulder at about 380 °C emerged for 25Cu–ASA-2, and its intensity continuously increased with an increase in the Si/Al molar ratio from 2 to 5. These observations indicate the varied distributions of acid sites depending on the Si/Al ratios of 25Cu–ASA-
y. Following common practice, relatively weaker and stronger acid sites were divided at the temperature of 300 °C based on the characteristics of the NH
3-TPD profiles, and the amount of acid sites was calculated. From the data shown in
Table 2, the total amount of acid sites was almost the same over 25Cu–ASA-1 and 25Cu–ASA-2, and it was slightly increased with an increase in the Si/Al ratio from 2 to 5. Where the acidity was concerned, the quantity of weaker acid sites relative to the total amount of acid sites of the 25Cu–ASA-
y samples was kept almost constant (58 ± 2%) irrespective of the Si/Al ratios, suggesting similar distributions of the Si and Al atoms over the catalysts.
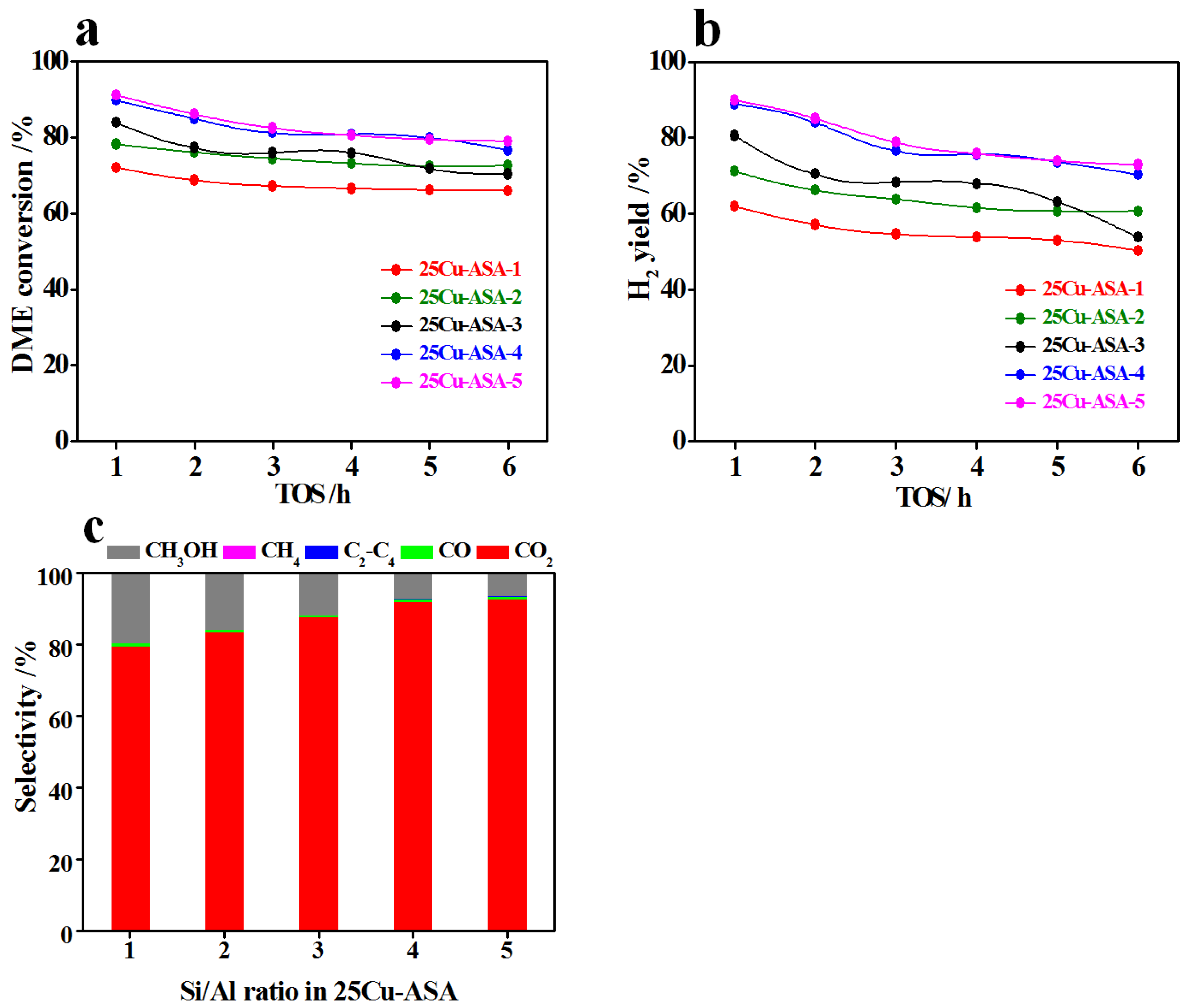
By keeping the same content of CuO for the bifunctional catalysts, the impact of acidity on the performance of 25Cu–ASA-
y for SRD was determined by adjusting the Si/Al ratios of ASA. From the catalytic results (
Figure 7), the SRD reaction for all of the catalysts occurred effectively, and the selectivity of hydrocarbons (CH
4 and C2-C4) was very low (less than 0.2%). Moreover, CO
2 was the dominant product, and the selectivity of CO was low (<1%), leading to the high H
2 yield. Specifically, as shown in
Figure 7a, the initial DME conversion decreased in the order of 25Cu–ASA-5 (91.1%) ≈ 25Cu–ASA-4 (~89.8%) > 25Cu–ASA-3 (83.9%) > 25Cu–ASA-2 (78.1%) > 25Cu–ASA-1 (72.0%). In the case of the initial H
2 yield (
Figure 7b), the change in the order was maintained, i.e., 25Cu–ASA-5 (89.9%) ≈ 25Cu–ASA-4 (~88.9%) > 25Cu–ASA-3 (80.5%) > 25Cu–ASA-2 (71.2%) > 25Cu–ASA-1 (62.0%). If the difference between the initial DME conversion and H
2 yield was compared, it was significantly decreased with an increase in the Si/Al ratio, i.e., 10% for 25Cu–ASA-1 vs. ~1% for 25Cu–ASA-4 and 25Cu–ASA-5. These results indicate the comparably higher efficiency of SRD for 25Cu–ASA-4 and 25Cu–ASA-5 in the production of hydrogen. This was directly supported by the selectivity of carbon-containing products at a TOS of 6 h (
Figure 7c), in which the selectivity of methanol as the main by-product continuously increased in the order of 25Cu–ASA-5 (6.5%) ≈ 25Cu–ASA-4 (~7.2%) < 25Cu–ASA-3 (11.8%) < 25Cu–ASA-2 (15.8%) < 25Cu–ASA-1 (19.5%). If the TOS results were examined (
Figure 7a,b, a slight and comparable decrease in both the DME conversion and H
2 yield was identified with increasing TOS from 1 to 6 h. If the very similar properties in the aspects of the CuO size, texture, and acidity (
Table 1 and
Table 2) were associated with the SRD results for 25Cu–ASA-1 and 25Cu–ASA-2, the presence of synergetic effects between Cu and acid sites could be perceived, which will be discussed in the next section together with the characterization results of the used catalysts.
2.3. Insights into the Synergetic Effects
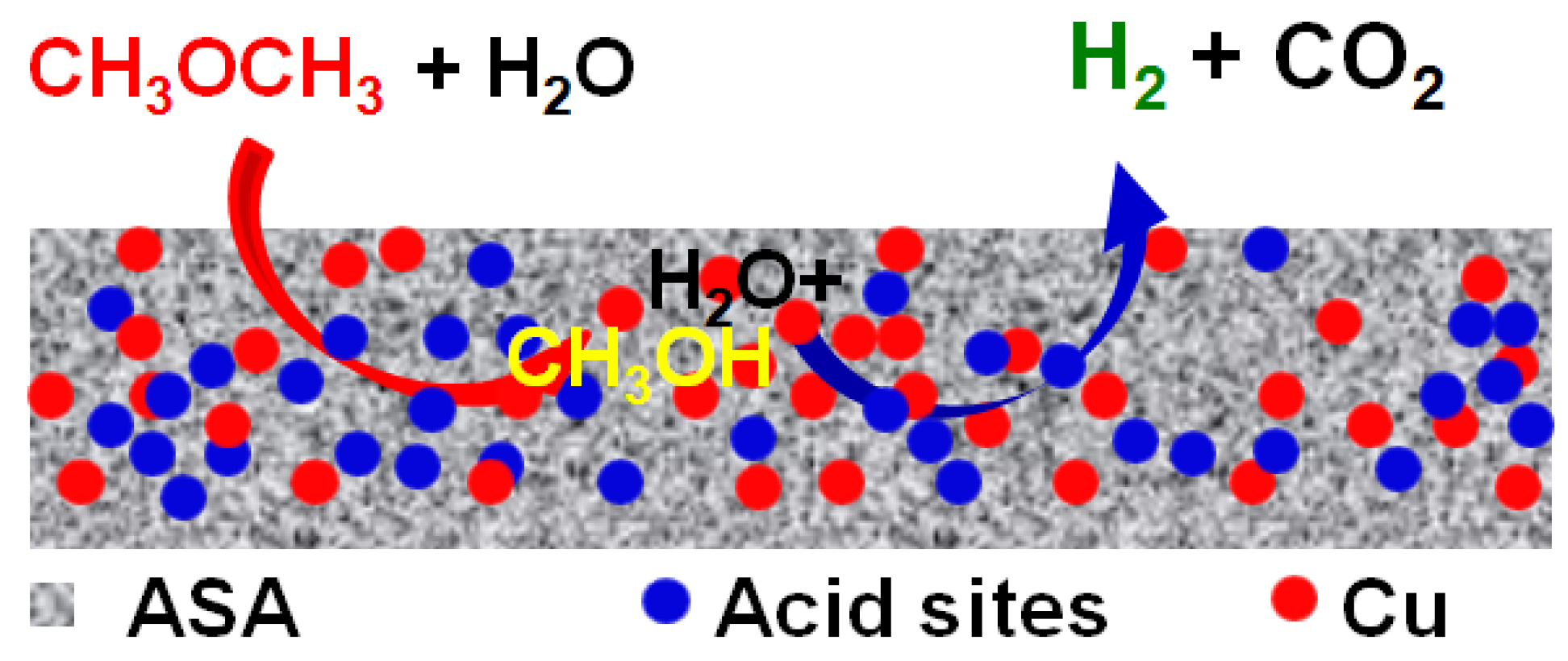
In comparison with the hybrid catalyst, the chemical and structural properties of Cu and the acid sites in the integrated bifunctional catalyst cannot be independently regulated to a relatively larger extent if both a reasonably high DME conversion and H2 yield are desired. This is exactly the case for the Cu–ASA catalyzed two-step consecutive SRD reaction. Due to the incorporation of Cu into ASA, the most obviously inevitable episode is the advent of inaccessible acid sites over ASA covered by the Cu species, the extent of which is expected to increase at a higher Cu content of the catalyst. Accordingly, the acid-catalyzed DME hydrolysis (Equation (2)) is reciprocally affected by Cu-catalyzed SRM (Equation (3)) during the SRD reaction of bifunctional Cu–ASA catalysts with various compositions. This analysis is consistent with the SRD results for the xCu–ASA-y catalysts.
The synergetic effects between Cu and ASA imposed on the SRD activity can be directly supported if the results for 25Cu/ASA-4 and 25Cu–ASA-4 are comparatively examined. Considering (1) the same synthesis procedure of ASA-4 and 25Cu–ASA-4 and (2) the same compositions and very similar textural properties of the two catalysts (
Table 1), analogous kinetics behaviors of 25Cu/ASA-4 and 25Cu–ASA-4 for the DME hydrolysis are reasonably expected. However, great variations between the initial DME conversion (90.9 vs. 48.8%) and H
2 yield (90.0 vs. 40.7%) were observed for the two catalysts (
Figure 5). If the two-step nature of the SRD reaction is taken into account, the large difference in the catalytic activity should be induced from varied rates of the second-step reaction of SRD, i.e., SRM. Recalling that Cu
0 is commonly accepted as the active site for SRM, the SRM rate of 25Cu/ASA-4 is expectedly lower than that for 25Cu–ASA-4 because of the larger Cu
0 size of 25Cu/ASA-4 compared with 25Cu–ASA-4 (35 vs. 21 nm,
Table 1). Thus, the synergetic effects between Cu and ASA play an important role in determining the SRD activity and the H
2 yield. This conclusion is further supported by the results for
xCu–ASA-4 if both the amount and diameter of Cu
0 particles are considered. Following this understanding, the results for 25Cu–ASA-
y, i.e., the gradually increased MeOH selectivity (
Figure 7c), the slightly decreased CuO size (
Table 1), and the trivially increased acidity with increasing Si/Al molar ratio, can be explained. Thus, as a result of the two-step consecutive reactions of SRD, the mutual effect between the acid sites for the DME hydrolysis and Cu
0 for SRM is crucial in determining the activity and H
2 yield of SRD for the Cu–ASA bifunctional catalyst, which is consistent with the results for the hybrid catalysts discussed in our previous work [
15].
As shown in
Figure 5 and
Figure 7, 25Cu–ASA-4 with optimal activity still showed slight decreases in both the DME conversion and H
2 yield when TOS was extended to only 6 h, indicating the lower stability of the
xCu–ASA-
y catalysts. For the Cu-based bifunctional SRD catalysts, the Cu sintering and coking are revealed as the main causes for the catalyst deactivation [
1,
15,
20,
22]. To find out the reason, the spent 25Cu–ASA-4 catalyst as a representative was characterized by the TG-DSC technique. As shown in
Figure 8a, the weight loss below 150 °C (~5.5%) was due to the removal of the physically adsorbed water, and coke deposited on the catalyst could not be unambiguously determined according to the TG and DSC patterns. However, if the XRD profiles of the reduced and spent 25Cu–ASA-4 catalyst were compared (
Figure 8b), the Cu (111) diffraction of the spent catalyst was apparently sharper, indicating the larger Cu
0 size. This was directly observed from the TEM images, i.e., the larger average Cu
0 size of 7.3 ± 3.0 nm with a wider distribution of the Cu
0 particles over the spent catalyst (
Figure 8c,d). Thus, the slight decreases in the catalytic activity of SRD over the
xCu–ASA-
y catalysts were mainly caused by Cu sintering, which is agreeable with the findings for the Cu-based hybrid catalysts [
22] or the monolithic anodic alumina-supported catalyst of Cu/γ-Al
2O
3/Al [
30]. In contrast, the coke deposition during SRD was revealed as the main reason for the deactivation of the mesoporous-structured Cu-SiO
2-Al
2O
3 catalyst [
20]. These results clearly indicate the complicated deactivation behavior of the bifunctional catalysts. If the SRD results of 10Cu–ASA-5 (
Figure 5) are analyzed, the significant decrease in the H
2 yield but slight decrease in DME conversion with increasing TOS can be reasonably ascribed as the quick deactivation of Cu in the SRM reaction, which is supported by the very high selectivity of methanol at the end of the test. Thus, the mutual effect of Cu
0 and acid sites over ASA also play a key role in determining the stability of the bifunctional catalyst, which essentially originated from the consecutive nature of the SRD reaction.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}