


Effects of Site Geometry and Local Composition on Hydrogenation of Surface Carbon to Methane on Ni, Co, and NiCo Catalysts

Abstract
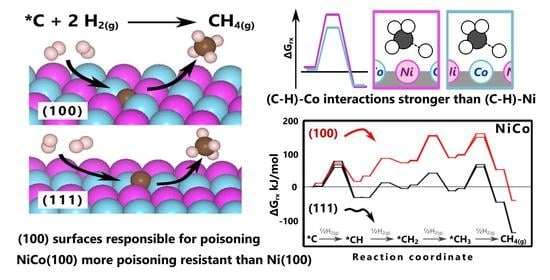
:
1. Introduction
2. Results and Discussion
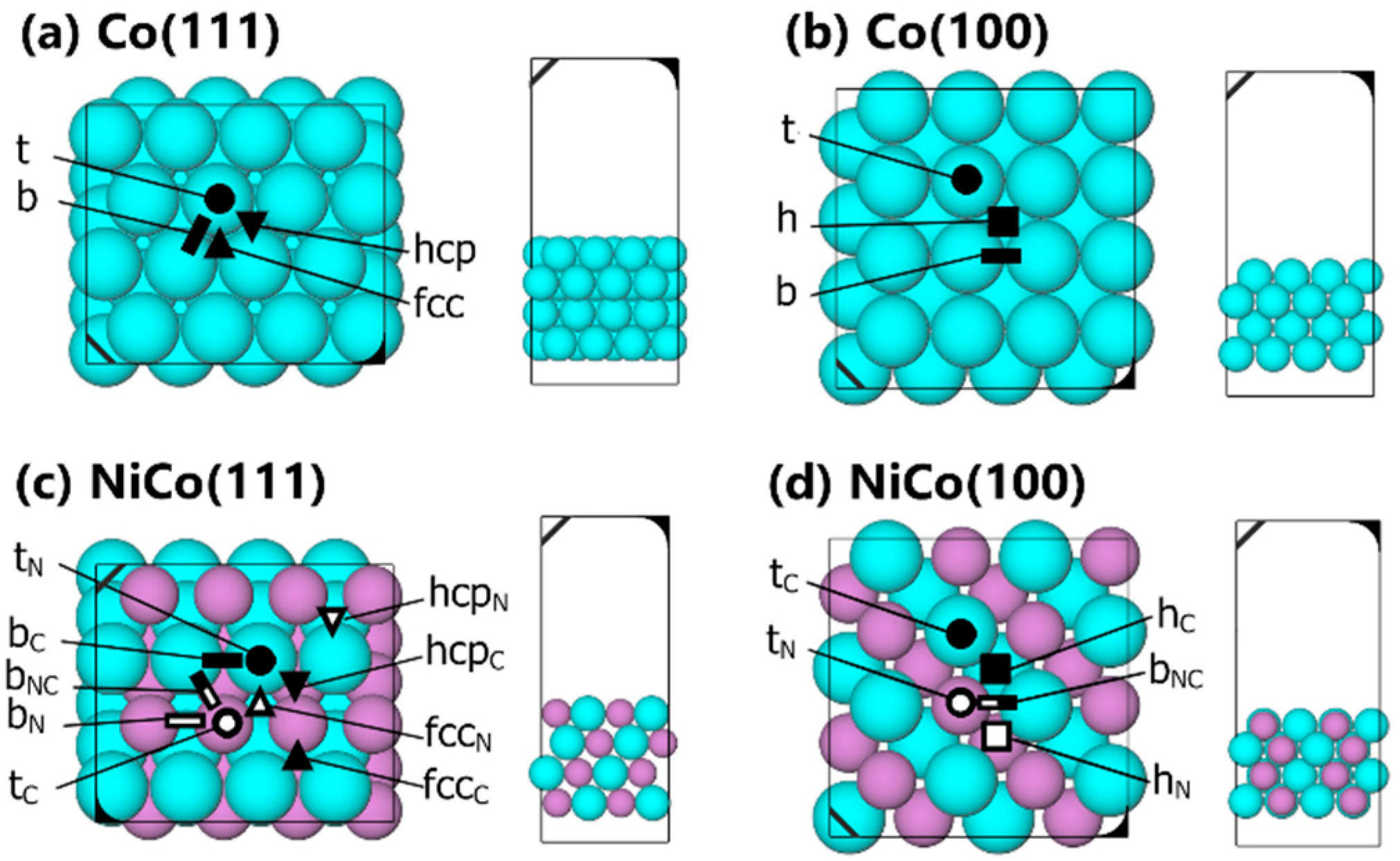
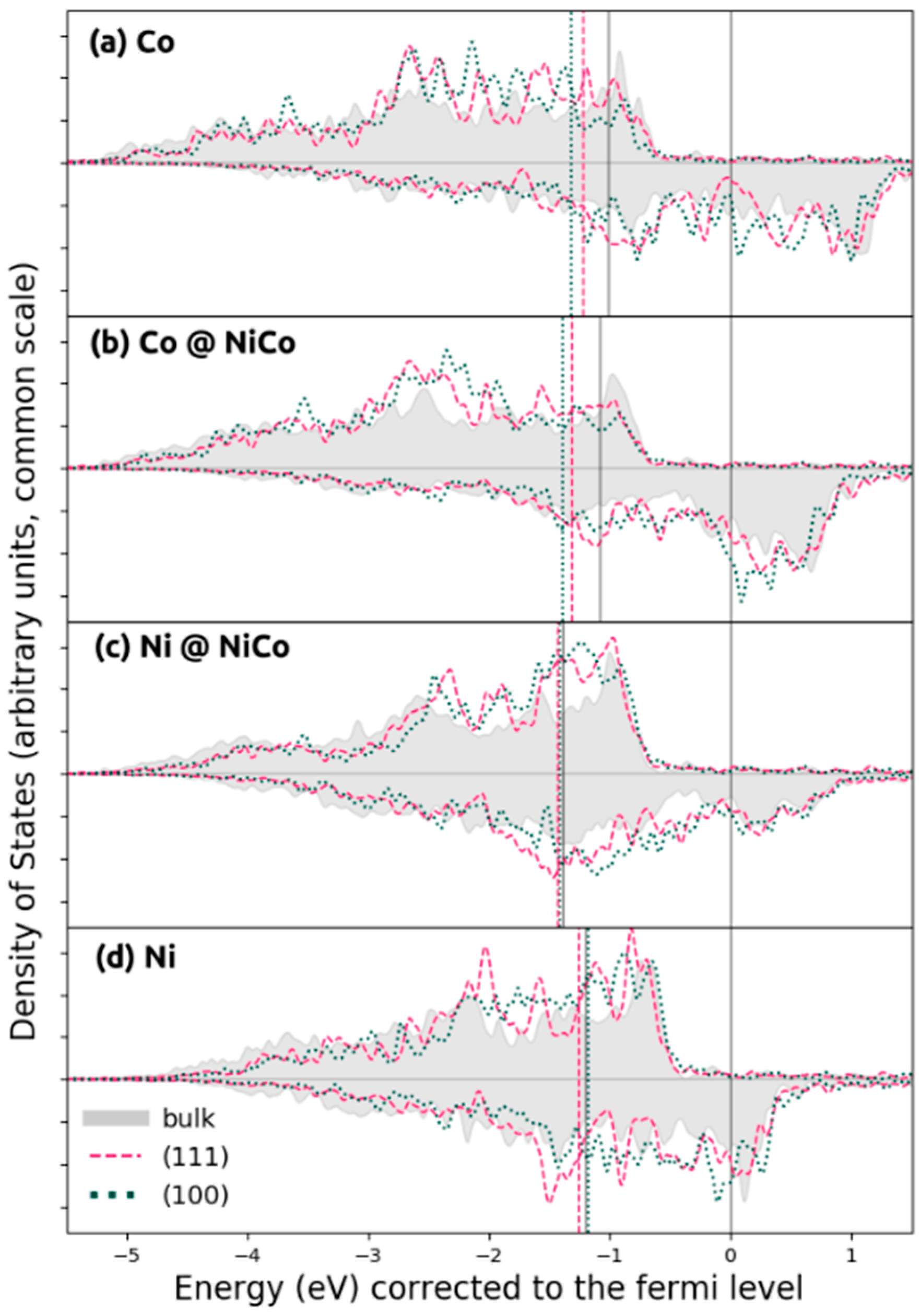
2.1. Structure and Electronic Properties of Ni, Co, and NiCo
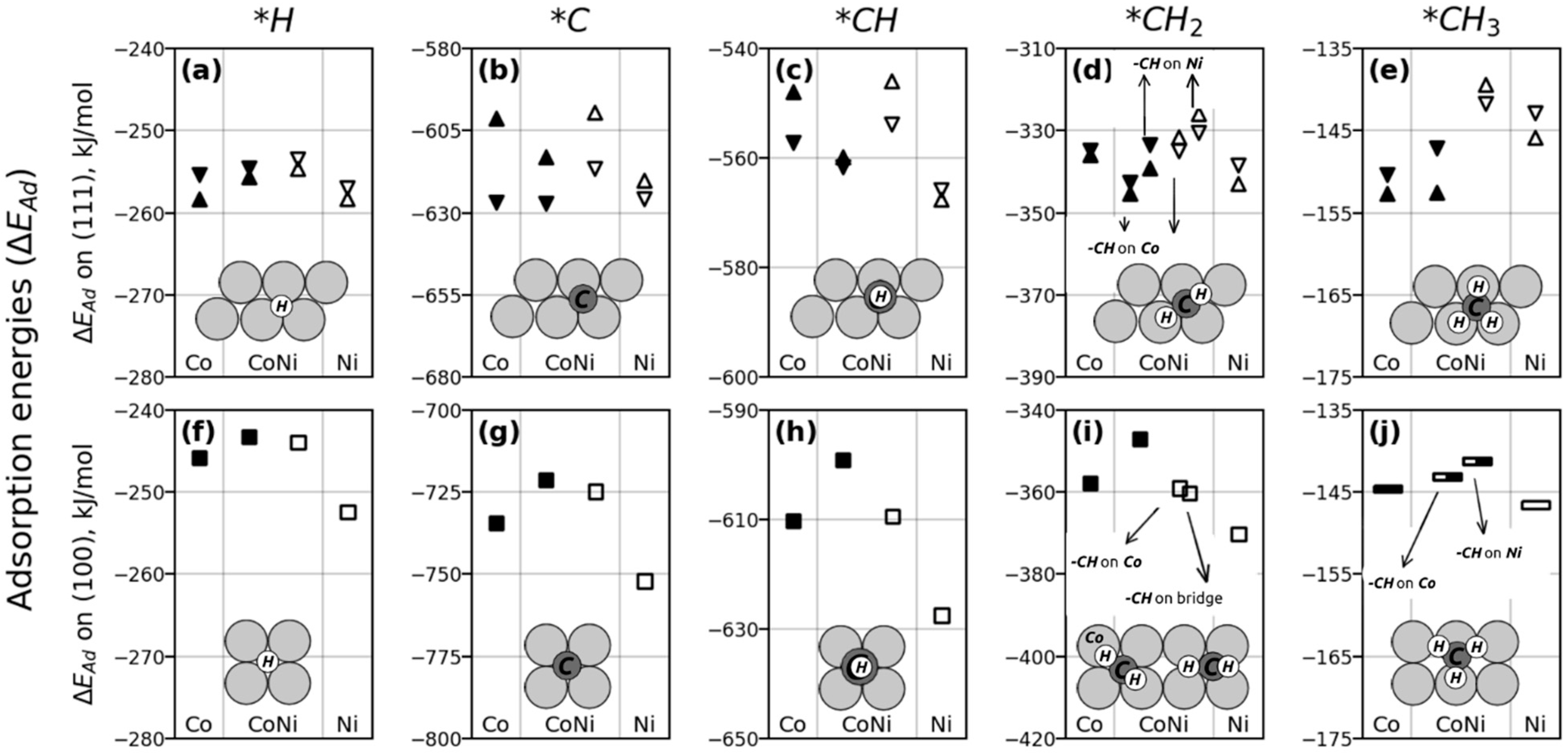
2.2. Geometries and Adsorption Energies of C, H, and CHx Species
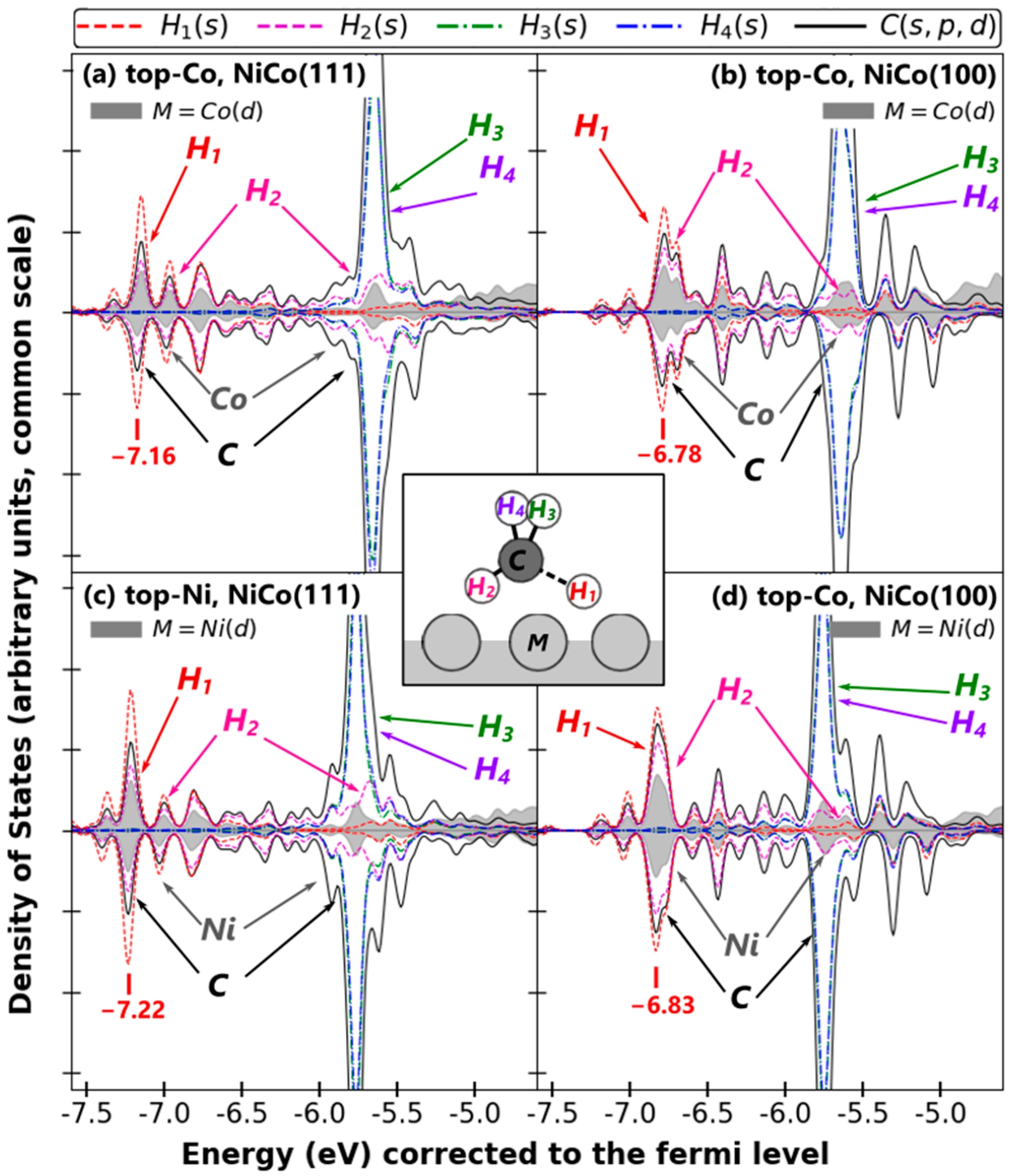
2.2.1. H and Cn Adsorptions
2.2.2. Adsorption of CHx Species
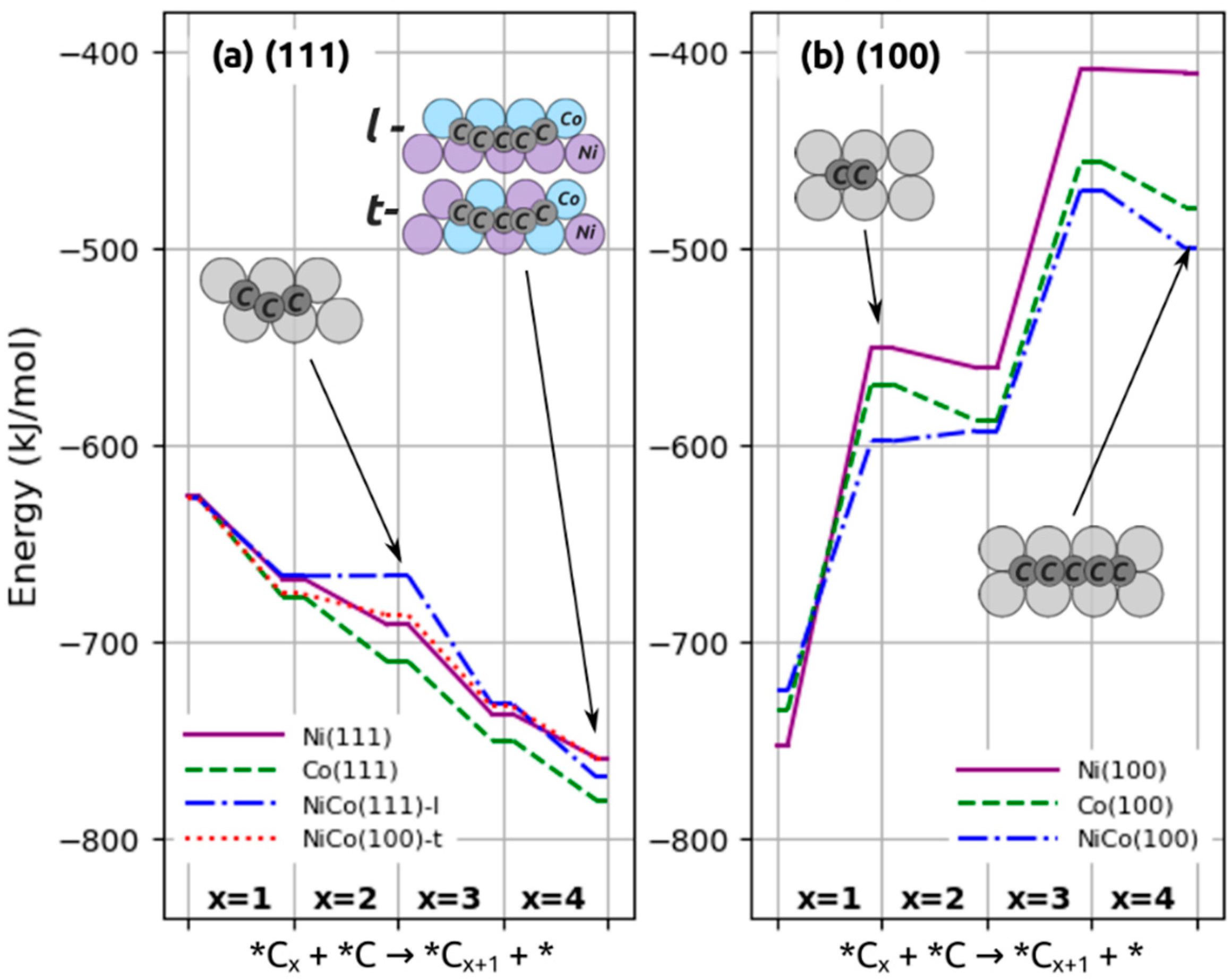
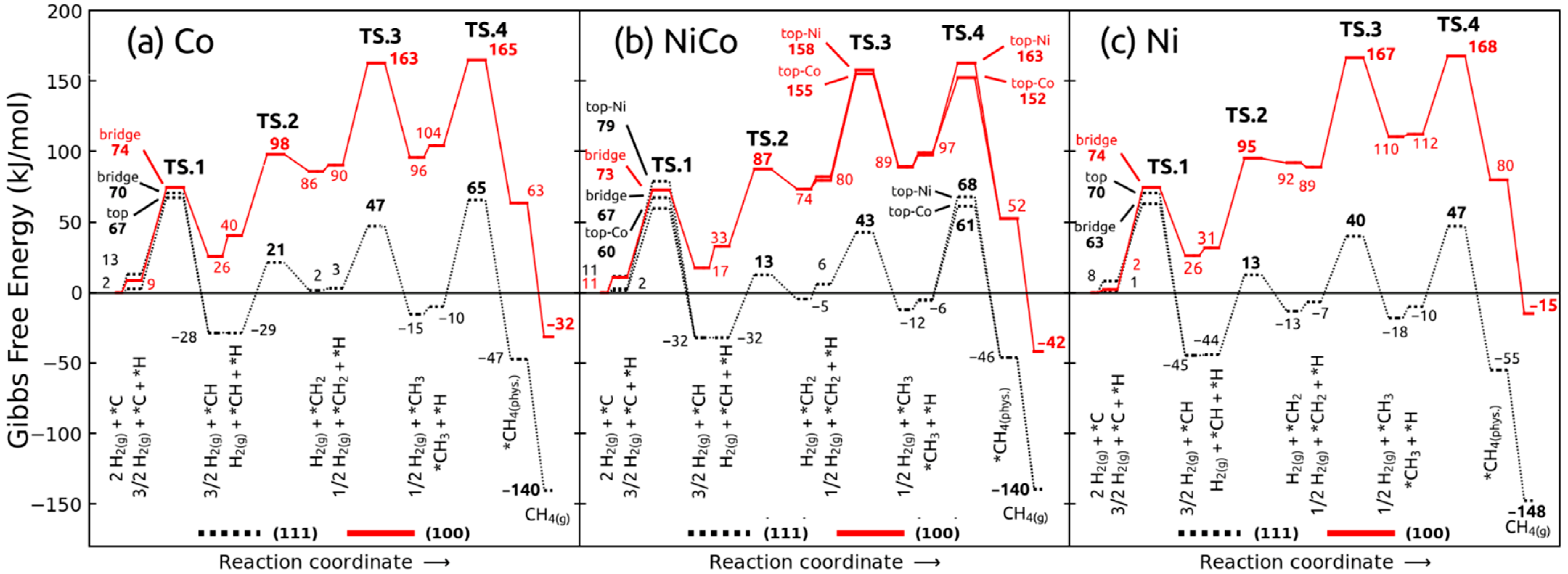
2.3. Elementary Steps and Energetics for Hydrogenation of Surface Carbon
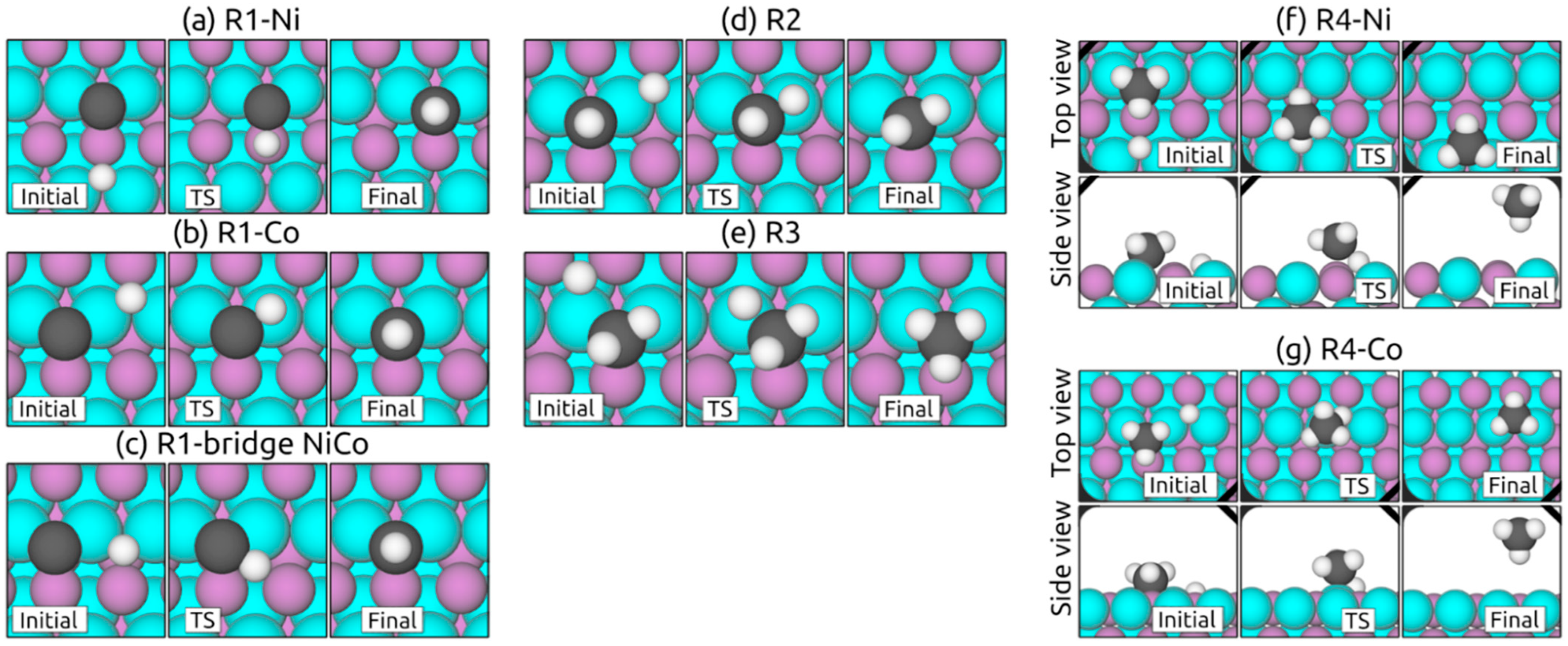
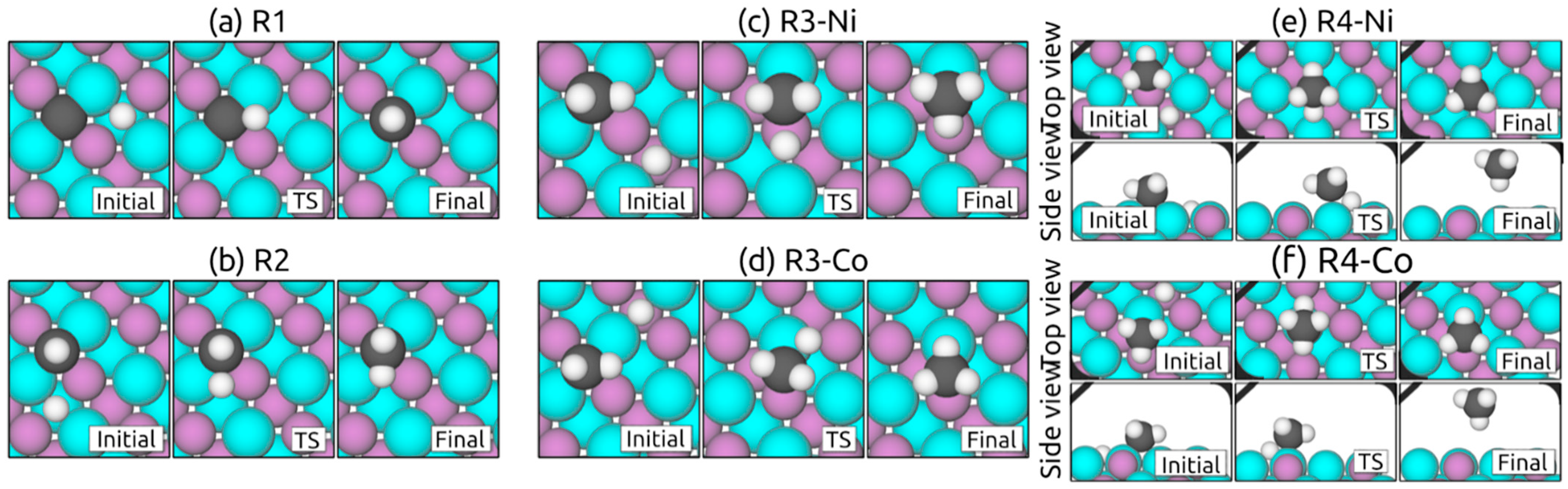
2.3.1. Hydrogenation Steps on (111) Surfaces
2.3.2. Hydrogenation Steps on (100) Surfaces
2.3.3. Effect of Density of States on Transition State Stabilization
3. Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, H.; Yu, Y.; Zhang, M. Mechanistic insight into methane dry reforming over cobalt: A density functional theory study. Phys. Chem. Chem. Phys. 2020, 22, 27320–27331. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Ran, J.; Niu, J.; Zhang, Z.; Deng, T.; He, Z.; Qin, C. Effect of active site and charge transfer on methane dehydrogenation over different Co doped Ni surfaces by density functional theory. Int. J. Hydrogen Energy 2020, 45, 31849–31862. [Google Scholar] [CrossRef]
- Ashok, A.; Kumar, A.; Bhosale, R.; Saad, M.A.S.; AlMomani, F.; Tarlochan, F. Study of ethanol dehydrogenation reaction mechanism for hydrogen production on combustion synthesized cobalt catalyst. Int. J. Hydrogen Energy 2017, 42, 23464–23473. [Google Scholar] [CrossRef]
- Takanabe, K.; Nagaoka, K.; Nariai, K.; Aika, K. Titania-supported cobalt and nickel bimetallic catalysts for carbon dioxide reforming of methane. J. Catal. 2005, 232, 268–275. [Google Scholar] [CrossRef]
- Kocić, S.; Corral Valero, M.; Schweitzer, J.-M.; Raybaud, P. Surface speciation of Co based Fischer-Tropsch catalyst under reaction conditions: Deactivation by coke or by oxidation? Appl. Catal. A Gen. 2020, 590, 117332. [Google Scholar]
- Liu, Y.; Yang, H.; Liu, Y.; Jiang, B.; Ding, J.; Woodward, R. Thermally induced fcc to hcp martensitic transformation in Co–Ni. Acta Mater. 2005, 53, 3625–3634. [Google Scholar] [CrossRef]
- Liu, H.; Wang, B.; Fan, M.; Henson, N.; Zhang, Y.; Towler, B.F.; Harris, H.G. Study on carbon deposition associated with catalytic CH4 reforming by using density functional theory. Fuel 2013, 113, 712–718. [Google Scholar] [CrossRef]
- Tu, W.; Ghoussoub, M.; Singh, C.V.; Chin, Y.-H.C. Consequences of Surface Oxophilicity of Ni, Ni-Co, and Co Clusters on Methane Activation. J. Am. Chem. Soc. 2017, 139, 6928–6945. [Google Scholar] [CrossRef]
- Li, K.; Jiao, M.; Wang, Y.; Wu, Z. CH4 dissociation on NiM(111) (M = Co, Rh, Ir) surface: A first-principles study. Surf. Sci. 2013, 617, 149–155. [Google Scholar] [CrossRef]
- Ray, K.; Sandupatla, A.S.; Deo, G. Activity and stability descriptors of Ni based alloy catalysts for dry reforming of methane: A density functional theory study. Int. J. Quantum Chem. 2020, 121, e26580. [Google Scholar] [CrossRef]
- Sprowl, L.H.; Adam, B.M.; Tucker, J.D.; Árnadóttir, L. First-principles study of the products of CO2 dissociation on nickel-based alloys: Trends in energetics with alloying element. Surf. Sci. 2018, 677, 219–231. [Google Scholar] [CrossRef]
- Turap, Y.; Wang, I.; Fu, T.; Wu, Y.; Wang, Y. Co–Ni alloy supported on CeO2 as a bimetallic catalyst for dry reforming of methane. Int. J. Hydrogen Energy 2020, 45, 6538–6548. [Google Scholar] [CrossRef]
- Ashok, A.; Kumar, A.; Ponraj, J.; Mansour, S.A.; Tarlochan, F. Effect of Ni incorporation in cobalt oxide lattice on carbon formation during ethanol decomposition reaction. Appl. Catal. B Environ. 2019, 254, 300–311. [Google Scholar] [CrossRef]
- Leba, A.; Yıldırım, R. Determining most effective structural form of nickel-cobalt catalysts for dry reforming of methane. Int. J. Hydrogen Energy 2020, 45, 4268–4283. [Google Scholar] [CrossRef]
- Guo, X.; Liu, H.; Wang, B.; Wang, Q.; Zhang, R. Insight into C + O(OH) reaction for carbon elimination on different types of CoNi(111) surfaces: A DFT study. RSC Adv. 2015, 5, 19970–19982. [Google Scholar] [CrossRef]
- Hao, X.; Wang, Q.; Li, D.; Zhang, R.; Wang, B. The adsorption and dissociation of methane on cobalt surfaces: Thermochemistry and reaction barriers. RSC Adv. 2014, 4, 43004–43011. [Google Scholar] [CrossRef]
- Li, J.; Croiset, E.; Ricardez-Sandoval, L. Methane dissociation on Ni (100), Ni (111), and Ni (553): A comparative density functional theory study. J. Mol. Catal. A Chem. 2012, 365, 103–114. [Google Scholar] [CrossRef]
- Mueller, J.E.; van Duin, A.C.T.; Goddard, W.A. Structures, Energetics, and Reaction Barriers for CHx Bound to the Nickel (111) Surface. J. Phys. Chem. C 2009, 113, 20290–20306. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhang, R.; Yan, R.; Wang, B.; Xie, K. CH4 dissociation on NiCo (111) surface: A first-principles study. Appl. Surf. Sci. 2011, 257, 8955–8964. [Google Scholar] [CrossRef]
- Chen, S.; Yang, B. Activity and stability of alloyed NiCo catalyst for the dry reforming of methane: A combined DFT and microkinetic modeling study. Catal. Today 2021, 400–401, 59–65. [Google Scholar] [CrossRef]
- Meltzman, H.; Chatain, D.; Avizemer, D.; Besmann, T.M.; Kaplan, W.D. The equilibrium crystal shape of nickel. Acta Mater. 2011, 59, 3473–3483. [Google Scholar] [CrossRef]
- Zhang, W.-B.; Chen, C.; Zhang, S.-Y. Equilibrium Crystal Shape of Ni from First Principles. J. Phys. Chem. C 2013, 117, 21274–21280. [Google Scholar] [CrossRef]
- Liu, J.-X.; Su, H.-Y.; Sun, D.-P.; Zhang, B.-Y.; Li, W.-X. Crystallographic Dependence of CO Activation on Cobalt Catalysts: HCP versus FCC. J. Am. Chem. Soc. 2013, 135, 16284–16287. [Google Scholar] [CrossRef] [PubMed]
- Andersson, M.; Abild-Pedersen, F.; Remediakis, I.; Bligaard, T.; Jones, G.; Engbæk, J.; Lytken, O.; Horch, S.; Nielsen, J.H.; Sehested, J. Structure sensitivity of the methanation reaction: H2-induced CO dissociation on nickel surfaces. J. Catal. 2008, 255, 6–19. [Google Scholar] [CrossRef]
- Bengaard, H.S.; Nørskov, J.K.; Sehested, J.; Clausen, B.S.; Nielsen, L.P.; Molenbroek, A.M.; Rostrup-Nielsen, J.R. Steam Reforming and Graphite Formation on Ni Catalysts. J. Catal. 2002, 209, 365–384. [Google Scholar] [CrossRef]
- Huang, H.; Yu, Y.; Zhang, M. Structure sensitivity of CH4 formation from successive hydrogenation of C on cobalt: Insights from density functional theory. Chem. Phys. Lett. 2019, 737, 136824. [Google Scholar] [CrossRef]
- Chen, S.; Zaffran, J.; Yang, B. Dry reforming of methane over the cobalt catalyst: Theoretical insights into the reaction kinetics and mechanism for catalyst deactivation. Appl. Catal. B Environ. 2020, 270, 118859. [Google Scholar] [CrossRef]
- Nandula, A.; Trinh, Q.T.; Saeys, M.; Alexandrova, A.N. Origin of Extraordinary Stability of Square-Planar Carbon Atoms in Surface Carbides of Cobalt and Nickel. Angew. Chem. Int. Ed. 2015, 54, 5312–5316. [Google Scholar] [CrossRef] [Green Version]
- Kitakami, O.; Sato, H.; Shimada, Y.; Sato, F.; Tanaka, M. Size effect on the crystal phase of cobalt fine particles. Phys. Rev. B 1997, 56, 13849–13854. [Google Scholar] [CrossRef]
- Pearson, W.B.; Thompson, L.T. The lattice spacings of nickel solid solutions. Can. J. Phys. 1957, 35, 349–357. [Google Scholar] [CrossRef]
- Wang, H.; Miller, J.T.; Shakouri, M.; Xi, C.; Wu, T.; Zhao, H.; Akatay, M.C. XANES and EXAFS studies on metal nanoparticle growth and bimetallic interaction of Ni-based catalysts for CO2 reforming of CH4. Catal. Today 2012, 207, 3–12. [Google Scholar] [CrossRef]
- Zhang, H.; Yao, T.; Sun, Z.; Li, Y.; Liu, Q.; Hu, F.; Pan, Z.; He, B.; Xie, Z.; Wei, S. Structural Study on Co−Ni Bimetallic Nanoparticles by X-ray Spectroscopy. J. Phys. Chem. C 2010, 114, 13596–13600. [Google Scholar] [CrossRef]
- Liu, X.; Sun, L.; Deng, W.-Q. Theoretical Investigation of CO2 Adsorption and Dissociation on Low Index Surfaces of Transition Metals. J. Phys. Chem. C 2018, 122, 8306–8314. [Google Scholar] [CrossRef]
- Peng, G.; Sibener, S.; Schatz, G.C.; Mavrikakis, M. CO2 Hydrogenation to Formic Acid on Ni(111). J. Phys. Chem. C 2012, 116, 3001–3006. [Google Scholar] [CrossRef]
- Watwe, R.; Bengaard, H.; Rostrup-Nielsen, J.; Dumesic, J.; Nørskov, J. Theoretical Studies of Stability and Reactivity of CHx Species on Ni(111). J. Catal. 2000, 189, 16–30. [Google Scholar] [CrossRef]
- Wang, S.-G.; Cao, D.-B.; Li, Y.-W.; Wang, J.; Jiao, H. CH4 dissociation on Ni surfaces: Density functional theory study. Surf. Sci. 2006, 600, 3226–3234. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Cheng, D.; Barcaro, G.; Charlier, J.-C.; Hou, M.; Fortunelli, A. Homogeneous Nucleation of Graphitic Nanostructures from Carbon Chains on Ni(111). J. Phys. Chem. C 2011, 115, 10537–10543. [Google Scholar] [CrossRef]
- Tan, K.F.; Xu, J.; Chang, J.; Borgna, A.; Saeys, M. Carbon deposition on Co catalysts during Fischer–Tropsch synthesis: A computational and experimental study. J. Catal. 2010, 274, 121–129. [Google Scholar] [CrossRef]
- Robinson, J.; Woodruff, D.P. The local adsorption geometry of CH3 and NH3 on Cu(111): A density functional theory study. Surf. Sci. 2002, 498, 203–211. [Google Scholar] [CrossRef]
- Abild-Pedersen, F.; Greeley, J.; Studt, F.; Rossmeisl, J.; Munter, T.R.; Moses, P.G.; Skúlason, E.; Bligaard, T.; Nørskov, J.K. Scaling Properties of Adsorption Energies for Hydrogen-Containing Molecules on Transition-Metal Surfaces. Phys. Rev. Lett. 2007, 99, 016105. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Mavrikakis, M. Surface and Subsurface Hydrogen: Adsorption Properties on Transition Metals and Near-Surface Alloys. J. Phys. Chem. B 2005, 109, 3460–3471. [Google Scholar] [CrossRef] [PubMed]
- Catapan, R.C.; Oliveira, A.A.M.; Chen, Y.; Vlachos, D.G. DFT Study of the Water–Gas Shift Reaction and Coke Formation on Ni(111) and Ni(211) Surfaces. J. Phys. Chem. C 2012, 116, 20281–20291. [Google Scholar] [CrossRef]
- Zhu, Y.-A.; Chen, D.; Zhou, X.-G.; Yuan, W.-K. DFT studies of dry reforming of methane on Ni catalyst. Catal. Today 2009, 148, 260–267. [Google Scholar] [CrossRef]
- Ren, J.; Guo, H.; Yang, J.; Qin, Z.; Lin, J.; Li, Z. Insights into the mechanisms of CO2 methanation on Ni(111) surfaces by density functional theory. Appl. Surf. Sci. 2015, 351, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Bian, Z.; Kawi, S. Highly carbon-resistant Ni–Co/SiO2 catalysts derived from phyllosilicates for dry reforming of methane. J. CO2 Util. 2017, 18, 345–352. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal--amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyuterev, V.G.; Vast, N. Murnaghan’s equation of state for the electronic ground state energy. Comput. Mater. Sci. 2006, 38, 350–353. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]
- Manz, T.A.; Limas, N.G. Introducing DDEC6 atomic population analysis: Part 1. Charge partitioning theory and methodology. RSC Adv. 2016, 6, 47771–47801. [Google Scholar] [CrossRef] [Green Version]
- Manz, T.A.; Sholl, D.S. Improved Atoms-in-Molecule Charge Partitioning Functional for Simultaneously Reproducing the Electrostatic Potential and Chemical States in Periodic and Nonperiodic Materials. J. Chem. Theory Comput. 2012, 8, 2844–2867. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Jónsson, H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- Liu, B.; Yuan, F.; Jin, K.; Zhang, Y.; Weber, W.J. Ab initio molecular dynamics investigations of low-energy recoil events in Ni and NiCo. J. Phys. Condens. Matter 2015, 27, 435006. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electronic Charges (e) | Spin Magnetic Moment (μB) | |||||
|---|---|---|---|---|---|---|
| Bulk | (111)surf. | (100)surf. | Bulk | (111)surf. | (100)surf. | |
| Co | 0 | −0.01 | +0.01 | 1.66 | 1.72 | 1.82 |
| Co(@ NiCo) | +0.04 | +0.04 | +0.10 | 1.72 | 1.81 | 1.89 |
| Ni(@ NiCo) | −0.04 | −0.05 | −0.09 | 0.65 | 0.64 | 0.65 |
| Ni | 0 | −0.01 | +0.01 | 0.65 | 0.67 | 0.70 |
| Bulk | (111)surf. | (100)surf. | |
|---|---|---|---|
| Co | −1.01 | −1.23 | −1.32 |
| Co@ NiCo | −1.08 | −1.32 | −1.38 |
| NiCo (average) | −1.24 | −1.38 | −1.40 |
| Ni @ NiCo | −1.39 | −1.43 | −1.41 |
| Ni | −1.21 | −1.26 | −1.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godoy, S.; Deshlahra, P.; Villagra-Soza, F.; Karelovic, A.; Jimenez, R. Effects of Site Geometry and Local Composition on Hydrogenation of Surface Carbon to Methane on Ni, Co, and NiCo Catalysts. Catalysts 2022, 12, 1380. https://doi.org/10.3390/catal12111380
Godoy S, Deshlahra P, Villagra-Soza F, Karelovic A, Jimenez R. Effects of Site Geometry and Local Composition on Hydrogenation of Surface Carbon to Methane on Ni, Co, and NiCo Catalysts. Catalysts. 2022; 12(11):1380. https://doi.org/10.3390/catal12111380
Chicago/Turabian StyleGodoy, Sebastian, Prashant Deshlahra, Francisco Villagra-Soza, Alejandro Karelovic, and Romel Jimenez. 2022. "Effects of Site Geometry and Local Composition on Hydrogenation of Surface Carbon to Methane on Ni, Co, and NiCo Catalysts" Catalysts 12, no. 11: 1380. https://doi.org/10.3390/catal12111380