Abstract
Heterogeneous electrochemical CO2 reduction has potential advantages with respect to the homogeneous counterpart due to the easier recovery of products and catalysts, the relatively small amounts of catalyst necessary for efficient electrolysis, the longer lifetime of the catalysts, and the elimination of solubility problems. Unfortunately, several disadvantages are also present, including the difficulty of designing the optimized and best-performing catalysts by the appropriate choice of the ligands as well as a larger heterogeneity in the nature of the catalytic site that introduces differences in the mechanistic pathway and in electrogenerated products. The advantages of homogeneous and heterogeneous systems can be preserved by anchoring intact organometallic molecules on the electrode surface with the aim of increasing the dispersion of active components at a molecular level and facilitating the electron transfer to the electrocatalyst. Electrode functionalization can be obtained by non-covalent or covalent interactions and by direct electropolymerization on the electrode surface. A critical overview covering the very recent literature on CO2 electroreduction by intact organometallic complexes attached to the electrode is summarized herein, and particular attention is given to their catalytic performances. We hope this mini review can provide new insights into the development of more efficient CO2 electrocatalysts for real-life applications.
1. Introduction
Global climate change generated by anthropogenic emission of CO2 represents a major challenge for our society. Among various approaches, including chemical, electrochemical, and photochemical techniques, the electrochemical conversion of carbon dioxide into added-value chemicals mediated by catalysts using renewable electricity has attracted growing interest in recent years. Particularly interesting is the production of several liquid fuels in which CO2 and hydrogen can be converted, such as methanol, light hydrocarbons, formic acid, dimethyl ether, etc. Recent decades have witnessed great advances in the field of electrochemical CO2 reduction reaction (CO2RR), but for applications on an industrial scale, many potential drawbacks should be taken into account: the excessive overpotential, the poor selectivity for the desired products, and, concerning the catalyst, limited efficiency, low stability, and high costs. Different categories of electrocatalysts have been investigated, including metal oxides, chalcogenides, nitrogen-doped carbons, and transition metal complexes [1,2,3,4,5,6,7,8,9,10,11,12].
The reduction products of CO2 are generally classified as C1, C2, …, Cn, where n indicates the number of carbon atoms in the product. Derivatives with n = 1 are by far the easiest to obtain in term of faradaic efficiency (FE), turnover number (TON), and turnover frequency (TOF) because a higher n means a higher number of multielectron transfers and multistep reactions. Thus, the CO2 reduction products can also be advantageously classified by considering only the number of electrons employed for the CO2 reduction (Equations (1)–(10)). The Cn products with n > 1 are generally very difficult to achieve, and even fewer examples are reported for carbon-based electrodes functionalized with molecular organometallic catalysts. A recent report from 2022 showed that copper phthalocyanine has the ability to interact with carbon (CuPc/C) after pyrolysis to exhibit an FE for C2H4 of 42.6 ± 0.5% at an extremely low potential of −0.4 V vs. RHE toward the electrochemical CO2RR, a process that was significantly boosted by the in situ oxygen reduction reaction (ORR) [13]. Generally, Cn products with n > 1 are more easily obtainable from metallic electrodes, as in the case of a recent report also published in 2022. This report provided a catalytic system composed of ultrathin CuO nanoplate arrays, which form Cu/Cu2O heterogeneous interfaces through self-evolution during electrocatalysis. The catalyst exhibited a high FEC2H4 of 84.5%, stable electrolysis for ~55 h in a flow cell using a neutral KCl electrolyte, and a full-cell ethylene energy efficiency of 27.6% at 200 mA cm−2 in a membrane-based electrochemical cell [14].
CO2 + 2 H+ + 2e− → CO + H2O
CO2 + 2 H+ + 2e− → HCOOH
CO2 + 4 H+ + 4e− → HCHO + H2O
CO2 + 6 H+ + 6e− → CH3OH + H2O
CO2 + 8 H+ + 8e− → CH4 + 2 H2O
2 CO2 + 8 H+ + 8e− → CH3COOH + 2 H2O
2 CO2 + 10 H+ + 10e− → CH3CHO + 3 H2O
2 CO2 + 12 H+ + 12e− → CH3CH2OH + 3 H2O
2 CO2 + 12 H+ + 12e− → C2H4 + 4 H2O
2 CO2 + 14 H+ + 14e− → C2H6 + 4 H2O
Regarding organometallic complexes, the local coordination and electronic environment of the metal center are key factors not only for the efficiency of CO2RR but also for a selective production of CO, HCOOH, CH3OH, CH4, etc. These catalysts cannot generally promote CO2 reduction beyond the two-electron process to generate more valuable products, although there are a few exceptions, e.g., cobalt phthalocyanines supported on carbon nanotubes (CNT) [15,16]. Design of the optimized and best-performing catalyst (high efficiency and tailored selectivity) can be addressed by the appropriate choice of ligands and the presence of second coordination sphere interactions and cocatalysts.
For example, the presence of local protons in tricarbonyl Mn complexes containing bpy-localized phenolic functionalities can afford not only CO but also formate via the CO2 insertion into a Mn hydride intermediate [17,18]. The hydride species for this category of Mn catalysts has been recently proven to be crucial for steering the selectivity towards formate when tertiary amines are strategically positioned on the bipyridine ligand [19]. The use of amines as additives with Mn(bpy)(CO)3Br has also been demonstrated to play a crucial role in directing the selectivity towards formate as a more desirable CO2 reduction product [20,21].
Excellent reviews have been published in recent years describing the use of transition metal complexes in homogeneous CO2RR in terms of key strategies to improve reaction rates, selectivity, and mechanistic insights [1,3,4,5,6,7,22,23].
One of the major advantages of homogeneous CO2RR is the use of single-atom transition metal complexes that have high selectivity and high performance, which can be obtained by careful optimization of the electronic and steric environment. The great benefit of the homogeneous approach is the use of in situ spectroelectrochemical techniques and intermediate isolation, which provide insights into the electroreduction mechanism such that catalyst performance can be improved with a clear materials design strategy. However, for real-life application, heterogenization of electrocatalysts represents a key step for increasing either their stability or durability and for increasing catalyst and product recovery at the end of catalytic cycles. There are additional potential advantages for heterogeneous electrocatalysts such as a more efficient electron transfer process to the attached or bonded catalyst compared with that in solution, the need for smaller amounts of catalyst for efficient electrolysis, and the possibility of using solvents such as water where the transition metal complexes would be normally completely insoluble. Solid catalysts in principle present a spectrum of active sites in which each of them possesses its own energetics, activity, and selectivity. In other words, atoms located at surface steps or kinks can be different both from one another and from other atoms situated at terrace sites or flat surfaces. This situation is encountered with all metals and alloys and with a very large number of other catalysts that are continuous solids. This situation reflects the variety of energetic conditions associated with the adsorbed species and the presence of different catalytic pathways. One strategy to preserve the advantages of homogeneous and heterogeneous systems is to anchor intact organometallic molecules on the electrode surface with the aim of increasing the molecular-level dispersion of the active components and the electrical conductivity of the electrocatalyst in the CO2RR.
The combination of the two approaches could make the catalytic conversion of the extremely stable CO2 molecule into usable fuels or chemical products by using renewable electricity and non-precious metals like manganese, cobalt, iron, nickel, copper, and zinc a very appealing process for widespread application at an industrial scale [24]. Isolated organometallic complexes anchored to electrode surfaces functioning as single-site catalysts have received considerable attention due to their known electronic structure and geometric configuration, 100% metal utilization efficiency, and outstanding activity. Here we present an overview of the most important recent advances in the field of carbon-based electrodes functionalized with intact organometallic complexes, focusing on their use as CO2 electroreduction catalysts.
2. Anchoring Strategies
Among the different techniques for immobilizing organometallic complexes onto the electrode surface, two main categories can be identified: (I) non-covalent immobilization (including insolubility immobilization, π-π interactions, and electrostatic interactions) and (II) covalent immobilization through chemical reactions and electropolymerization. The second approach has recently gained interest because of its appealing aspects, which might overcome some of the limitations arising from the non-covalent immobilization strategy. If the catalyst is anchored on the electrode surface via weak Van der Waals interactions, leaching of the catalyst in the electrocatalytic solution may occur, especially during long-term measurements. Additionally, non-covalent anchoring may not provide efficient electron transfer between the molecular catalyst and the substrate. A third approach is the periodic assembly method, which includes use of a metal–organic framework or porous organic framework. Herein, we are going to focus our review on the main results for CO2RR mediated by heterogenized molecular electrocatalysts on carbon-based electrodes because the different immobilization techniques have already been widely discussed in other reviews [2,25,26,27]. Graphitic electrodes, graphene derivatives, carbon cloth, carbon black, carbon papers, and carbon nanotubes (CNT) are among the most widely employed carbon-based materials in the field of CO2RR due to their high stability and conductive surface area. The interaction of these supports with the molecular catalysts affects many variables such as electron transfer, transport of species, and durability. For instance, MWCNTs (multi-walled carbon nanotubes) display high stability, high surface area, and high electrical conductivity. Therefore, many electrocatalysts have been immobilized on CNTs.
Our research group published a review in 2016 about recent advances in catalytic CO2 reduction mediated by organometallic complexes anchored on electrodes [28]; in this review we are going to report the findings in the field from then on.
2.1. Non-Covalent Bonding
The non-covalent immobilization technique might enable a facile screening of heterogenized electrocatalysts since normally no further functionalization of the ligand is required. However, it must be considered that metal complexes should display relevant attractive Van Der Waals interactions between (typically) the ligand of the metal complex and the carbon-based substrate. This includes, for example, easily polarizable systems with a high degree of aromaticity and significant delocalized electrons. Porphyrin and phthalocyanine ligands possess a highly conjugated electronic structure that can be directly immobilized via π-π interactions. For ligands that are non-aromatic in nature and exhibit a low content of delocalized structure, such as bipyridines, pincer derivatives, and macrocycles (e.g., cyclam), a conjugated fragment such as pyrene needs to be embedded in the molecular structure to effectively immobilize the catalyst onto the substrate via π-π interactions involving the surface and the pyrene anchoring units (Figure 1).
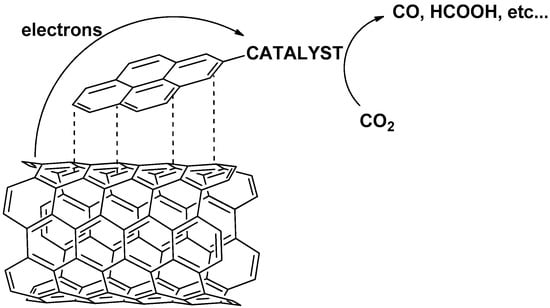
Figure 1.
Representative scheme of non-covalent bonding via pyrene units on CNTs.
The most representative reports about bipyridine–pyrene immobilization include the anchoring of the well-known Lehn catalyst [29,30], fac-Re(bpy)(CO)3Cl, on a graphite support [31] (Figure 2a); to the best of our knowledge, this was the first report of the heterogenization on carbon-based electrodes of this highly selective CO2-to-CO electrocatalyst. They observed that the catalytic activity was increased compared with the same dissolved catalyst in a homogeneous solution. Surendranath et al. [32] published a powerful strategy in which fac-Re(5,6-diamino-1,10-phenanthroline)(CO)3Cl was condensed onto o-quinone edge defects (Figure 2b) on graphitic carbon surfaces to generate graphite-conjugated rhenium (GCC-Re) catalysts that are highly active for CO2 reduction to CO in acetonitrile electrolyte. GCC-Re species on glassy carbon surfaces display catalytic currents greater than 50 mA cm−2 with 96 ± 3% faradaic efficiency for CO production (Table 1).
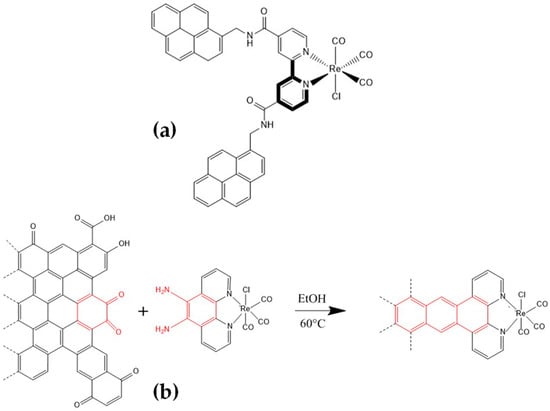
Figure 2.
Chemical sketches of the modified fac-Re(bpy)(CO)3Cl for further non-covalent immobilization. (a,b) were adapted with permission from ref. [31,32], respectively.

Table 1.
Figures of merits of selected molecular electrocatalysts anchored on carbon-based electrodes via non-covalent immobilization.
In 2022, Costentin, Chardon-Noblat, and co-workers [33] investigated the effect of conjugated and non-conjugated substituents, which are structurally similar to anchoring compounds previously used to immobilize a rhenium carbonyl bipyridyl molecular electrocatalyst. A drastic effect on the catalytic activity of the complex toward CO2 electroreduction was observed for carboxylic ester groups, which mimic the anchoring of the catalyst via carboxylate binding to the surface. Through spectroelectrochemical investigations, they showed that the reducing equivalents mainly accumulated on the electron-withdrawing ester on the bipyridine ligand, which prevented the formation of the rhenium(0) center and its interaction with CO2. Conversely, they were able to show that alkyl-phosphonic ester substituents not conjugated with the bpy ligand, which mimic the anchoring of the catalyst via phosphonate binding to the surface, preserved the catalytic activity of the complex.
In 2011, Deronzier et al. [34] replaced Re with the cheaper and more abundant fac-Mn(bpy)(CO)3Br. The electrochemical mechanism of Mn(bpy)(CO)3Br has been extensively studied [34,35,36,37,38,39,40]. The proposed mechanism in a homogeneous solution is shown in Scheme 1. From the depicted mechanism, it is clear that Mn requires addition of external proton sources to activate catalysis. Once CO2 binds the metal center of the five coordinate species [Mn(bpy)(CO)3]−, a sequence of protonation and reduction steps occurs. Two possible catalytic pathways focused on the different sequences of the reduction/protonation steps for evolution of the abovementioned metal carboxylic adduct have been proposed. The protonation-first pathway starts with heterolytic C–OH bond cleavage of the [Mn(bpy)(COOH)] species facilitated by a proton donor leading to the formation of H2O and [Mn(bpy)(CO)4]+, which is further reduced to generate [Mn(bpy)(CO)4]●. In contrast, the reduction-first pathway proceeds with reduction of [Mn(bpy)(COOH)] followed by heterolytic C–OH bond cleavage, which yields H2O and the common [Mn(bpy)(CO)4]● intermediate. Stepwise or concerted one-electron reduction and CO evolution steps complete the catalytic cycle, regenerating the active catalyst. Notably, Scheme 1 also indicates the possible formation of the Mn hydride species, which is considered to be the main intermediate for formate release through the formation of the formato complex (MnO(CO)H). New mechanistic insights have been provided by Daasbjerg and co-workers [41]. They found that the dimer complex, Mn2(bpy)2(CO)6, is not, as is often assumed, formed by dimerization of the singly reduced manganese complex, [Mn(bpy)(CO)3]●, but that it is instead produced from its reduction to [Mn(bpy)(CO)3]−. The formation of the dimer by this parent–child reaction can be suppressed by employing bulky ligands or immobilize Mn(bpy)(CO)3Br on surfaces.
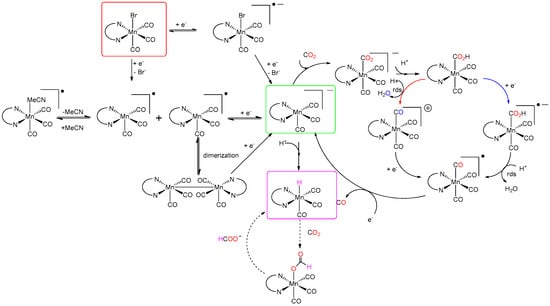
Scheme 1.
A common set of pathways that may occur after one- and two-electron reduction of [Mn(bpy)(CO)3Br] (also valid for [Re(bpy)(CO)3Cl]) in CH3CN and in the presence of CO2 and protons, resulting in the catalytic reduction of CO2 to CO and formate by two different pathways: reduction-first (blue arrows) and protonation-first (red arrows). (rds = rate-determining step). Adapted from ref. [39].
In 2014, Cowan and co-workers [42] casted Mn(bpy)(CO)3Br in a Nafion membrane and subsequently added MWCNTs, whose presence led to a ∼10-fold current increase in CO2 electroreduction to CO in neutral aqueous electrolyte (Figure 3a and Table 1). The same research group further investigated films composed of Mn-bpy-type catalysts in Nafion/MWCNT on glassy carbon electrodes together with immobilization on TiO2 [43,44]. Similarly, in 2017, Reisner et al. [45] reported the assembly of a [MnBr(2,2′bipyridine)(CO)3] complex anchored to a carbon nanotube electrode (namely CNT/Mn) via a pyrene unit (Figure 3b and Table 1). Immobilization of this molecular catalyst allows the use of water as solvent for the electrolysis. A deep mechanistic study by means of in situ IR spectroscopy coupled with electrochemistry revealed how the product selectivity can be tuned by altering the catalyst loading on the nanotube surface. At high-catalyst loadings, the main product is CO, whereas at low loadings, the dominant product is formate. The reason for this behaviour is the involvement of the different catalytic species mentioned earlier. At higher surface loadings, the predominant species formed is the Mn0 dimer, which is mainly responsible for the CO2-to-CO pathway. At lower surface loadings, the generation of a Mn hydride intermediate is suggested to greatly enhance formate production.
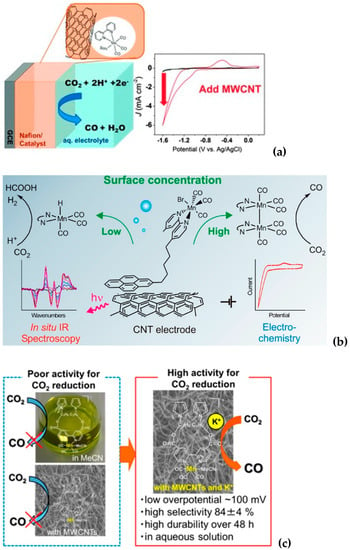
Figure 3.
Non-covalent immobilization of Mn-bpy-type catalysts on carbon nanotubes. (a) Reproduced with permission from ref. [42], (b) reproduced with permission from ref. [45], (c) reproduced with permission from ref. [46].
This seminal work paved the way for other reports on the immobilization of Mn bipyridine-type complexes. Sato and co-workers [46] reported that very low energy CO2 electroreduction can be achieved combining the use of Mn-bpy-type catalyst immobilized on MWCNT with K+ cations in solution (Figure 3c and Table 1). In this paper, the authors mostly investigated the synergistic effect of the MWCNT and K+ cations on lowering the overpotential for CO2 reduction in aqueous solution. A new Mn complex electrocatalyst, [Mn{4,4′-di(1H-pyrrolyl-3-propyl carbonate)2,2′-bipyridine}(CO)3MeCN]+(PF6)− ([Mn-MeCN]), was developed and loaded onto conductive MWCNTs ([Mn-MeCN]/MWCNT). Experiments verified that bare [Mn-MeCN] yielded minimal amounts of CO2 reduction products in acetonitrile with added water (5%) even in the presence of K+ cations. Therefore, the effect of the Lewis acid is negligible. On the other hand, the electrode tested without MWCNTs did not catalyze CO2 reduction even in the presence of K+ cations. These observations indicated that the presence of both MWCNTs and K+ cations is important for a very high CO2 reduction rate at a low overpotential. Their findings indicated that CO2 reduction can proceed through a one-electron-reduced species (OER) of the Mn catalyst. Generally, OER species of metal complex catalysts with electron-withdrawing groups do not readily react with CO2 molecules due to their very low LUMO potentials. They confirmed that [Mn-MeCN] cannot act as a CO2 reduction catalyst without the MWCNT support and K+ cations. Their work further highlights the importance of the nature of the carbonaceous support and of the catalyst’s microenvironment.
In a similar fashion, a pyrene pincer ligand was used to immobilize an Iridium complex onto a gas diffusion electrode via carbon nanotubes [47]. The most important class of homogeneous electrocatalysts for formate production is the one of Ir(III) PCP-type pincer complexes, mainly developed by Meyer’s group [47,48,49,50,51]. The generally accepted mechanism for this category of catalysts proceeds through a two-electron, one-proton process converting the precursor into the dihydride species, which rapidly binds CO2 to form a η1-formate adduct via an insertion into the Ir-H bond. Formate is finally released, which is followed by regeneration of the starting species (Figure 4a) [50]. Specifically, an iridium pincer dihydride catalyst was immobilized on carbon nanotube-coated gas diffusion electrodes (GDEs) (Figure 4b). These GDEs are efficient, selective, durable, gas-permeable electrodes used for electrocatalytic reduction of CO2 to formate. High turnover numbers (ca. 54,000) and turnover frequencies (ca. 15 s−1) were obtained in aqueous solutions saturated in CO2 with added bicarbonates (Table 1).
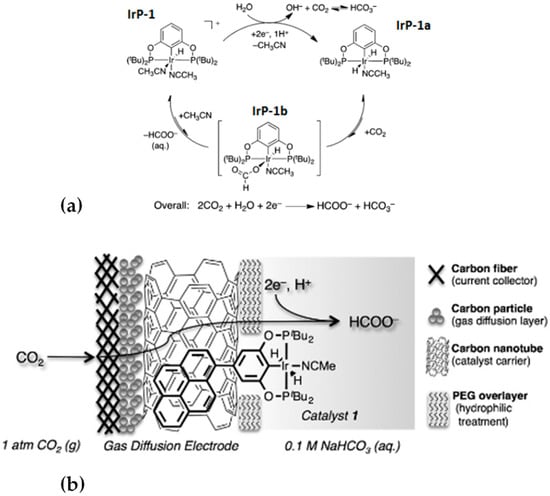
Figure 4.
(a) Proposed mechanism for CO2 water-assisted reduction mediated by IrP-1. Reproduced with permission from ref. [42], (b) carbon nanotube-coated gas diffusion electrode with surface-bound Ir pincer dihydride catalyst for electrochemical reduction of CO2 to formate. Reproduced with permission from ref. [47].
One of the most popular macrocyclic ligands that has been used for decades in coordination chemistry is 1,4,8,11-tetraazatetradecane, universally known as cyclam. Ni (II) cyclam is a remarkable example for which the CO2-to-CO conversion occurs very efficiently and selectively (high TON and high FE) in water (pH 4 to 5) at a mercury electrode. Replacing the mercury pool with an inert glassy carbon electrode enabled a high catalytic activity for CO production to be reached in a 1:4 water-acetonitrile solution, although it occurred at a slower rate [52,53,54,55]. Kubiak and collaborators were the first to attempt covalent heterogenization of Ni-cyclam, although they had little success [56]. More recently, an original pyrene–cyclam derivative and the corresponding Ni–cyclam complex have been synthesized (Figure 5a). There was no precedent for its non-covalent immobilization on a carbon-based nanostructured electrode [57]. The as-prepared electrode is efficient, selective, and robust for electrocatalytic reduction of CO2 to CO, (Table 1 for its figures of merits). Very high turnover numbers (ca. 61,460) and turnover frequencies (ca. 4.27 s−1) were enabled by the use of the novel electrode material in organic solvent–water mixtures saturated with CO2. Similarly, Cowan and co-workers recently reported a [Ni(CycPy)]2+ = Ni(1-(4-(pyren-1-yl)butyl)-1,4,8,11-tetraazacyclotetradecane)-modified GDE that was tested for the first time in an aqueous electrolyte (0.5 M KHCO3), where it was shown to be active towards CO production despite the fact that the catalytic activity decreased over time [58]. The non-covalent interaction between the pyrene group and the carbon surface does not prevent desorption of the catalyst in aqueous conditions, in which the complex can easily dissolve. In a GDE structure, the wetting of the catalyst layer is limited by the additional polytetrafluoroethylene (PTFE) added to the catalyst ink used when preparing the electrode, but desorption still occurs. Fontecave and collaborators published a very recent paper in which they showed the ability of a heterogenized C-substituted Ni-cyclam catalyst to electro-reduce CO2 to CO in water [59]. The interaction between the cyclam ligand and the electrode surface was demonstrated to significantly affect the efficiency of Ni-cyclam catalysts. N-functionalization seemed to result in increased structural hindrance when attached to the electrode, as it prevented the ligand from maintaining a flat structure that favors selectivity for CO. Thus, this was the first time the Fontecave group placed the pyrene substituent on a carbon atom of the cyclam ligand (Figure 5b). After its immobilization on MWCNTs and deposition on a Gas Diffusion Layer (GDL), the modified electrode is very active, stable, and highly selective for CO2 electroreduction to CO, retaining excellent selectivity for CO (FE up to 87%) in aqueous medium (Table 1).
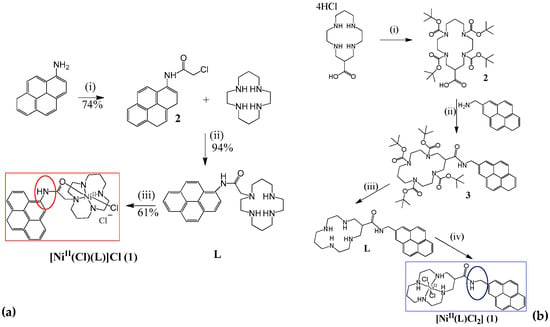
Figure 5.
(a) Synthetical steps for a cyclam derivative N-functionalized with a pyrene moiety. Adapted from ref. [57], (b) Synthetical steps for a cyclam derivative C-functionalized with pyrene. Adapted from ref. [59].
As mentioned earlier, metal phthalocyanines can be easily adsorbed on surfaces because of their scarce solubility in organic solvents and water. Commonly employed techniques for functionalizing these molecules are dip-coating and drop-casting. Dip-coating is a simple and low-cost method that involves the deposition of a wet liquid film by simply immersing the electrode in a solution containing the metal catalysts (i.e., solutions of cobalt phthalocyanine in dimethylformamide). After drying the substrate, a homogeneous liquid film is formed on the substrate’s surface. This technique allows acquisition of very reproducible results in comparison with the drop-casting method. The latter is very commonly employed for producing chemically modified electrodes in which the modifying layer is composed of particles such as nanotubes or nanoparticles. The major drawback of drop-casted surfaces is their scarce reproducibility [60,61]. A very recent and detailed review about immobilization strategies for porphyrin-based molecular catalysts for the electroreduction of CO2 was published in 2022 by Burdyny et al. [62]. In this review, the authors listed the majority of the variously substituted Fe tetraphenyl porphyrin that have been shown to be the most active homogeneous catalysts for CO2-to-CO conversion in aprotic solvents (DMF, MeCN) reported in the literature [2,63,64,65,66,67,68,69,70,71]. Few examples will be cited herein; for a more extensive screening for this category of catalysts, the reader is redirected to ref. [62]. One of the earliest works on heterogenization of porphyrins reported a pyrene-appended iron triphenyl porphyrin bearing six pendant OH groups on the phenyl rings in all ortho and ortho′ positions that was immobilized on carbon nanotubes via non-covalent interactions and that was further deposited on glassy carbon. This modified electrode was found to be active for electroreduction of CO2 to CO in water (pH 7.3) with 480 mV overpotential [72]. A comparative study of amino functionalized iron-tetraphenylporphyrins (amino-Fe-TPPs) immobilized onto carbonaceous materials in both H-cells and flow cells was conducted to selectively reduce CO2 to CO. In a flow cell set up operating in alkaline media, the resulting hybrid catalyst exhibited 87% faradaic efficiency with extraordinary current density (j) of 119 mA/cm2 and turnover frequency of 14 s−1 at −1.0 V vs. RHE. By combining the heterogenization approach with a flow cell design it was possible to obtain a significant catalytic activity and an effective strategy for future approaches toward CO2RRs. Figure 6 depicts an operational scheme for common immobilization of metal tetraphenyl porphyrins on CNTs.
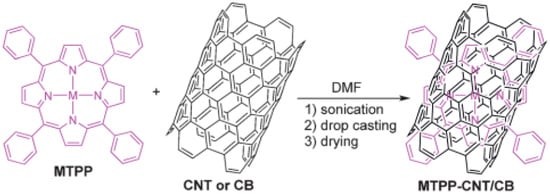
Figure 6.
Immobilization of the catalyst–carbon composite on GC. (M = Co or Fe, TPP = tetraphenylporphyrin, CB = Carbon Black). Reproduced with permission from ref. [73].
Among the most recent and original results, cobalt phthalocyanines modified with -NH2 groups were dispersed on CNTs and were found to catalyze the electrochemical conversion of carbon dioxide to methanol (Figure 7a) [15,16]. In the same year, a cobalt phthalocyanine bearing a trimethyl ammonium group appended to the phthalocyanine macrocycle adsorbed on MWCNTs was found to be able to catalyze the reduction of CO2 to CO in water with 95% selectivity, good stability, and a maximum partial current density of 165 mA cm−2 (at −0.92 V vs. RHE) [16]. Conversely, a Co porphyrin catalyst was previously adhered on CNT supporting material, which greatly enhanced catalytic activities and enabled catalysis in water, which was otherwise impossible. However, the main reduction product still remained CO [73,74].
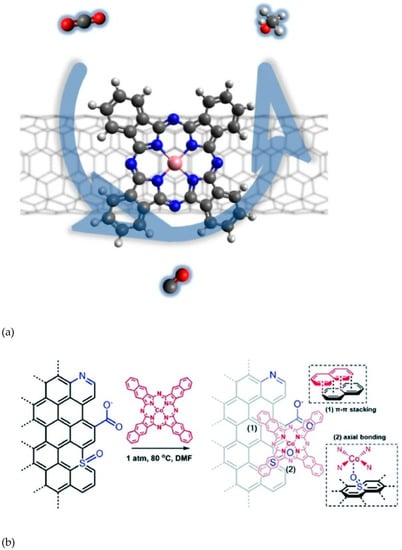
Figure 7.
(a) Co phtalocyanines as CO2 to MeOH electrocatalysts. Reproduced with permission from ref. [15], (b) NapCo for CO2-to-CO electroreduction on doped graphene. Reproduced with permission from ref. [75].
In a relatively recent paper, planar Co(II)-2,3-naphthalocyanine (NapCo) complexes were immobilized onto doped graphene, which resulted in a heterogenous catalytic system for the CO2-to-CO reductive pathway (Figure 7b) [75]. A systematic study revealed the importance of the dopants employed; compared to carboxyl dopants, the sulfoxide dopants further improved the electron communication between NapCo and graphene, which increased the turnover frequency of Co sites by ~3 times for CO production with a faradaic efficiency up to 97%.
Another strategy for effective anchoring of intact organometallic molecules on carbonaceous supports through π-π interactions is the use of the sono-deposition method [76]. Sono-immobilized CoPc on carbon cloth exhibited a high selectivity for CO production in aqueous media over a wide range of potentials from −0.5 to −0.9 V vs. RHE. Interestingly, the CO faradaic efficiency is over 80% in the studied potentials range, and the conversion efficiency to CO production reached 96% at −0.9 V. The CoPc/CC catalyst retained its selectivity with no sign of degradation for 10 h of electrolysis, making it one of the most robust catalysts reported so far.
Focusing on a metal other than cobalt, Kraatz and co-workers. [77] reported a modified 1,3-ditert-butylimidazolin-2-ylidenamino nickel porphyrin for anchoring onto CNTs. With respect to the homogeneous counterpart, they were able to show that there was a dramatic enhancement of product selectivity and catalytic activity at a much more positive potential of −0.5 V versus RHE.
2.2. Covalent Bonding
The formation of covalent bonds between the catalyst and the electrode is beneficial in terms of the stability and durability of the catalyst. It is a more robust alternative to non-covalent immobilization, which can lead to some catalyst leaching after several hours of operation, especially when using solvents in which the molecular catalyst itself can be dissolved. This strategy can also provide a more efficient electron transfer between the electrode and the molecular catalyst. Covalent functionalization can proceed through chemical reactions or through electrochemical reactions, i.e., electropolymerization. The number of reports for covalent immobilization is significantly lower than the non-covalent counterpart. This might be due to a more challenging synthetic procedure that should be followed in order to precisely tune the ligand structure and the electrode surface.
2.2.1. Formation of Chemical Bonds
Many classical organic reactions, including C-C couplings, C-N couplings, and click chemistry can be used for covalent grafting. The major results of the covalently anchored electrocatalysts that will be considered in the remainder of the review are listed in Table 2.
Table 2.
Figures of merits of selected molecular electrocatalysts anchored on carbon-based electrodes via covalent immobilization.
A recent paper reported high activity for the reduction of CO2 to CO of a cobalt(II) phtalocyanine (CoPc)-COOH/(CNT)-NH2 hybrid catalyst that was rationally designed by clicking CoPc-COOH molecules onto the surface of CNT-NH2 through an amidation reaction [79]. The superior activity of this novel hybrid catalyst (Figure 8a) was ascribed to a better charge transfer induced by the presence of -COOH and NH2 moieties on the catalyst and on the CNT, respectively. A similar chemistry was employed in 2012 in the case of an alkyne-substituted cobalt porphyrin that reacted with an azide-functionalized diamond surface through a copper(I)-catalyzed alkyne–azide cycloaddition reaction to fabricate a monolayer heterogeneous molecular catalyst [80].
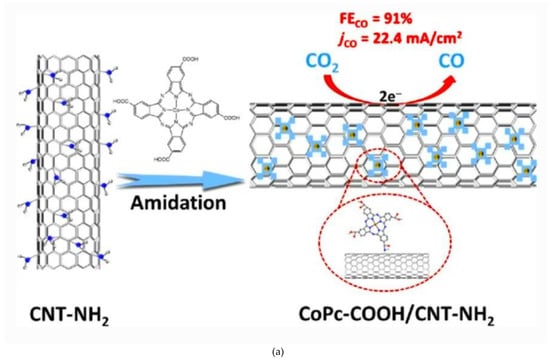
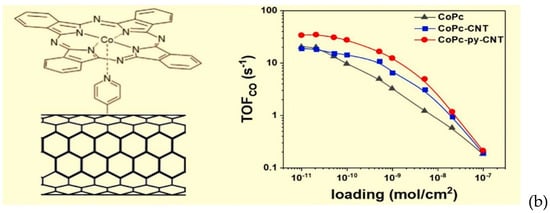
Figure 8.
(a) Scheme of the click reaction for the formation of (CoPc)-COOH/carbon nanotube (CNT)-NH2 hybrid electrode. Reproduced with permission from ref. [79], (b) Pyridine functionalized CNT/CoPc. Reproduced with permission from ref. [81].
Molecular catalysts such as porphyrins and phtalocyanines have the tendency to aggregate at high loadings. Therefore, to improve catalyst dispersion, the strategy of strengthening the interaction through covalent bonds between supports and molecular catalyst is advantageous. This was realized by Zhu et al. [81] in 2019, who prepared carbon nanotubes functionalized with pyridine (Figure 8b), which could then coordinate the metal centers. This novel hybrid catalyst exhibited a high activity (TOFCO: 34.5 s−1 at −0.63 V vs. RHE) and selectivity (FECO > 98%) for electrochemical CO2 reduction. In the same year, McCrory et al. provided evidence that the axial coordination from the pyridyl moieties in poly-4-vinylpyridine to the cobalt–phthalocyanine complex changes the rate-determining step in the CO2 reduction mechanism, thus accounting for the increased activity in the catalyst–polymer composite [82].
In 2021, an active and stable bifunctional electrocatalyst was elaborately designed to achieve the integration of CO2 reduction and anodic non-classical reaction to efficiently yield products of high value at both electrodes. The system was built by anchoring novel multi-azido-group-bearing nickel phthalocyanine onto carbon nanotubes at the single-molecule level via strong interactions. The obtained heterojunction-type electrocatalyst exhibited ultrahigh activity for completely selective CO2-to-CO conversion with a 100% faradaic efficiency in a wide potential window, large partial current density (>200 mA cm−2) and turnover frequency, and remarkable stability [83] (Figure 9a).
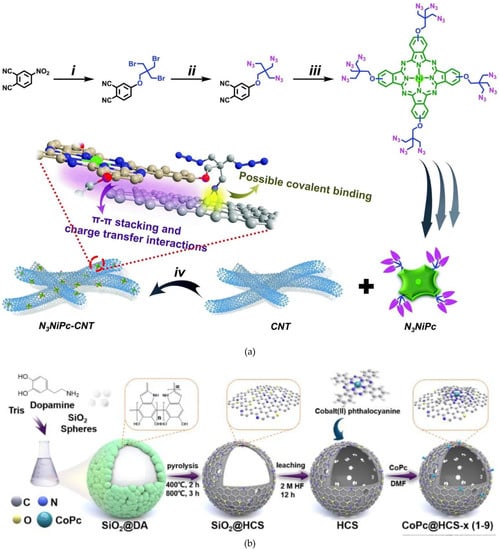
Figure 9.
(a) Schematic illustration of the overall synthetic procedure for N3NiPc and N3NiPc–CNT: (i) DMF, K2CO3, r.t., 72 h; (ii) DMF, NaN3, 80 °C; (iii) nickel acetate after dehydration, DBU, 135 °C, 48 h, Ar gas; and (iv) THF, 80 °C, 24 h. Reproduced with permission from ref. [83], (b) Illustration of the preparation procedure of CoPc@HCS-x (x = 1–9) heterogeneous molecular catalysts. Reproduced with permission from ref. [84].
Another original work involving Co phtalocyanines was recently published by Lv et al. [84]. In this study, the authors synthesized ultra-thin (~5 nm) nitrogen-doped hollow carbon spheres with high specific surface area to anchor CoPc via the coordination affinity between pyrrolic N species and the central atom of CoPc (Figure 9b). By adjusting the loading of CoPc, they systematically explored the influence of the existence formation of CoPc in the hybrid catalyst on CO2RR. The content of Co in the optimal existence formation of CoPc was c.a. 0.49 wt%, i.e., CoPc@HCS-6. This hybrid catalyst exhibited a good turnover number for CO, a good turnover frequency, an energy efficiency of 57.6%, and a CO-production rate of 0.232 mmol h−1 cm−2. Additionally, its FECO remained above 90% at the potential ranging from −0.67 to −0.87 V vs. RHE, which is dramatically higher than pure CoPc at the same potentials.
In 2016, Maurin and Robert reported the grafting of an Fe porphyrin through a covalent bond between the peripheral carboxylic acid group and the surface amine group of modified carbon nanotubes, for which they obtained a high CO selectivity and turnover at 500 mV overpotential (Figure 10a) [85].
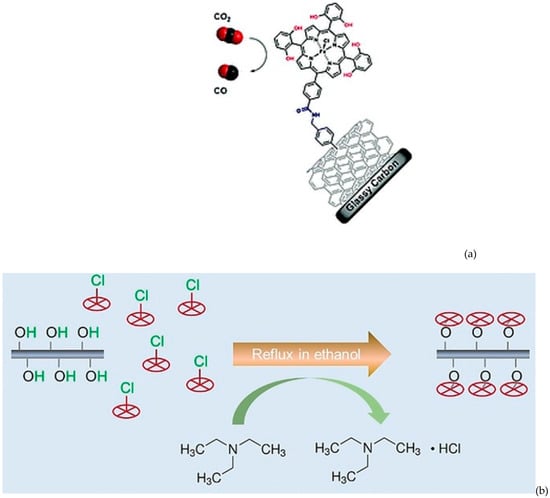
Figure 10.
(a) Scheme of the grafting of Fe porphyrin onto CNT surface through amidation. Reproduced with permission from ref. [85]. (b) Preparation of chemically grafted cobalt porphyrins on CNT-OH. Reproduced with permission from ref. [86].
As mentioned earlier, direct grafting of the catalyst through covalent bonding can be achieved through the reaction between surface functional groups of the support and peripheral functionalities. One of the major disadvantages of this approach might be the high distance between the metal and the electrode as a consequence of poor electron-conducting bonds, which could result in delayed electron transfer. Therefore, in 2019, a new strategy to prepare highly dispersed molecular electrocatalysts was developed (Figure 10b) [86]. In this case, metal centers (cobalt from cobalt porphyrins) were directly grafted onto carbon nanotubes through covalent bonds, leading to much improved catalytic performance compared with those prepared via traditional physical mixing methods. To synthesize the grafted catalysts, protoporphyrin IX cobalt chloride (CoPPCl) was refluxed with hydroxyl-functionalized carbon nanotubes (CNT-OH) in ethanol with the addition of triethylamine. A covalent bond is formed between Co and a surface O atom. As a result, the turnover frequency for CO formation was improved by a factor of three compared with traditional physically mixed catalysts with the same cobalt content. This led to an outstanding overall current density of 25.1 mA/cm2, a faradaic efficiency of 98.3% at 490 mV overpotential, and excellent long-term stability.
Another example of covalent immobilization was reported by Jiang and co-workers (Figure 11a) [87]. They presented their concept of “molecular wire” to covalently anchor Co porphyrins on carbon cloth. The strategy employed was the in situ reduction of diazonium salt. Formation of CO in a neutral aqueous electrolyte at –1.05 V vs. NHE (η = 500 mV) occurred with a TOF of 8.3 s−1. Enhanced catalytic activity was achieved, with the total CO production being about 2.5 times higher than that of the non-covalent counterpart after a 4 h of electrocatalysis. However, the performance was still far from meeting industrial requirements because of the relatively modest intrinsic activity of CoTPP. Additionally, the use of diazonium salts for the modification of powder-like CNTs requires a large excess of expensive porphyrin substrate, and a more atom-efficient method would be beneficial. For all these reasons, the same research group recently published a paper about covalent grafting of cobalt aminoporphyrin-based electrocatalysts onto carbon nanotubes for excellent activity in CO2 reduction [88].
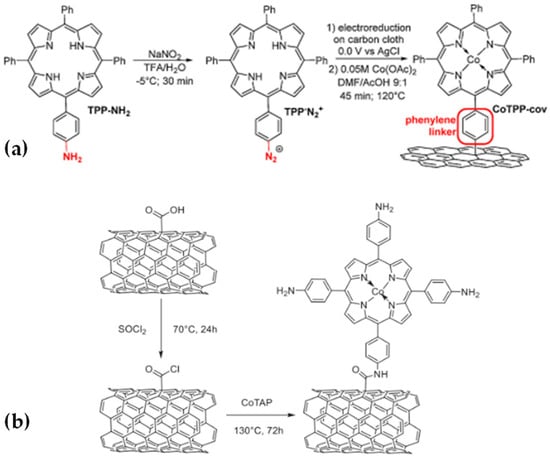
Figure 11.
(a) Preparation of covalently immobilized Co tetraphenylporphyrin (CoTPP-cov). Reproduced with permission from ref. [87]. (b) Covalent ligation of CoTAP on CNTs (CoTAP-cov). Reproduced with permission from ref. [88].
They combined the benefits of both approaches by covalently immobilizing a porphyrin-based catalyst bearing donating substituents onto the surface of MWCNTs. Co tetrakis-(4-aminophenyl) porphyrin (CoTAP) was chosen as a catalyst, and its covalent grafting was achieved via nucleophilic substitution of Cl− within the in situ-generated -COCl-modified CNTs (Figure 11b). The covalently grafted complex CoTAP-cov exhibited a turnover frequency for CO formation (TOFco) of 6.0 s−1, and the faradaic efficiency to CO (FEco) was ~100% at the overpotential of 550 mV, making it one of the best catalysts to date. In contrast, the non-covalently immobilized counterpart CoTAP-noncov showed a more moderate TOFco of 2.3 s−1 and a lower FEco of 85%.
The addition of the donating -NH2 group into the structure of porphyrin greatly enhanced the electron density on the cobalt center thus boosting its intrinsic activity to TOFco of 36.6 s−1. They concluded that the amide bond could act as a molecular wire to enhance the electron transfer from CNTs to the cobalt center.
Clearly CO2 electroreduction mediated by metalloporphyrins and metallophtalocyanines anchored on carbon-based electrodes, both via the covalent and non-covalent approach, represents the majority of reports present in the literature. Additionally, most of these hybrid electrocatalysts are selective for reduction of CO2 to CO as the major product.
Our research group has significant expertise in covalently anchoring Re and Mn bipyridines on carbon-based electrodes. Lately, we have mostly focused on Mn because it is a better replacement than Re, as it is cheaper and more abundant.
In 2017, we reported the catalytic activities of fac-Mn(apbpy)(CO)3Br and fac-Re(apbpy)(CO)3Cl (where apbpy = 4(4-aminophenyl)-2,2′-bipyridine), Figure 12a, anchored on glassy carbon electrodes [89]. The functionalization was performed via two different immobilization strategies: (a) direct electrochemical oxidation of the amino group with the formation of C-N bonds and (b) electrochemical reduction of the corresponding diazonium salts with the formation of C-C bonds.
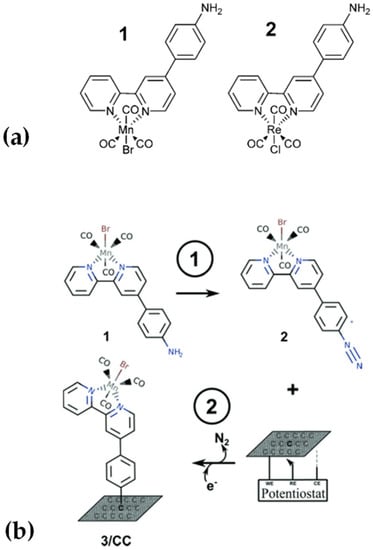
Figure 12.
(a) Chemical sketch of the complexes under study. (b) Preparation of the fac-Mn(apbpy)(CO)3Br/CC (3/CC) electrode via diazonium salt reduction; (1) in situ preparation of the diazonium salt; and (2) direct electrochemical reduction on carbon cloth.
For both metals, the functionalized electrodes obtained by reducing the diazonium salts displayed better durability than the ones obtained by oxidizing the amino moiety, a fact that was mostly evident for Mn. Both electrodes were tested for CO2 electroreduction in acetonitrile to better compare their performances with their homogeneous counterparts.
In this work, we synthesized and isolated the corresponding diazonium salts of the complexes for their subsequent electroreduction, and we simplified this procedure in 2019 by chemically bonding the Mn complex to carbon cloth (CC) [90]. This was done via direct in situ generation of the corresponding diazonium salt (employing amyl nitrite) and immediate electrochemical reduction (Figure 12b).
Carbon cloth was chosen as support because of its low electrical resistance, relatively low cost, and large surface area. This hybrid electrode was tested as a CO2 electrocatalyst in aqueous conditions, and the performance was very promising. It displayed high durability (after 10 h the electrocatalyst was still very active), it was capable of producing 4.5 Nl h−1 of CO per electrode square meter at −1.35 V (vs. RHE), and it had a faradaic efficiency (of CO) of 60% with the rest being H2. Thus, it appears to be an ideal catalyst for syngas production. The cumulative TON for CO production reached more than 33,000 after 10 h (Table 2).
The extremely low metal loading of the electrocatalyst (0.45 mgMn cm−2 geometric surface area), corresponding to a monolayer of the organometallic complex and a mass activity of 870 NlCO h−1 gMn−1, demonstrated a very high metal atom specific efficiency.
The same Mn electrocatalyst has been anchored (also through diazonium salt reduction) on a GDL. Ref. [91] This decorated GDL electrode was able to convert CO2 into CO and HCOOH with faradaic efficiencies of 76% and 10%, respectively, with a CO-productivity close to 70 Nl min−1gMn−1 and with CO2RR turnover numbers reaching up to 1.6·105. This result largely outperformed a state-of-the-art gold nanoparticle electrocatalyst (Au/C 10 wt%) operating in the same cell conditions.
Unlike in half-cell systems, a complete gas-fed electrochemical cell is potentially scalable by increasing the electrode surface. The electrocatalyst operating at the liquid–gas interface showed a TON value which was almost four times greater than the value reported in a half-cell (CO2-saturated electrolyte) study, suggesting that the enhanced mass transport due to CO2 gas feeding plays an important role in the overall electrocatalytic performance, as was already reported for metal nanoparticles. Furthermore, no formate was detected after 10 h of operation at the same cathodic potential in the previous half-cell study. Conversely, a FEHCOOH of 10% was observed in the gas-fed system. Such a selectivity difference was also observed by Reisner et al. [45] (see Section 2.1) at different electrocatalyst loadings for [MnBr(2,2′-bipyridine)(CO)3] in aqueous conditions. However, when comparing gas-fed full-cell experiments with half-cell experiments, the difference cannot be ascribed to a different loading of the electrocatalyst, as the preparation of the electrocatalyst is identical. Hence the difference in selectivity can be hypothesized to be related to a lower interfacial pH due to the presence of pure CO2 in the GDL that buffers the pH by consuming OH− formed both by CO2RR and HER to produce HCO3−. It is reported that more acidic conditions in homogeneous Mn-bpy solutions increase the production of the hydride and hence formate.
Intrigued by this difference in selectivity, we tested the same Mn system as a CO2 electrocatalyst in methanol and in the presence of amines [21]. Amines can play many different roles in the field of CO2RR [92], including acting as proton sources. With our recent paper, we demonstrated that this role can be played by methanol and that the adduct formed between CO2 and the amine can act as an effector or inhibitor toward the catalyst, thereby enhancing or reducing the production of formate. Pentamethyldiethylentriamine (PMDETA), identified as the best effector in the series of studied amines, converted CO2 in wet methanolic solution into bisammonium bicarbonate. Computational studies revealed that this adduct was responsible for a barrierless transformation of CO2 to formate by the reduced form of the Mn catalyst covalently bonded to the electrode surface. As a consequence, selectivity can be switched on demand from CO to formate anion, and in the case of (PMDETA), an impressive TONHCOO− of 2.8 × 104 can be reached. This new valuable knowledge of an integrated capture and utilization system paves the way toward a more efficient transformation of CO2 into liquid fuel. Formate is a more appealing reduction product than CO, and this hybrid Mn electrocatalyst was found to be one of the rare examples among the covalently bonded molecular complexes listed in this review that is able to reduce CO2 to formate.
2.2.2. Electropolymerization
An alternative approach for preparing functionalized electrodes containing highly dispersed catalysts for CO2 electroreduction is the electropolymerization of polymerizable functional groups bound to the metal [93,94,95].
This approach may represent an alternative pathway to support the desired intact organometallic catalyst on an electrode surface. Activity and lifetime of the solid-supported catalysts can be greatly enhanced with respect to the corresponding homogeneous counterparts.
The electropolymerization process is based on deposition of a polymer onto the surface of a solid electrode material that involves the formation of cationic radical by the oxidation of the monomer on the electrode. In general, electropolymerization is a cost-effective and easy-to-use method for the preparation of electrosynthetic conducting polymer films. Electrodes functionalized by electropolymerization show unique morphological, electronic, and electrochemical properties. Electropolymerization is initiated by the oxidation of a monomer in an electrochemical cell, followed by the growth of the polymer film on the surface of the working electrode, which may be a carbonaceous, metallic, or conducting glass material. Interesting catalytic performances in electrocatalysis, photocatalysis, and organocatalysis have been obtained by the use of atomically isolated metal catalysts that maximize atom utilization efficiency and tuneable coordination microenvironment [93,94].
The polymeric films obtained are electroactive, stable, and reasonably uniform. Furthermore, they can contain, by their nature, a redox center in each repeating unit of the polymer. Continued growth of the polymer film can be facilitated by the redox conductivity since the outer boundary of the film can act as an electron transfer mediator at the film–solution interface. For example, in the case of [(vbpy)Re(CO)3Cl] (where vbpy is 4-Me-4′-vinylbpy) and its nitrile derivative, the formation of vinyl-linked polymers on the surface is obtained by reduction of the complexes to form films of the catalyst at the electrode at potentials that are sufficiently negative to reduce the ligand before diffusional loss from the surface [96,97].
Electropolimerization of Re bpy complexes bearing bisphenylethynyl [98,99] or thiophene [100] moieties produce electron-conducting films. In the case of thiophene [100], the chemically modified electrodes show significant catalytic activities for CO2 reduction and higher relative stabilities when compared with the homogeneous solution counterparts.
Wu and co-workers [101] were able to obtain porphyrinic triazine frameworks through the polymerization of the monomers 5,10,15,20-tetrakis(4-cyanophenyl)-porphyrin (TPPCN) or [5,10,15,20-tetrakis(4-cyanophenyl)-porphyrinato]-Ni (Ni-TPPCN) with different molar ratios in the presence of ZnCl2 (see Figure 13).
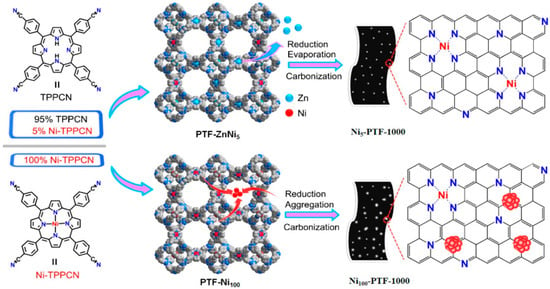
Figure 13.
Schematic views of the fabrication of Ni5-PTF-1000 catalyst with atomically isolated nickel sites from PTF-ZnNi5 via spatial sites separation strategy (Top) and the Ni100-PTF-1000 with Ni NPs from PTF-Ni100 (Bottom). Reproduced with permission from Ref. [101].
Isolated nickel species anchored in porous nitrogen-doped carbons showed high efficiency as electrocatalysts for CO2RR (91% CO faradaic efficiency between −0.6 and −1.0 V and high TOF of 20,180 h−1 at −1.0 V). Interestingly, the distance of the neighbouring Ni-N4 units in the porphyrinic CTF precursors can be controlled by varying the molar ratio of the porphyrin monomers Ni-TPPCN and TPPCN.
Metallopolymers obtained by electropolymerization exhibit a well-defined coordination environment, which can be applied in the development of new smart materials [102,103,104].
This is the case, for example, for the electropolymerization of a Pd(salen) complex onto a Pt disc electrode that produces an extended three-dimensional network formed by axial coordination of the d orbitals of the central metal with the π orbital of the phenolate moiety of the adjacent complex [105].
Electrocatalytic reduction of CO2 in an aqueous solution by a platinum electrode coated with poly-Pd(salen) showed that the cathodic current increased for the modified electrode, requiring 0.15 V less overpotential than the corresponding unmodified platinum electrode. The hexafluorophosphate counterion used in the electropolymerization process of the metal–salen complex introduces molecular spaces into the polymer of the appropriate size to stabilize carbon dioxide, which improves the efficiency of the coupling reaction. Teixeira and co-workers were able to demonstrate that the CO2RR process on the metallopolymer occurred in three steps: (a) reductive adsorption of carbon dioxide by the central metal, (b) radial reduction of carbon dioxide to adsorbed CO, and (c) reduction of adsorbed CO to adsorbed -CHO.
3. Conclusions
Within this mini review, the most recent literature regarding functionalization of carbon-based electrodes with organometallic complexes has been summarized. Significant technological advances have been reached in the field of CO2 electroreduction, although highly active, durable, and selective electrocatalysts should be developed to potentially pave the way for industrial applications. Carefully evaluating the organometallic candidates, the category of metallo-porphyrins and phthalocyanines has achieved unprecedent results as evidenced by the incredibly high number of reports present in the literature. These electrocatalysts are mostly selective for CO formation, while a major interest is devoted to the generation of liquid fuels (formate, methanol, etc.). Therefore, porphyrins and phtalocyanines might be integrated in a cascade approach considering their well-known activity for the CO2-to-CO pathway. As can be observed in Table 1 and Table 2, very few examples are reported for direct CO2 electroreduction to formate, especially with high selectivity. Our research group has just recently demonstrated that Mn bipyridine-type catalysts can display high stability and selectivity for formate production when grafted onto carbon cloth via covalent bonding.
One of the major drawbacks of anchoring electrocatalysts on surfaces, especially via covalent bonding, is the necessity of elaborated modification of the organic ligands. On the other hand, among one of the major advantages of the heterogenized catalysts over their homogeneous counterparts is the possibility to work in aqueous conditions, which is normally not applicable for the catalysts themselves.
The non-covalent bonding of molecules is simply achieved by immersion of the solid electrode in a solution of the molecular organometallic complex (i.e., dip coating) or by drop casting the catalyst solution or suspension on the electrode surface. This works well whenever the molecular catalyst is not soluble in the solvent, the electrode surface is not wetted by the solvent, and the molecular catalyst has functional groups suitable for strong non-covalent interactions (occasionally, functionalization of the ligand is required, i.e., the pyrene unit). Indeed, the strength of non-covalent interactions is crucial for avoiding leaking of the catalysts in solution due to mechanical removal (i.e., whenever flow electrochemical cells are employed). Thus, covalent functionalization requires a ligand modification strategy, as well as the adoption of a solid material that allows formation of covalent bonds between the electrode and the molecular catalyst. However, covalent functionalization guarantees much stronger adhesion to the electrode surface and fast heterogeneous electron transfer. Overall, it can be concluded that, by passing from non-covalent to covalent bonding, higher current densities, longer operational times, and higher catalytic activities can be reached. Another important factor in the field of CO2RR is the role of the electrolyte. By looking at both Table 1 and Table 2, one of the major pieces of evidence is the employment of aqueous electrolytes, which are normally not suitable for the same organometallic catalysts in homogeneous conditions. The solubility of CO2 is much lower with respect to organic solvents, but it allows for a more environmentally friendly strategy. Careful optimization of the pH is another important parameter for steering the selectivity of the reduction products, and it is important to keep in mind that HER (hydrogen evolution reaction) is competitive in aqueous solutions. It has been shown that K+ cations are essential for catalysis for a Mn-bpy-type catalyst. A non-negligible role can be played by cations and anions of the electrolyte (i.e., KHCO3 vs. NaHCO3); the Lewis acid affect is essential, and varying the cation size induces a change in the outer Helmholtz plane (OHP) potential. Larger cations will adsorb more easily on the electrode surface. A detailed comparison of the electrolyte employed is beyond the scope of this mini review, and interested readers are redirected to a useful review about electrolyte effects for CO2RR [106].
As mentioned in the introduction, other types of catalysts can be used, including metal cathodes [107]. They are stable, can be modeled in almost any geometrical form, and can reach very high current densities. However, their catalytic properties are intrinsically difficult to tune because they generally suffer from low selectivity and low faradaic efficiencies. Molecular organometallic complexes have much greater opportunities for tuning their electronic and steric properties toward the electrosynthesis of selected CO2 reduction products. Computational modeling and hence mechanistic interpretations are much easier, thus opening the possibility of faster development. Nevertheless, molecular organometallic complexes require careful design of the ligands and sometimes, as mentioned, require challenging synthetic modifications. On the other hand, they also require a suitable electrode surface and chemical approach for heterogeneous functionalization and may generally undergo much easier catalyst degradation with lower stabilities.
Promising results have been achieved in recent years, and these are summarized in this mini review. We hope this review will further stimulate original research in the field not only for the development of new catalysts but also looking for a method of combined CO2 capture and conversion.
Author Contributions
Conceptualization, R.G. and C.N.; data curation all the authors; writing L.R., R.G. and C.N.; figures A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Perazio, A.; Lowe, G.; Gobetto, R.; Bonin, J.; Robert, M. Light-driven catalytic conversion of CO2 with heterogenized molecular catalysts based on fourth period transition metals. Coord. Chem. Rev. 2021, 443, 214018. [Google Scholar] [CrossRef]
- Franco, F.; Rettenmaier, C.; Sang Jeon, H.; Cuenya, B.R. Transition metal-based catalysts for the electrochemical CO2 reduction: From atoms and molecules to nanostructured materials. Chem. Soc. Rev. 2020, 49, 6884–6946. [Google Scholar] [CrossRef]
- Franco, F.; Fernández, S.; Lloret-Fillol, J. Advances in the electrochemical catalytic reduction of CO2 with metal complexes. Curr. Opin. Electrochem. 2019, 15, 109–117. [Google Scholar] [CrossRef]
- Dalle, K.E.; Warnan, J.; Leung, J.J.; Reuillard, B.; Karmel, I.S.; Reisner, E. Electro- and Solar-Driven Fuel Synthesis with First Row Transition Metal Complexes. Chem. Rev. 2019, 119, 2752–2875. [Google Scholar] [CrossRef]
- Elgrishi, N.; Chambers, M.B.; Wang, X.; Fontecave, M. Molecular polypyridine-based metal complexes as catalysts for the reduction of CO2. Chem. Soc. Rev. 2017, 46, 761–796. [Google Scholar] [CrossRef]
- Takeda, H.; Cometto, C.; Ishitani, O.; Robert, M. Electrons, Photons, Protons and Earth-Abundant Metal Complexes for Molecular Catalysis of CO2 Reduction. ACS Catal. 2017, 7, 70–88. [Google Scholar] [CrossRef]
- Rotundo, L.; Gobetto, R.; Nervi, C. Electrochemical CO2 reduction with earth-abundant metal catalysts. Curr. Opin. Green Sust. Chem. 2021, 31, 100509. [Google Scholar] [CrossRef]
- Rosen, J.; Hutchings, G.S.; Lu, Q.; Rivera, S.; Zhou, Y.; Vlachos, D.G.; Jiao, F. Mechanistic Insights into the Electrochemical Reduction of CO2 to CO on Nanostructured Ag Surfaces. ACS Catal. 2015, 5, 4293–4299. [Google Scholar] [CrossRef]
- Marshall-Roth, T.; Libretto, N.J.; Wrobel, A.T.; Anderton, K.J.; Pegis, M.L.; Ricke, N.D.; Voorhis, T.V.; Miller, J.T.; Surendranath, Y. A pyridinic Fe-N4 macrocycle models the active sites in Fe/N-doped carbon electrocatalysts. Nat. Commun. 2020, 11, 5283. [Google Scholar] [CrossRef]
- Chen, C.; Sun, X.; Yan, X.; Wu, Y.; Liu, H.; Zhu, Q.; Bediako, B.B.A.; Han, B. Boosting CO2 Electroreduction on N,P-Co-doped Carbon Aerogels. Angew. Chem. Int. Ed. 2020, 59, 11123–11129. [Google Scholar] [CrossRef]
- Jiang, Z.; Wang, Y.; Zhang, X.; Zheng, H.; Wang, X.; Liang, Y. Revealing the hidden performance of metal phthalocyanines for CO2 reduction electrocatalysis by hybridization with carbon nanotubes. Nano Res. 2019, 12, 2330–2334. [Google Scholar] [CrossRef]
- Manbeck, G.F.; Fujita, E. A review of iron and cobalt porphyrins, phthalocyanines and related complexes for electrochemical and photochemical reduction of carbon dioxide. J. Porphyr. Phthalocyanines 2015, 19, 45–64. [Google Scholar] [CrossRef]
- Li, M.; Li, T.; Wang, R.; Sun, C.; Zhang, N.; Gao, R.; Song, Y. Heat-treated copper phthalocyanine on carbon toward electrochemical CO2 conversion into ethylene boosted by oxygen reduction. Chem. Commun. 2022, 58, 12192–12195. [Google Scholar] [CrossRef]
- Liu, W.; Zhai, P.; Li, A.; Wei, B.; Si, K.; Wei, Y.; Wang, X.; Zhu, G.; Chen, Q.; Gu, X.; et al. Electrochemical CO2 reduction to ethylene by ultrathin CuO nanoplate arrays. Nat. Commun. 2022, 13, 1877. [Google Scholar] [CrossRef]
- Wu, Y.; Jiang, Z.; Lu, X.; Liang, Y.; Wang, H. Domino electroreduction of CO2 to methanol on a molecular catalyst. Nature 2019, 575, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Torbensen, K.; Salvatore, D.; Ren, S.; Joulie, D.; Dumoulin, F.; Mendoza, D.; Lassalle-Kaiser, B.; Isci, U.; Berlinguette, C.P.; et al. CO2 electrochemical catalytic reduction with a highly active cobalt phthalocyanine. Nat. Commun. 2019, 10, 3602. [Google Scholar] [CrossRef] [PubMed]
- Franco, F.; Cometto, C.; Vallana, F.F.; Sordello, F.; Priola, E.; Minero, C.; Nervi, C.; Gobetto, R. A local proton source in a [Mn(bpy-R)(CO)3Br]-type redox catalyst enables CO2 reduction even in the absence of Brønsted acids. Chem. Commun. 2014, 50, 14670–14673. [Google Scholar] [CrossRef] [PubMed]
- Franco, F.; Cometto, C.; Nencini, L.; Barolo, C.; Sordello, F.; Minero, C.; Fiedler, J.; Robert, M.; Gobetto, R.; Nervi, C. Local Proton Source in Electrocatalytic CO2 Reduction with [Mn(bpy-R)(CO)3 Br] Complexes. Chem. Eur. J. 2017, 23, 4782–4793. [Google Scholar] [CrossRef] [PubMed]
- Rønne, M.H.; Cho, D.; Madsen, M.R.; Jakobsen, J.B.; Eom, S.; Escoudé, É.; Hammershøj, H.C.D.; Nielsen, D.U.; Pedersen, S.U.; Baik, M.-H.; et al. Ligand-Controlled Product Selectivity in Electrochemical Carbon Dioxide Reduction Using Manganese Bipyridine Catalysts. J. Am. Chem. Soc. 2020, 142, 4265–4275. [Google Scholar] [CrossRef]
- Madsen, M.R.; Rønne, M.H.; Heuschen, M.; Golo, D.; Ahlquist, M.S.G.; Skrydstrup, T.; Pedersen, S.U.; Daasbjerg, K. Promoting Selective Generation of Formic Acid from CO2 Using Mn(bpy)(CO)Br as Electrocatalyst and Triethylamine/Isopropanol as Additives. J. Am. Chem. Soc. 2021, 143, 20491–20500. [Google Scholar] [CrossRef]
- Stuardi, F.M.; Tiozzo, A.; Rotundo, L.; Leclaire, J.; Gobetto, R.; Nervi, C. Efficient Electrochemical Reduction of CO2 to Formate in Methanol Solutions by Mn-Functionalized Electrodes in the Presence of Amines. Chem. Eur. J. 2022, 28, e202104377. [Google Scholar] [CrossRef]
- Francke, R.; Schille, B.; Roemelt, M. Homogeneously Catalyzed Electroreduction of Carbon Dioxide—Methods, Mechanisms, and Catalysts. Chem. Rev. 2018, 118, 4631–4701. [Google Scholar] [CrossRef]
- Fujita, E.; Grills, D.C.; Manbeck, G.F.; Polyansky, D.E. Understanding the Role of Inter- and Intramolecular Promoters in Electro- and Photochemical CO2 Reduction Using Mn, Re, and Ru Catalysts. Acc. Chem. Res. 2022, 55, 616–628. [Google Scholar] [CrossRef]
- Zhang, S.; Fan, Q.; Xia, R.; Meyer, T.J. CO2 Reduction: From Homogeneous to Heterogeneous Electrocatalysis. Acc. Chem. Res. 2020, 53, 255–264. [Google Scholar] [CrossRef]
- Sun, L.; Reddu, V.; Fisher, A.C.; Wang, X. Electrocatalytic reduction of carbon dioxide: Opportunities with heterogeneous molecular catalysts. Energy Environ. Sci. 2020, 13, 374–403. [Google Scholar] [CrossRef]
- Feng, D.-M.; Zhu, Y.-P.; Chen, P.; Ma, T.-Y. Recent Advances in Transition-Metal-Mediated Electrocatalytic CO2 Reduction: From Homogeneous to Heterogeneous Systems. Catalysts 2017, 7, 373. [Google Scholar] [CrossRef]
- Bullock, R.M.; Das, A.K.; Appel, A.M. Surface Immobilization of Molecular Electrocatalysts for Energy Conversion. Chemistry 2017, 23, 7626–7641. [Google Scholar] [CrossRef]
- Sun, C.; Gobetto, R.; Nervi, C. Recent advances in catalytic CO2 reduction by organometal complexes anchored on modified electrodes. New J. Chem 2016, 40, 5656–5661. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Electrocatalytic reduction of carbon dioxide mediated by Re(bipy)(CO)3Cl (bipy = 2,2′-bipyridine). J. Chem. Soc. Chem. Commun. 1984, 328–330. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Photochemical and Electrochemical Reduction of Carbon Dioxide to Carbon Monoxide Mediated by (2,2’-Bipyridine) tricarbonylchlororhenium(I) and Related Complexes as Homogeneous Catalysts. Helv. Chim. Acta 1986, 69, 1990–2012. [Google Scholar] [CrossRef]
- Blakemore, J.D.; Gupta, A.; Warren, J.J.; Brunschwig, B.S.; Gray, H.B. Noncovalent immobilization of electrocatalysts on carbon electrodes for fuel production. J. Am. Chem. Soc. 2013, 135, 18288–18291. [Google Scholar] [CrossRef]
- Oh, S.; Gallagher, J.R.; Miller, J.T.; Surendranath, Y. Graphite-Conjugated Rhenium Catalysts for Carbon Dioxide Reduction. J. Am. Chem. Soc. 2016, 138, 1820–1823. [Google Scholar] [CrossRef]
- Guyot, M.; Lalloz, M.-N.; Aguirre-Araque, J.S.; Rogez, G.; Costentin, C.; Chardon-Noblat, S. Rhenium Carbonyl Molecular Catalysts for CO2 Electroreduction: Effects on Catalysis of Bipyridine Substituents Mimicking Anchorage Functions to Modify Electrodes. Inorg. Chem. 2022, 61, 16072–16080. [Google Scholar] [CrossRef]
- Bourrez, M.; Molton, F.; Chardon-Noblat, S.; Deronzier, A. [Mn(bipyridyl)(CO)3Br]: An Abundant Metal Carbonyl Complex as Efficient Electrocatalyst for CO2 Reduction. Angew. Chem. Int. Ed. 2011, 50, 9903–9906. [Google Scholar] [CrossRef]
- Sampson, M.D.; Kubiak, C.P. Manganese Electrocatalysts with Bulky Bipyridine Ligands: Utilizing Lewis Acids To Promote Carbon Dioxide Reduction at Low Overpotentials. J. Am. Chem. Soc. 2016, 138, 1386–1393. [Google Scholar] [CrossRef]
- Riplinger, C.; Sampson, M.D.; Ritzmann, A.M.; Kubiak, C.P.; Carter, E.A. Mechanistic Contrasts between Manganese and Rhenium Bipyridine Electrocatalysts for the Reduction of Carbon Dioxide. J. Am. Chem. Soc. 2014, 136, 16285–16298. [Google Scholar] [CrossRef]
- Smieja, J.M.; Sampson, M.D.; Grice, K.A.; Benson, E.E.; Froehlich, J.D.; Kubiak, C.P. Manganese as a Substitute for Rhenium in CO2 Reduction Catalysts: The Importance of Acids. Inorg. Chem. 2013, 52, 2484–2491. [Google Scholar] [CrossRef]
- Keith, J.A.; Grice, K.A.; Kubiak, C.P.; Carter, E.A. Elucidation of the Selectivity of Proton-Dependent Electrocatalytic CO2 Reduction by fac-Re(bpy)(CO)3Cl. J. Am. Chem. Soc. 2013, 135, 15823–15829. [Google Scholar] [CrossRef]
- Grills, D.C.; Ertem, M.Z.; McKinnon, M.; Ngo, K.T.; Rochford, J. Mechanistic aspects of CO2 reduction catalysis with manganese-based molecular catalysts. Coord. Chem. Rev. 2018, 374, 173–217. [Google Scholar] [CrossRef]
- Siritanaratkul, B.; Eagle, C.; Cowan, A.J. Manganese Carbonyl Complexes as Selective Electrocatalysts for CO2 Reduction in Water and Organic Solvents. Acc. Chem. Res. 2022, 55, 955–965. [Google Scholar] [CrossRef]
- Rønne, M.H.; Madsen, M.R.; Skrydstrup, T.; Pedersen, S.U.; Daasbjerg, K. Mechanistic Elucidation of Dimer Formation and Strategies for Its Suppression in Electrochemical Reduction of fac-Mn(bpy)(CO)3Br. ChemElectroChem 2021, 8, 2108–2114. [Google Scholar] [CrossRef]
- Walsh, J.J.; Neri, G.; Smith, C.L.; Cowan, A.J. Electrocatalytic CO2 reduction with a membrane supported manganese catalyst in aqueous solution. Chem. Commun. 2014, 50, 12698–12701. [Google Scholar] [CrossRef]
- Walsh, J.J.; Smith, C.L.; Neri, G.; Whitehead, G.F.S.; Robertson, C.M.; Cowan, A.J. Improving the efficiency of electrochemical CO2 reduction using immobilized manganese complexes. Faraday Discuss. 2015, 183, 147–160. [Google Scholar] [CrossRef]
- Walsh, J.J.; Forster, M.; Smith, C.L.; Neri, G.; Potter, R.J.; Cowan, A.J. Directing the mechanism of CO2 reduction by a Mn catalyst through surface immobilization. Phys. Chem. Chem. Phys. 2018, 20, 6811–6816. [Google Scholar] [CrossRef]
- Reuillard, B.; Ly, K.H.; Rosser, T.E.; Kuehnel, M.F.; Zebger, I.; Reisner, E. Tuning Product Selectivity for Aqueous CO2 Reduction with a Mn(bipyridine)-pyrene Catalyst Immobilized on a Carbon Nanotube Electrode. J. Am. Chem. Soc. 2017, 139, 14425–14435. [Google Scholar] [CrossRef]
- Sato, S.; Saita, K.; Sekizawa, K.; Maeda, S.; Morikawa, T. Low-Energy Electrocatalytic CO2 Reduction in Water over Mn-Complex Catalyst Electrode Aided by a Nanocarbon Support and K+ Cations. ACS Catal. 2018, 8, 4452–4458. [Google Scholar] [CrossRef]
- Kang, P.; Zhang, S.; Meyer, T.J.; Brookhart, M. Rapid Selective Electrocatalytic Reduction of Carbon Dioxide to Formate by an Iridium Pincer Catalyst Immobilized on Carbon Nanotube Electrodes. Angew. Chem. Int. Ed. 2014, 53, 8709–8713. [Google Scholar] [CrossRef]
- Kang, P.; Cheng, C.; Chen, Z.; Schauer, C.K.; Meyer, T.J.; Brookhart, M. Selective electrocatalytic reduction of CO2 to formate by water-stable iridium dihydride pincer complexes. J. Am. Chem. Soc. 2012, 134, 5500–5503. [Google Scholar] [CrossRef]
- Cao, L.; Sun, C.; Sun, N.; Meng, L.; Chen, D. Theoretical mechanism studies on the electrocatalytic reduction of CO2 to formate by water-stable iridium dihydride pincer complex. Dalton Trans. 2013, 42, 5755–5763. [Google Scholar] [CrossRef]
- Ahn, S.T.; Bielinski, E.A.; Lane, E.M.; Chen, Y.; Bernskoetter, W.H.; Hazari, N.; Palmore, G.T.R. Enhanced CO2 electroreduction efficiency through secondary coordination effects on a pincer iridium catalyst. Chem. Commun. 2015, 51, 5947–5950. [Google Scholar] [CrossRef]
- Kang, P.; Meyer, T.J.; Brookhart, M. Selective electrocatalytic reduction of carbon dioxide to formate by a water-soluble iridium pincer catalyst. Chem. Sci. 2013, 4, 3497–3502. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.-P.; Ruppert, R.; Sauvage, J.-P. Nickel(II)-cyclam: An extremely selective electrocatalyst for reduction of CO2 in water. J. Chem. Soc. Chem. Commun. 1984, 1315–1316. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.P.; Ruppert, R.; Sauvage, J.P. Electrocatalytic reduction of carbon dioxide by nickel cyclam2+ in water: Study of the factors affecting the efficiency and the selectivity of the process. J. Am. Chem. Soc. 1986, 108, 7461–7467. [Google Scholar] [CrossRef] [PubMed]
- Collin, J.P.; Jouaiti, A.; Sauvage, J.P. Electrocatalytic properties of (tetraazacyclotetradecane)nickel2+ and Ni2(biscyclam)4+ with respect to carbon dioxide and water reduction. Inorg. Chem. 1988, 27, 1986–1990. [Google Scholar] [CrossRef]
- Balazs, G.B.; Anson, F.C. Effects of CO on the electrocatalytic activity of Ni (cyclam)2+ toward the reduction of CO2. J. Electroanal. Chem. 1993, 361, 149–157. [Google Scholar] [CrossRef]
- Zhanaidarova, A.; Moore, C.E.; Gembicky, M.; Kubiak, C.P. Covalent attachment of [Ni(alkynyl-cyclam)]2+catalysts to the glassy carbon electrodes. Chem. Commun. 2018, 54, 4116–4119. [Google Scholar] [CrossRef] [PubMed]
- Pugliese, S.; Huan, N.T.; Forte, J.; Grammatico, D.; Zanna, S.; Su, B.-L.; Li, Y.; Fontecave, M. Functionalization of Carbon Nanotubes with Nickel Cyclam for the Electrochemical Reduction of CO2. ChemSusChem 2020, 13, 6449–6456. [Google Scholar] [CrossRef]
- Greenwell, F.; Neri, G.; Piercy, V.; Cowan, A.J. Noncovalent immobilization of a nickel cyclam catalyst on carbon electrodes for CO2 reduction using aqueous electrolyte. Electrochim. Acta 2021, 392, 139015. [Google Scholar] [CrossRef]
- Pugliese, S.; Huan, N.T.; Solé-Daura, A.; Li, Y.; Rivera de la Cruz, J.-G.; Forte, J.; Zanna, S.; Krief, A.; Su, B.-L.; Fontecave, M. CO2 Electroreduction in Water with a Heterogenized C-Substituted Nickel Cyclam Catalyst. Inorg. Chem. 2022, 61, 15841–15852. [Google Scholar] [CrossRef]
- Corbin, N.; Zeng, J.; Williams, K.; Manthiram, K. Heterogeneous molecular catalysts for electrocatalytic CO2 reduction. Nano Res. 2019, 12, 2093–2125. [Google Scholar] [CrossRef]
- Huai, M.; Yin, Z.; Wei, F.; Wang, G.; Xiao, L.; Lu, J.; Zhuang, L. Electrochemical CO2 reduction on heterogeneous cobalt phthalocyanine catalysts with different carbon supports. Chem. Phys. Lett. 2020, 754, 137655. [Google Scholar] [CrossRef]
- Abdinejad, M.; Tang, K.; Dao, C.; Saedy, S.; Burdyny, T. Immobilization strategies for porphyrin-based molecular catalysts for the electroreduction of CO2. J. Mater. Chem. A 2022, 10, 7626–7636. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Hiratsuka, K.; Sasaki, H.; Toshima, S. Electrocatalytic behavior of metal porphyrins in the reduction of carbon dioxide. Chem. Lett. 1979, 8, 305–308. [Google Scholar] [CrossRef]
- Boutin, E.; Merakeb, L.; Ma, B.; Boudy, B.; Wang, M.; Bonin, J.; Anxolabehere-Mallart, E.; Robert, M. Molecular catalysis of CO2 reduction: Recent advances and perspectives in electrochemical and light-driven processes with selected Fe, Ni and Co aza macrocyclic and polypyridine complexes. Chem. Soc. Rev. 2020, 49, 5772–5809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.-Z.; Wu, B.-Y.; Li, Q.; Lu, L.-L.; Shi, W.; Cheng, P. Design strategies and mechanism studies of CO2 electroreduction catalysts based on coordination chemistry. Coord. Chem. Rev. 2020, 422, 213436. [Google Scholar] [CrossRef]
- Costentin, C.; Savéant, J.-M. Towards an intelligent design of molecular electrocatalysts. Nat. Chem. Rev. 2017, 1, 1–8. [Google Scholar] [CrossRef]
- Costentin, C.; Robert, M.; Saveant, J.-M. Current Issues in Molecular Catalysis Illustrated by Iron Porphyrins as Catalysts of the CO2-to-CO Electrochemical Conversion. Acc. Chem. Res. 2015, 48, 2996–3006. [Google Scholar] [CrossRef]
- Costentin, C.; Saveant, J.-M. Homogeneous Molecular Catalysis of Electrochemical Reactions: Catalyst Benchmarking and Optimization Strategies. J. Am. Chem. Soc. 2017, 139, 8245–8250. [Google Scholar] [CrossRef]
- Azcarate, I.; Costentin, C.; Robert, M.; Saveant, J.-M. Through-Space Charge Interaction Substituent Effects in Molecular Catalysis Leading to the Design of the Most Efficient Catalyst of CO2-to-CO Electrochemical Conversion. J. Am. Chem. Soc. 2016, 138, 16639–16644. [Google Scholar] [CrossRef]
- Martin, D.J.; Mayer, J.M. Oriented Electrostatic Effects on O2 and CO2 Reduction by a Polycationic Iron Porphyrin. J. Am. Chem. Soc. 2021, 143, 11423–11434. [Google Scholar] [CrossRef]
- Martin, D.J.; Mercado, B.Q.; Mayer, J.M. All Four Atropisomers of Iron Tetra(o-N,N,N-trimethylanilinium)porphyrin in Both the Ferric and Ferrous States. Inorg. Chem. 2021, 60, 5240–5251. [Google Scholar] [CrossRef] [PubMed]
- Maurin, A.; Robert, M. Noncovalent Immobilization of a Molecular Iron-Based Electrocatalyst on Carbon Electrodes for Selective, Efficient CO2-to-CO Conversion in Water. J. Am. Chem. Soc. 2016, 138, 2492–2495. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.-M.; Rønne, M.H.; Pedersen, S.U.; Skrydstrup, T.; Daasbjerg, K. Enhanced Catalytic Activity of Cobalt Porphyrin in CO2 Electroreduction upon Immobilization on Carbon Materials. Angew. Chem. Int. Ed. 2017, 56, 6468–6472. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hu, X.-M.; Daasbjerg, K.; Ahlquist, M.S.G. Understanding the Enhanced Catalytic CO2 Reduction upon Adhering Cobalt Porphyrin to Carbon Nanotubes and the Inverse Loading Effect. Organometallics 2020, 39, 1634–1641. [Google Scholar] [CrossRef]
- Wang, J.; Huang, X.; Xi, S.; Lee, J.-M.; Wang, C.; Du, Y.; Wang, X. Linkage Effect in the Heterogenization of Cobalt Complexes by Doped Graphene for Electrocatalytic CO2 Reduction. Angew. Chem. Int. Ed. 2019, 58, 13532–13539. [Google Scholar] [CrossRef]
- BaQais, A.; Ait Ahsaine, H. β-Cobalt phthalocyanine sono-immobilized on carbon cloth for efficient electrochemical reduction of CO2-to-CO. Mater. Lett. 2022, 324, 132614. [Google Scholar] [CrossRef]
- Abdinejad, M.; Wilm, L.F.B.; Dielmann, F.; Kraatz, H.B. Electroreduction of CO2 Catalyzed by Nickel Imidazolin-2-ylidenamino-Porphyrins in Both Heterogeneous and Homogeneous Molecular Systems. ACS Sustain. Chem. Eng. 2021, 9, 521–530. [Google Scholar] [CrossRef]
- Abdinejad, M.; Dao, C.; Zhang, X.-A.; Kraatz, H.B. Enhanced electrocatalytic activity of iron amino porphyrins using a flow cell for reduction of CO2 to CO. J. Energy Chem. 2021, 58, 162–169. [Google Scholar] [CrossRef]
- Xu, H.; Cai, H.; Cui, L.; Yu, L.; Gao, R.; Shi, C. Molecular modulating of cobalt phthalocyanines on aminofunctionalized carbon nanotubes for enhanced electrocatalytic CO2 conversion. Nano Res. 2022, in press. [Google Scholar] [CrossRef]
- Yao, S.A.; Ruther, R.E.; Zhang, L.; Franking, R.A.; Hamers, R.J.; Berry, J.F. Covalent attachment of catalyst molecules to conductive diamond: CO2 reduction using “smart” electrodes. J. Am. Chem. Soc. 2012, 134, 15632–15635. [Google Scholar] [CrossRef]
- Zhu, M.; Chen, J.; Guo, R.; Xu, J.; Fang, X.; Han, Y.-F. Cobalt phthalocyanine coordinated to pyridine-functionalized carbon nanotubes with enhanced CO2 electroreduction. Appl. Catal. B 2019, 251, 112–118. [Google Scholar] [CrossRef]
- Liu, Y.; McCrory, C.C.L. Modulating the mechanism of electrocatalytic CO2 reduction by cobalt phthalocyanine through polymer coordination and encapsulation. Nat. Commun. 2019, 10, 1683. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.-D.; Han, S.-G.; Cao, C.; Wei, W.; Li, X.; Chen, B.; Wu, X.-T.; Zhu, Q.-L. Bifunctional single-molecular heterojunction enables completely selective CO2-to-CO conversion integrated with oxidative 3D nano-polymerization. Energy Environ. Sci. 2021, 14, 1544–1552. [Google Scholar] [CrossRef]
- Gong, S.; Wang, W.; Xiao, X.; Liu, J.; Wu, C.; Lv, X. Elucidating influence of the existence formation of anchored cobalt phthalocyanine on electrocatalytic CO2-to-CO conversion. Nano Energy 2021, 84, 105904. [Google Scholar] [CrossRef]
- Maurin, A.; Robert, M. Catalytic CO2-to-CO conversion in water by covalently functionalized carbon nanotubes with a molecular iron catalyst. Chem. Commun. 2016, 52, 12084–12087. [Google Scholar] [CrossRef]
- Zhu, M.; Chen, J.; Huang, L.; Ye, R.; Xu, J.; Han, Y.-F. Covalently Grafting Cobalt Porphyrin onto Carbon Nanotubes for Efficient CO2 Electroreduction. Angew. Chem. Int. Ed. 2019, 58, 6595–6599. [Google Scholar] [CrossRef]
- Marianov, A.N.; Jiang, Y. Covalent ligation of Co molecular catalyst to carbon cloth for efficient electroreduction of CO2 in water. Appl. Catal. B 2019, 244, 881–888. [Google Scholar] [CrossRef]
- Gu, S.; Marianov, A.N.; Jiang, Y. Covalent grafting of cobalt aminoporphyrin-based electrocatalyst onto carbon nanotubes for excellent activity in CO2 reduction. Appl. Catal. B 2022, 300, 120750. [Google Scholar] [CrossRef]
- Sun, C.; Rotundo, L.; Garino, C.; Nencini, L.; Yoon, S.S.; Gobetto, R.; Nervi, C. Electrochemical CO2 Reduction at Glassy Carbon Electrodes Functionalized by Mn I and Re I Organometallic Complexes. ChemPhysChem 2017, 18, 3219–3229. [Google Scholar] [CrossRef]
- Rotundo, L.; Filippi, J.; Gobetto, R.; Miller, H.A.; Rocca, R.; Nervi, C.; Vizza, F. Electrochemical CO2 reduction in water at carbon cloth electrodes functionalized with a fac-Mn(apbpy)(CO)3Br complex. Chem. Commun. 2019, 55, 775–777. [Google Scholar] [CrossRef]
- Filippi, J.; Rotundo, L.; Gobetto, R.; Miller, H.A.; Nervi, C.; Lavacchi, A.; Vizza, F. Turning manganese into gold: Efficient electrochemical CO2 reduction by a fac-Mn(apbpy)(CO)3Br complex in a gas–liquid interface flow cell. Chem. Eng. J. 2021, 416, 129050. [Google Scholar] [CrossRef]
- Jakobsen, J.B.; Rønne, M.H.; Daasbjerg, K.; Skrydstrup, T. Are Amines the Holy Grail for Facilitating CO2 Reduction? Angew. Chem. Int. Ed. 2021, 60, 9174–9179. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, T.R.; Margerum, L.D.; Westmoreland, T.D.; Vining, W.J.; Murray, R.W.; Meyer, T.J. Electrocatalytic reduction of CO2 at a chemically modified electrode. J. Chem. Soc. Chem. Commun. 1985, 1416–1417. [Google Scholar] [CrossRef]
- O’Toole, T.R.; Sullivan, B.P.; Bruce, M.R.-M.; Margerum, L.D.; Murray, R.W.; Meyer, T.J. Electrocatalytic reduction of CO2 by a complex of rhenium in thin polymeric films. J. Electroanal. Chem. Interfacial Electrochem. 1989, 259, 217–239. [Google Scholar] [CrossRef]
- Guadalupe, A.R.; Usifer, D.A.; Potts, K.T.; Hurrell, H.C.; Mogstad, A.E.; Abruna, H.D. Novel chemical pathways and charge-transport dynamics of electrodes modified with electropolymerized layers of [Co(v-terpy)2]2+. J. Am. Chem. Soc. 1988, 110, 3462–3466. [Google Scholar] [CrossRef]
- Denisevich, P.; Abruna, H.D.; Leidner, C.R.; Meyer, T.J.; Murray, R.W. Electropolymerization of vinylpyridine and vinylbipyridine complexes of iron and ruthenium: Homopolymers, copolymers, reactive polymers. Inorg. Chem. 1982, 21, 2153–2161. [Google Scholar] [CrossRef]
- Calvert, J.M.; Schmehl, R.H.; Sullivan, B.P.; Facci, J.S.; Meyer, T.J.; Murray, R.W. Synthetic and mechanistic investigations of the reductive electrochemical polymerization of vinyl-containing complexes of iron(II), ruthenium(II), and osmium(II). Inorg. Chem. 1983, 22, 2151–2162. [Google Scholar] [CrossRef]
- Portenkirchner, E.; Oppelt, K.; Ulbricht, C.; Egbe, D.A.M.; Neugebauer, H.; Knoer, G.; Sariciftci, N.S. Electrocatalytic and photocatalytic reduction of carbon dioxide to carbon monoxide using the alkynyl-substituted rhenium(I) complex (5,5’-bisphenylethynyl-2,2’-bipyridyl)Re(CO)3Cl. J. Organomet. Chem. 2012, 716, 19–25. [Google Scholar] [CrossRef]
- Portenkirchner, E.; Gasiorowski, J.; Oppelt, K.; Schlager, S.; Schwarzinger, C.; Neugebauer, H.; Knör, G.; Sariciftci, N.S. Electrocatalytic Reduction of Carbon Dioxide to Carbon Monoxide by a Polymerized Film of an Alkynyl-Substituted Rhenium(I) Complex. ChemCatChem 2013, 5, 1790–1796. [Google Scholar] [CrossRef]
- Sun, C.; Prosperini, S.; Quagliotto, P.; Viscardi, G.; Yoon, S.S.; Gobetto, R.; Nervi, C. Electrocatalytic reduction of CO2 by thiophene-substituted rhenium(I) complexes and by their polymerized films. Dalton Trans. 2016, 45, 14678–14688. [Google Scholar] [CrossRef]
- Wu, Q.; Liang, J.; Xie, Z.-L.; Huang, Y.-B.; Cao, R. Spatial Sites Separation Strategy to Fabricate Atomically Isolated Nickel Catalysts for Efficient CO2 Electroreduction. ACS Mater. Lett. 2021, 3, 454–461. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Jones, R.A.; Holliday, B.J. Recent advances in the functional applications of conducting metallopolymers. Coord. Chem. Rev. 2018, 377, 237–258. [Google Scholar] [CrossRef]
- Whittell, G.R.; Hager, M.D.; Schubert, U.S.; Manners, I. Functional soft materials from metallopolymers and metallosupramolecular polymers. Nat. Mater. 2011, 10, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Elmas, S.; Macdonald, T.J.; Skinner, W.; Andersson, M.; Nann, T. Copper Metallopolymer Catalyst for the Electrocatalytic Hydrogen Evolution Reaction (HER). Polymers 2019, 11, 110. [Google Scholar] [CrossRef]
- Teixeira, M.; Olean-Oliveira, A.; Anastácio, F.; David-Parra, D.; Cardoso, C. Electrocatalytic Reduction of CO2 in Water by a Palladium-Containing Metallopolymer. Nanomaterials 2022, 12, 1193. [Google Scholar] [CrossRef] [PubMed]
- Moura de Salles Pupo, M.; Kortlever, R. Electrolyte Effects on the Electrochemical Reduction of CO2. ChemPhysChem 2019, 20, 2926–2935. [Google Scholar] [CrossRef]
- Hori, Y. Electrochemical CO2 Reduction on Metal Electrodes. In Modern Aspects of Electrochemistry; Vayenas, C.G., White, R.E., Gamboa-Aldeco, M.E., Eds.; Springer: New York, NY, USA, 2008; pp. 89–189. ISBN 978-0-387-49489-0. [Google Scholar]















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).