3.2. Synthesis of the Lanthionine 5
3.2.1. 2-Methyl 1-(2-(Trimethylsilyl)ethyl) (2R,3R)-3-methylaziridine-1,2-dicarboxylate (2)
To a solution of D-threonine (5.00 g, 42.0 mmol, 1.0 equiv) in MeOH (150 mL) was added SOCl2 (15.3 mL, 210 mmol, 5.0 equiv) dropwise at 0 °C, and the mixture was stirred at the same temperature for 30 min. After being stirred at reflux in an oil bath for 12 h, the reaction mixture was cooled to room temperature, and concentrated in vacuo. The resulting crude methyl ester was used for the next reaction without further purification.
To a solution of the crude amine in dry CH2Cl2 (150 mL) were added Et3N (14.6 mL, 105 mmol, 2.5 equiv) and TrtCl (11.7 g, 42.0 mmol, 1.0 equiv) at 0 °C under an argon atmosphere. After being stirred at room temperature for 43 h, the reaction mixture was washed with 10% aqueous citric acid, saturated aqueous NaHCO3 and brine, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo, and the resulting crude N-Trt amine was used for the next reaction without further purification.
To a solution of the crude alcohol in dry THF (120 mL) were added Et3N (14.6 mL, 105 mmol, 2.5 equiv) and MsCl (3.6 mL, 46.2 mmol, 1.1 equiv) at 0 °C under an argon atmosphere, and the mixture was stirred at the same temperature for 30 min. After being stirred at reflux in an oil bath for 72 h, the reaction mixture was concentrated in vacuo to remove THF. The resulting residue was diluted with EtOAc, and the organic layer was washed with 10% aqueous citric acid and saturated aqueous NaHCO3, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo, and the resulting crude aziridine was used for next reaction without further purification.
To a solution of the crude N-Trt aziridine in dry CH2Cl2 (150 mL) were added dry MeOH (2.6 mL, 63.0 mmol, 1.5 equiv) and TFA (6.5 mL, 84.0 mmol, 2.0 equiv) at 0 °C under an argon atmosphere. After being stirred at the same temperature for 1 h, the reaction mixture was basified by Et3N (20.5 mL, 147 mmol, 3.5 equiv). TeocOSu (10.9 g, 42.0 mmol, 1.0 equiv) was then added to the above mixture at 0 °C. After being stirred at room temperature for 19 h, the reaction mixture was washed with 10% aqueous citric acid and saturated aqueous NaHCO3, dried over MgSO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was suspended in CH2Cl2/MeOH. The suspension was filtered through a pad of Celite®, and the filtrate was concentrated in vacuo. The resulting residue was purified by column chromatography on silica gel (eluted with hexane/EtOAc = 4:1) to afford the N-Teoc aziridine 2 (5.98 g, 23.0 mmol, 55% in 4 steps) as a colorless oil. [α]22D +64 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 4.18–4.22 (m, 2H), 3.79 (s, 3H), 3.15 (d, 1H, J = 6.8 Hz), 2.77–2.82 (m, 1H), 1.35 (d, 3H, J = 6.4 Hz), 1.00–1.04 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 167.7, 161.8, 65.3, 52.2, 39.7, 38.7, 17.4, 12.9, −1.5; IR (neat) 2955, 1756, 1729, 1442, 1425, 1285, 1252, 1201, 1181, 1081, 1038, 860, 838 cm−1; HRMS [ESI] m/z calcd for C11H21NO4SiNa [M+Na]+ 282.1132, found 282.1131.
3.2.2. Fmoc-Cys-OH (3)
To a solution of L-cystine (5.00 g, 20.8 mmol, 1.0 equiv) in 1,4-dioxane (90 mL) were added a solution of Na
2CO
3 (6.62 g, 62.4 mmol, 3.0 equiv) in water (60 mL) and a solution of FmocOSu (14.0 g, 41.6 mmol, 2.0 equiv) in 1,4-dioxane (90 mL) at 0 °C. After being stirred at the room temperature for 15 h, the reaction mixture was concentrated in vacuo to remove 1,4-dioxane. The aqueous layer was acidified with 6 M aqueous HCl until pH1, and extracted three times with CH
2Cl
2. The combined organic layers were dried over Na
2SO
4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was suspended in Et
2O. The white precipitate was filtered, and dried under vacuum to afford the
N-Fmoc amine (12.7 g, 18.5 mmol, 89%) as a white solid. The spectral data of synthetic compound were in good agreement with those of reported [
47]. Mp 153–154 °C [lit. 149–151 °C]; [α]
20D −89 (
c 1.2, MeOH) [lit. [α]
24D −87.1 (
c 1.0, MeOH)];
1H NMR (400 MHz, DMSO-
d6, rotamer mixture)
δ 13.0 (s, 1H), 7.87 (d, 2H,
J = 7.5 Hz), 7.78 (d, 1H,
J = 7.5 Hz), 7.69 (d, 2H,
J = 7.5 Hz), 7.37–7.41 (m, 2H), 7.30 (t, 2H,
J = 7.5 Hz), 4.26–4.31 (m, 3H), 4.21 (dd, 1H,
J = 12.6, 5.6 Hz), 3.16 (dd, 1H,
J = 13.5, 3.9 Hz), 2.94 (dd, 1H,
J = 13.5, 10.3 Hz);
13C{
1H} NMR (100 MHz, DMSO-
d6, rotamer mixture)
δ 172.2, 156.0, 143.8, 143.7, 140.7, 127.6, 127.1, 125.3, 125.2, 120.1, 65.8, 53.0, 46.6, 39.1; IR (neat) 1696, 1515, 1448, 1331, 1228, 1049, 758, 739 cm
−1; HRMS [FAB]
m/
z calcd for C
36H
33N
2O
8S
2 [M+H]
+ 685.1673, found 685.1690.
To a solution of the disulfide (14.0 g, 20.4 mmol, 1.0 equiv) in dry THF (70 mL) were added 1 M aqueous HCl (70 mL) and activated zinc dust (4.00 g, 61.2 mmol, 3.0 equiv) at 0 °C. After being stirred at the room temperature for 30 min, the reaction mixture was filtered through of a pad of Celite
®. The filtrate was concentrated in vacuo, and the resulting residue was diluted with 1 M aqueous HCl. The aqueous layer was extracted with CH
2Cl
2. The organic layer was dried over Na
2SO
4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was suspended in CH
2Cl
2/hexane. The precipitate was filtered and dried under vacuum to afford the thiol
3 (10.7 g, 31.3 mmol, 77%) as a white solid. The spectral data of synthetic compound were in good agreement with those of reported [
48]. Mp 119–123 °C; [α]
20D −5.7 (
c 0.93, MeOH);
1H NMR (400 MHz, DMSO-
d6)
δ 12.9 (s, 1H), 7.88 (d, 2H,
J = 7.5 Hz), 7.69–7.73 (m, 3H), 7.41 (t, 2H,
J = 7.5 Hz), 7.32 (t, 2H,
J = 7.5 Hz), 4.29–4.31 (m, 2H), 4.23 (t, 1H,
J = 7.0 Hz), 4.14 (dt, 1H,
J = 8.3, 4.3 Hz), 2.89–2.92 (m, 1H), 2.71–2.78 (m, 1H);
13C{
1H} NMR (100 MHz, DMSO-
d6)
δ 171.8, 156.0, 143.8, 140.7, 127.6, 127.1, 125.2, 120.1, 65.7, 56.5, 46.6, 25.4; IR (neat) 3314, 1694, 1536, 1476, 1447, 1418, 1230, 1103, 1047, 756, 736, 620 cm
−1; HRMS [FAB]
m/
z calcd for C
18H
18NO
4S [M+H]
+ 344.0951, found 344.0942.
3.2.3. The Lanthionine 4
To a solution of the aziridine 2 (4.62 g, 17.8 mmol, 2.0 equiv) in dry Et2O (90 mL) were added Fmoc-Cys-OH (3) (3.06 g, 8.91 mmol, 1.0 equiv) and InCl3 (788 mg, 3.56 mmol, 0.4 equiv) at room temperature under an argon atmosphere. After being stirred at the same temperature for 18 h, the reaction mixture was quenched with water. The organic layer was separated, and aqueous layer was extracted three times with EtOAc. The combined organic layers were dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by column chromatography on silica gel (eluted with CH2Cl2/MeCN = 1:1) to afford the lanthionine 4 (2.89 g, 4.79 mmol, 54%) as a white amorphous solid. [α]24D +2.3 (c 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 7.89 (d, 2H, J = 7.5 Hz), 7.71–7.73 (m, 3H), 7.41 (t, 2H, J = 7.5 Hz), 7.31–7.33 (m, 3H), 4.21–4.34 (m, 4H), 4.09–4.15 (m, 1H), 4.04–4.06 (m, 2H), 3.65 (s, 3H), 3.21–3.24 (m, 1H), 2.95 (dd, 1H, J = 13.6, 4.7 Hz), 2.74 (dd, 1H, J = 13.5, 9.4 Hz), 1.21 (d, 3H, J = 7.0 Hz), 0.91–0.93 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 172.0, 170.8, 156.2, 155.9, 143.7, 140.7, 127.6, 127.0, 125.2, 120.0, 65.7, 62.2, 58.4, 54.0, 51.9, 46.6, 41.6, 32.1, 18.8, 17.3, −1.6; IR (neat) 3327, 3019, 2953, 1720, 1513, 1478, 1449, 1338, 1249, 1213, 1080, 1049, 859, 837, 758, 740 cm−1; HRMS [ESI] m/z calcd for C29H38N2O8NaSSi [M+Na]+ 625.2010, found 625.2007.
3.2.4. The MOM Ester 5
To a solution of the carboxylic acid 4 (2.89 g, 4.79 mmol, 1.0 equiv) in dry acetone (90 mL) were added KHCO3 (1.20 g, 12.0 mmol, 2.5 equiv) and MOMCl (437 μL, 5.75 mmol, 1.2 equiv) at room temperature under an argon atmosphere. After being stirred at the same temperature for 15 h, the reaction mixture was concentrated in vacuo to remove acetone. The resulting residue was diluted with EtOAc. The organic layer was washed with saturated aqueous NaHCO3, dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1) to afford the MOM ester 5 (3.06 g, 4.73 mmol, 99%) as a white amorphous solid. [α]23D −16 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.74 (d, 2H, J = 7.5 Hz), 7.57–7.59 (m, 2H), 7.38 (t, 2H, J = 7.5 Hz), 7.29 (d, 2H, J = 7.5 Hz), 5.66 (d, 1H, J = 6.8 Hz), 5.41 (d, 1H, J = 8.5 Hz), 5.31 (d, 1H, J = 5.6 Hz), 5.27 (d, 1H, J = 5.6 Hz), 4.58–4.61 (m, 1H), 4.51 (d, 1H, J = 7.5 Hz), 4.36–4.44 (m, 2H), 4.22 (t, 1H, J = 6.9 Hz), 4.11–4.16 (m, 2H), 3.72 (s, 3H), 3.44–3.47 (m, 4H), 3.03 (dd, 1H, J = 13.4, 3.7 Hz), 2.91 (dd, 1H, J = 13.4, 5.4 Hz), 1.31 (d, 3H, J = 7.0 Hz), 0.95–0.97 (m, 2H), −0.01 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.1, 170.0, 156.7, 155.7, 143.7, 141.3, 127.7, 127.0, 125.0, 120.0, 91.8, 67.2, 63.7, 58.1, 58.0, 53.8, 52.5, 47.1, 43.7, 33.5, 19.4, 17.6, −1.6; IR (neat) 3335, 2953, 1722, 1511, 1450, 1339, 1249, 1210, 1157, 1081, 1047, 994, 928, 859, 837, 759, 741 cm−1; HRMS[ESI] m/z calcd for C31H42N2O9NaSSi [M+Na]+ 669.2272, found 669.2280.
3.3. Synthesis of the Tripeptide 1 by Solution-Phase Peptide Synthesis
3.3.1. The Tripeptide 1a
To a solution of the N-Fmoc-amine 5 (3.06 g, 4.73 mmol, 1.0 equiv) in dry MeCN (40 mL) was added Et2NH (10 mL) at room temperature under an argon atmosphere. After being stirred at the same temperature for 40 min, the reaction mixture was concentrated in vacuo. The resulting residue was azeotroped three times with MeCN to remove Et2NH, and the resulting crude amine was used for the next reaction without further purification.
To a solution of the crude amine in dry CH2Cl2 (50 mL) were added DIEA (1.7 mL, 9.46 mmol, 2.0 equiv), Cbz-Phe-OH (1.70 g, 5.68 mmol, 1.2 equiv), HOBt (773 mg, 5.68 mmol, 1.2 equiv) and EDCI·HCl (1.09 g, 5.68 mmol, 1.2 equiv) at 0 °C under an argon atmosphere. After being stirred at room temperature for 13 h, the reaction mixture was diluted with CH2Cl2. The organic layer was washed with 10% aqueous citric acid and saturated aqueous NaHCO3, dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1) to afford the tripeptide 6a (2.88 g, 4.08 mmol, 86% in 2 steps) as a white amorphous solid. [α]23D −25 (c 0.90, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.20–7.27 (m, 10H), 6.73 (d, 1H, J = 6.5 Hz), 5.45 (d, 1H, J = 9.2 Hz), 5.38 (d, 1H, J = 5.8 Hz), 5.29 (d, 1H, J = 5.8 Hz), 5.26 (d, 1H, J = 5.8 Hz), 5.08 (s, 2H), 4.72–4.74 (m, 1H), 4.48–4.50 (m, 2H), 4.15–4.17 (m, 2H), 3.75 (s, 3H), 3.48 (s, 3H), 3.37–3.41 (m, 1H), 3.00–3.18 (m, 3H), 2.84 (dd, 1H, J =13.9, 5.9 Hz), 1.27 (d, 3H, J = 7.0 Hz), 0.96–1.01 (m, 2H), 0.02 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.1, 171.0, 169.5, 156.6, 155.9, 136.2, 136.1, 129.3, 128.6, 128.4, 128.1, 127.9, 127.0, 91.7, 67.0, 63.7, 58.1, 57.9, 56.0, 52.5, 52.2, 43.3, 38.1, 32.8, 19.2, 17.6, −1.6; IR (neat) 3314, 3030, 2953, 1721, 1519, 1454, 1338, 1249, 1215, 1155, 1083, 1048, 931, 860, 837, 750, 699 cm−1; HRMS [ESI] m/z calcd for C33H47N3O10NaSSi [M+Na]+ 728.2644, found 728.2650.
To a solution of the MOM ester 6a (2.88 g, 4.08 mmol, 1.0 equiv) in 1,4-dioxane (30 mL) was added 4 M HCl/1,4-dioxane (10 mL) at 0 °C under argon atmosphere. After being stirred at room temperature for 1 h, the reaction mixture was concentrated in vacuo. The resulting residue was purified by column chromatography on silica gel (eluted with CH2Cl2/MeOH = 50:1) to afford the carboxylic acid 1a (2.35 g, 3.55 mmol, 87%) as a white amorphous solid. [α]23D −13 (c 1.1, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 12.9 (brs, 1H), 8.38 (d, 1H, J = 7.7 Hz), 7.46 (d, 1H, J = 8.7 Hz), 7.19–7.31 (m, 11 H), 4.93 (s, 2H), 4,25–4.43 (m, 3H), 4.03–4.05 (m, 2H), 3.64 (s, 3H), 3.25–3.31 (m, 1H), 2.96–3.03 (m, 2H), 2.70–2.80 (m, 2H), 1.21 (d, 3H, J = 6.8 Hz), 0.90–0.92 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 171.7, 171.6, 170.8, 156.2, 155.7, 138.0, 136.9, 129.2, 128.2, 128.0, 127.6, 127.3, 126.2, 65.1, 62.2, 58.5, 55.9, 52.2, 51.9, 41.8, 37.4, 32.1, 18.7, 17.3, −1.5; IR (neat) 3315, 3064,3030, 2953, 1721, 1518, 1454, 1439, 1340, 1287, 1250, 1215, 1180, 1081, 1050, 860, 837, 753, 698 cm−1; HRMS [ESI] m/z calcd for C31H43N3O9NaSSi [M+Na]+ 684.2381, found 684.2391.
3.3.2. The Tripeptide 1b
Compound 6b was prepared from the N-Fmoc-amine 5 (248 mg, 383 µmol) according to the procedure above described for 6a, and obtained in 69% yield (178 mg, 265 µmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1). [α]22D −17 (c 1.3, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.18–7.28 (m, 5H), 6.75 (d, 1H, J = 7.2 Hz), 5.40 (d, 1H, J = 8.5 Hz), 5.26 (d, 1H, J = 5.8 Hz), 5.22 (d, 1H, J = 5.8 Hz), 5.01–5.03 (m, 1H), 4.69–4.71 (m, 1H), 4.39–4.46 (m, 2H), 4.13–4.15 (m, 2H), 3.74 (s, 3H), 3.44 (s, 3H), 3.35–3.37 (m, 1H), 3.11 (dd, 1H, J = 14.2, 5.8 Hz), 2.99–3.02 (m, 2H), 2.82 (dd, 1H, J = 13.8, 6.3 Hz), 1.36 (s, 9H), 1.26 (d, 3H, J = 7.2 Hz), 0.95–0.97 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.4, 171.1, 169.6, 156.7, 155.3, 136.5, 129.3, 128.7, 127.0, 91.7, 80.3, 63.7, 58.1, 58.0, 55.8, 52.5, 52.3, 43.5, 38.1, 33.1, 28.2, 19.3, 17.7, −1.5; IR (neat) 3317, 2953, 1719, 1510, 1454, 1366, 1339, 1249, 1210, 1168, 1086, 1048, 933, 860, 837, 776, 699 cm−1; HRMS[ESI] m/z calcd for C30H49N3O10NaSSi [M+Na]+ 650.2800, found 650.2819.
To a solution of the MOM ester 6b (158 mg, 235 μmol, 1.0 equiv) in 1,4-dioxane (4.20 mL) was added 4 M HCl/1,4-dioxane (0.6 mL) at 0 °C under argon atmosphere. After being stirred at room temperature for 2.5 h, the reaction mixture was concentrated in vacuo. The resulting residue was purified by column chromatography on silica gel (eluted with CH2Cl2/MeOH = 10:1) to afford the carboxylic acid 1b (109 mg, 173 μmol, 74%) as a white amorphous solid. [α]21D −12 (c 1.0, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, 1H, J = 7.2 Hz), 7.20–7.36 (m, 5H), 7.14–7.20 (m, 1H), 6.84 (d, 1H, J = 8.5 Hz), 4.36–4.45 (m, 1H), 4.16–4.35 (m, 2H), 3.99–4.09 (m, 2H), 3.64 (s, 3H), 3.19–3.28 (m, 1H), 2.90–3.06 (m, 2H), 2.66–2.83 (m, 2H), 1.27 (s, 9H), 1.21 (d, 3H, J = 6.5 Hz), 0.89–0.94 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 171.7, 170.8, 156.2, 155.1, 138.0, 129.2, 127.9, 126.1, 78.0, 66.2, 58.4, 55.6, 52.0, 51.9, 41.8, 37.4, 32.2, 28.1, 18.7, 17.3, –1.5; IR (neat) 3320, 2954, 1721, 1512, 1453, 1367, 1339, 1249, 1170, 1080, 1049, 859, 837, 699 cm−1; HRMS[ESI] m/z calcd for C28H45N3O9NaSSi [M+Na]+ 650.2538, found 650.2546.
3.3.3. The Tripeptide 1c
Compound 6c was prepared from the N-Fmoc-amine 5 (367 mg, 568 μmol) according to the procedure above described for 6a, and obtained in 86% yield (388 mg, 488 μmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1). [α]20D −28 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.73 (d, 2H, J = 7.5 Hz), 7.50 (t, 2H, J = 7.5 Hz), 7.37 (t, 2H, J = 7.5 Hz), 7.21–7.29 (m, 7H), 6.70–6.82 (m, 1H), 5.44 (d, 2H, J = 8.5 Hz), 5.23–5.26 (m, 2H), 4.71–4.72 (m, 1H), 4.46–4.48 (m, 2H), 4.39–4.42 Hz (m, 1H), 4.27–4.30 (m, 1H), 4.13–4.17 (m, 3H), 3.71 (s, 3H), 3.43 (s, 3H), 3.34–3.40 (m, 1H), 2.98–3.10 Hz (m, 3H), 2.81–2.85 (m, 1H), 1.24 (d, 3H, J = 7.1 Hz), 0.95–0.97 (m, 2H), −0.01 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.1, 171.0, 169.5, 156.6, 155.9, 143.71, 143.66, 141.2, 136.2, 129.3, 128.7, 127.7, 127.0, 125.0, 119.9, 91.7, 77.2, 67.1, 63.7, 58.0, 52.5, 52.2, 47.0, 43.3, 38.2, 32.8, 29.2, 19.2, 17.6, −1.6; IR (neat) 3310, 3064, 3025, 2953, 1719, 1670, 1517, 1450, 1412, 1381, 1338, 1287, 1249, 1217, 1154, 1084, 1047, 932, 860, 837, 757, 742, 700 cm−1; HRMS[ESI] m/z calcd for C40H51N3O10NaSSi [M+Na]+ 816.2957, found 816.2960.
Compound 1c was prepared from the MOM ester 6c (340 mg, 428 µmol) according to the procedure above described for 1a, and obtained in 85% yield (272 mg, 362 µmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with CH2Cl2/MeOH = 50:1). [α]24D −14 (c 0.96, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 8.37 (d, 1H, J = 7.5 Hz), 7.87 (d, 2H, J = 7.5 Hz), 7.60–7.64 (m, 3H), 7.16–7.42 (m, 10H), 4.25–4.40 (m, 3H), 4.09–4.16 (m, 3H), 4.03–4.05 (m, 2H), 3.64 (s, 3H), 3.25–3.26 (m, 1H), 2.95–3.03 (m, 2H), 2.78–2.80 (m, 2H), 1.19 (d, 3H, J = 7.0 Hz), 0.90–0.92 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 171.8, 171.5, 170.8, 156.2, 155.6, 143.7, 143.6, 140.6, 138.0, 129.2, 127.9, 127.5, 127.0, 126.1, 125.23, 125.17, 120.0, 65.6, 62.2, 58.4, 56.0, 52.3, 51.8, 46.5, 41.7, 37.4, 32.2, 18.7, 17.3, −1.6; IR (neat) 3313, 3064, 3028, 2953, 1721, 1516, 1450, 1338, 1249, 1216, 1180, 1081, 1048, 859, 837, 757, 742, 699 cm−1; HRMS [ESI] m/z calcd for C38H47N3O9NaSSi [M+Na]+ 772.2694, found 772.2715.
3.3.4. The Tripeptide 1d
Compound 6d was prepared from the N-Fmoc-amine 5 (480 mg, 742 μmol) according to the procedure above described for 6a, and obtained in 75% yield (344 mg, 558 μmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1 to hexane/acetone = 3:1). [α]23D −16 (c 1.1, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.25–7.32 (m, 5H), 7.01 (s, 1H), 5.67 (s, 1H), 5.48 (d, 1H, J = 9.2 Hz), 5.28 (d, 1H, J = 5.8 Hz), 5.23 (d, 1H, J = 5.8 Hz), 5.10 (s, 2H), 4.77 (dt, 1H, J = 6.3, 5.6 Hz), 4.48 (dd, 1H, J = 8.8, 3.0 Hz), 4.12–4.15 (m, 2H), 3.92 (d, 2H, J = 5.6 Hz), 3.72 (s, 3H), 3.36–3.44 (m, 4H), 3.03 (dd, 1H, J = 13.6, 4.0 Hz), 2.86 (dd, 1H, J = 13.9, 6.2 Hz), 1.27 (d, 3H, J = 7.2 Hz), 0.95–0.97 (m, 2H), −0.01 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.1, 169.8, 169.2, 156.6, 156.5, 136.1, 128.5, 128.1, 128.0, 91.7, 77.2, 67.1, 63.7, 57.9, 52.5, 52.1, 44.4, 43.3, 32.7, 19.1, 17.6, −1.6; IR (neat) 3325, 2953, 1723, 1515, 1453, 1381, 1339, 1249, 1156, 1088, 1048, 927, 860, 837, 754, 698 cm−1; HRMS[ESI] m/z calcd for C26H41N3O10NaSSi [M+Na]+ 638.2174, found 638.2180.
Compound 1d was prepared from the MOM ester 6d (215 mg, 349 µmol) according to the procedure above described for 1a, and obtained in 45% yield (89.6 mg, 157 µmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with CH2Cl2/MeOH = 50:1 to 10:1). [α]24D +2.7 (c 0.95, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 12.9 (brs, 1H), 8.18 (d, 1H, J = 8.0 H), 7.30–7.44 (m, 7H), 5.02 (s, 2H), 4.37–4.40 (m, 1H), 4.23 (dd, 1H, J = 8.0, 6.0 Hz), 4.03–4.05 (m, 2H), 3.66 (d, 2H, J = 6.3 Hz), 3.63 (s, 3H), 3.18–3.20 (m, 1H), 2.91 (dd, 1H, J = 13.5, 5.1 Hz), 2.72 (dd, 1H, J = 13.5, 8.1 Hz), 1.18 (d, 3H, J = 6.8 Hz), 0.90–0.93 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 171.7, 170.7, 169.0, 156.4, 156.2, 137.0, 128.2, 127.7, 127.6, 65.4, 62.2, 58.4, 51.9, 51.8, 43.2, 41.8, 32.3, 18.6, 17.3, −1.6; IR (neat) 3327, 2953, 1722, 1523, 1453, 1438, 1340, 1249, 1081, 1050, 860, 837, 697 cm−1; HRMS [ESI] m/z calcd for C24H37N3O9NaSSi [M+Na]+ 594.1912, found 594.1924.
3.3.5. The Tripeptide 1e
To a solution of N-Fmoc-amine 5 (359 mg, 554 μmol, 1.0 equiv) in dry MeCN (4.4 mL) was added Et2NH (1.1 mL) at room temperature under an argon atmosphere. After being stirred at the same temperature for 1.5 h, the reaction mixture was concentrated in vacuo. The resulting residue was azeotroped three times with MeCN, and the resulting crude amine was used for the next reaction without further purification.
To a solution of the crude amine in dry CH2Cl2 (5.5 mL) were added DIEA (193 μL, 1.11 mmol, 2.0 equiv), Cbz-Ile-OH (177 mg, 665 μmol, 1.2 equiv) and HATU (253 mg, 665 μmol, 1.2 equiv) at 0 °C under an argon atmosphere. After being stirred at room temperature for 7 h, the reaction mixture was diluted with CH2Cl2. The organic layer was washed with 10% aqueous citric acid and saturated aqueous NaHCO3, dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1) to afford the tripeptide 6e (373 mg, 539 mmol, 97% in 2 steps) as a yellowish amorphous solid. [α]23D −19 (c 0.98, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.29–7.34 (m, 5H), 6.82 (d, 1H, J = 6.5 Hz), 5.51 (d, 1H, J = 9.2 Hz), 5.43 (d, 1H, J = 8.7 Hz), 5.32 (d, 1H, J = 5.8 Hz), 5.27 (d, 1H, J = 5.8 Hz), 5.11 (s, 2H), 4.78–4.80 (m, 1H), 4.52 (dd, 1H, J = 9.3, 3.0 Hz), 4.14–4.17 (m, 3H), 3.75 (s, 3H), 3.48 (s, 3H), 3.37–3.48 (m, 1H), 3.04 (dd, 1H, J = 13.8, 4.3 Hz), 2.89 (dd, 1H, J = 13.8, 6.0 Hz), 1.89–1.91 (m, 1H), 1.60–1.70 (m, 1H) 1.30 (d, 3H, J = 7.0 Hz), 1.07–1.26 (m, 1H), 0.90–1.01 (m, 8H), 0.03 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ 171.4, 171.2, 169.7, 156.7, 156.2, 136.2, 128.5, 128.1, 128.0, 91.7, 67.1, 63.7, 59.6, 58.1, 58.0, 52.5, 52.0, 43.5, 37.4, 32.9, 24.6, 19.3, 17.6, 15.4, 11.3, −1.6; IR (neat) 3311, 2959, 1723, 1666, 1524, 1454, 1382, 1339, 1284, 1248, 1156, 1087, 1045, 932, 860, 837, 697 cm−1; HRMS[ESI] m/z calcd for C30H49N3O10NaSSi [M+Na]+ 694.2800, found 694.2814.
Compound 1e was prepared from the MOM ester 6e (373 mg, 539 µmol) according to the procedure above described for 1a, and obtained in 67% yield (228 mg, 362 µmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with CH2Cl2/MeCN = 3:1 to CH2Cl2/MeOH = 20:1). [α]21D −6.5 (c 0.99, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 12.8 (brs, 1H), 8.21 (d, 1H, J = 7.7 Hz), 7.24–7.33 (m, 7H), 5.02 (s, 2H), 4.35–4.38 (m, 1H), 4.25 (dd, 1H, J = 7.4, 5.7 Hz), 4.03–4.05 (m, 2H), 3.91–3.99 (m, 1H), 3.62–3.64 (m, 3H), 3.21 (s, 1H), 2.92 (dd, 1H, J = 13.0, 5.1 Hz), 2.72 (dd, 1H, J = 13.0, 8.2 Hz), 1.63–1.82 (m, 1H), 1.31–1.49 (m, 1H), 1.03–1.23 (m, 4H), 0.72–0.96 (m, 8H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 171.7, 171.2, 170.8, 156.2, 155. 9, 137.0, 128.2, 127.6, 127.5, 65.3, 62.2, 59.0, 58.4, 51.90, 51.85, 41.6, 36.6, 32.1, 24.2, 18.6, 17.3, 15.2, 10.8, −1.5; IR (neat) 3316, 2959, 1721, 1666, 1517, 1454, 1340, 1286, 1249, 1216, 1179, 1082, 1045, 859, 837 cm−1; HRMS [ESI] m/z calcd for C28H45N3O9NaSSi [M+Na]+ 650.2538, found 650.2551.
3.3.6. The Tripeptide 1f
Compound 6f was prepared from the N-Fmoc-amine 5 (268 mg, 414 μmol) according to the procedure above described for 6a, and obtained in 74% yield (242 mg, 307 μmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with hexane/EtOAc = 1:1). [α]22D −21 (c 1.2, CHCl3); 1H NMR (400 MHz, CDCl3) δ 7.25–7.32 (m, 5H), 6.92–6.94 (m, 1H), 5.47–5.49 (m, 2H), 5.30 (d, 1H, J = 5.8 Hz), 5.23 (d, 1H, J = 5.8 Hz), 5.08 (s, 2H), 4.74 (dt, 1H, J = 6.8, 5.4 Hz), 4.65 (brs, 1H), 4.48 (dd, 1H, J = 8.7, 2.4 Hz), 4.21–4.22 (m, 1H), 4.13–4.15 (m, 2H), 3.73 (s, 3H), 3.37–3.43 (m, 4H), 3.02–3.04 (m, 3H), 2.86 (dd, 1H, J = 13.2, 5.9 Hz), 1.25–1.85 (m, 18H), 0.95–0.97 (m, 2H), −0.01 (s, 9H); 13C{1H} NMR (100 MHz, CDCl3) δ171.8, 171.2, 169.7, 156.7, 156.2, 156.1, 136.2, 128.4, 128.1, 128.0, 91.7, 79.0, 67.0, 63.7, 58.1, 57.9, 54.7, 52.5, 52.1, 43.3, 39.8, 32.7, 31.9, 29.6, 28.4, 22.3, 19.2, 17.6, −1.6; IR (neat) 3319. 2952, 1714, 1511, 1454, 1365, 1339, 1249, 1169, 1086, 1046, 860, 837 cm−1; HRMS[ESI] m/z calcd for C35H58N4O12NaSSi [M+Na]+ 809.3433, found 809.3434.
Compound 1f was prepared from the MOM ester 6f (215 mg, 279 µmol) according to the procedure above described for 1b, and obtained in 87% yield (180 mg, 242 µmol) as a white amorphous solid after purification by column chromatography on silica gel (eluted with CH2Cl2/MeCN = 1:1 to CH2Cl2/MeOH = 10:1). [α]24D −13 (c 1.1, CHCl3); 1H NMR (400 MHz, DMSO-d6) δ 7.91 (d, 1H, J = 7.2 Hz), 7.28–7.32 (m, 5H), 7.11–7.12 (m, 1H), 6.90–6.91 (m, 1H), 6.47–6.48 (m, 1H), 5.02 (s, 2H), 4.32–4.37 (m, 1H), 4.23 (dd, 1H, J = 8.1, 5.9 Hz), 3.99–4.06 (m, 3H), 3.63 (s, 3H), 3.19–3.25 (m, 1H), 2.93–3.02 (m, 3H), 2,74 (dd, 1H, J = 13.4, 7.6 Hz), 1.61–1.64 (m, 1H), 1.51–1.53 (m, 1H), 1.13–1.37 (m, 16H), 0.90–0.93 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (100 MHz, DMSO-d6) δ 171.8, 171.7, 170.7, 156.1, 155.8, 155.4, 136.9, 128.2, 127.6, 127.5, 66.2, 65.3, 62.1, 58.4, 54.5, 52.0, 51.7, 41.6, 32.2, 31.6, 29.1, 28.1, 27.8, 22.6, 18.6, 17.2, −1.6; IR (neat) 3325, 2953, 1713, 1515, 1453, 1411, 1391, 1366, 1340, 1249, 1214, 1172, 1081, 1047, 860, 837, 754, 697 cm−1; HRMS [ESI] m/z calcd for C33H54N4O11NaSSi [M+Na]+ 765.3171, found 765.3179.
3.4. The Photocatalytic AviMeCys Formation Using 1
3.4.1. The β-Thioenamides (Z)-9a and (E)-9a
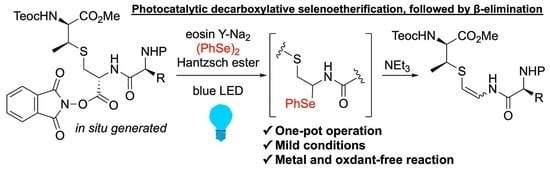
To a solution of the carboxylic acid 6a (66.6 mg, 100 µmol, 1.0 equiv) and N-hydroxyphthalimide (18.1 mg, 110 µmol, 1.1 equiv) in dry CH2Cl2 (1.0 mL) was added DIC (17.3 µL, 110 µmol, 1.1 equiv) at room temperature under an argon atmosphere, and the mixture was stirred at the same temperature for 30 min. After complete consumption of 6a (monitored by TLC analysis), the reaction mixture was cooled to −40 °C. A solution of eosin Y-Na2 (0.7 mg, 1.00 μmol, 1 mol%), Hantzsch ester (25.5 mg, 100 µmol, 1.0 equiv) and diphenyl diselenide (62.8 mg, 200 µmol, 2.0 equiv) in dry DMF (1.5 mL, used immediately after freeze-pump-thaw cycling) was then added to the above mixture at −40 °C. After being stirred at the same temperature for 30 min under irradiated Blue LEDs, Et3N (250 μL, 1.79 mmol, 18 equiv) was added to the solution at −40 °C. After being stirred at room temperature for 3 h, the reaction mixture was quenched with saturated aqueous NaHCO3, and stirred for 12 h. The aqueous layer was extracted three times with Et2O. The combined organic layers were dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by preparative TLC (eluted with hexane/EtOAc = 7:2) to afford the β-thioenamide (Z)-9a (26.2 mg, 42.5 µmol, 42%) as a white amorphous solid and the β-thioenamide (E)-9a (19.6 mg, 31.8 µmol, 32%) as a white amorphous solid. (Z)-9a: [α]24D −41 (c 1.1, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.42 (d, 1H, J = 10.6 Hz), 7.19–7.34 (m, 10H), 7.15 (dd, 1H, J = 10.6, 7.2 Hz), 5.49–5.50 (m, 1H), 5.40–5.41 (m, 1H), 5.32 (d, 1H, J = 7.2 Hz), 5.07–5.09 (m, 2H), 4.56–4.58 (m, 1H), 4.51 (dd, 1H, J = 9.2, 3.9 Hz), 4.14–4.15 (m, 2H), 3.65 (s, 3H), 3.31–3.32 (m, 1H), 3.19 (dd, 1H, J = 13.9, 6.2 Hz), 3.09–3.11 (m, 1H), 1.32 (d, 3H, J = 7.2 Hz), 0.95–0.97 (m, 2H), 0.00 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.3, 168.9, 156.8, 156.2, 136.2, 136.1, 129.7, 129.3, 128.9, 128.6, 128.3, 128.2, 127.2, 99.7, 67.3, 63.9, 58.9, 56.4, 52.6, 45.8, 38.1, 18.3, 17.8, −1.4; IR (neat) 3310, 3064, 3031, 2952, 1697, 1628, 1498, 1455, 1380, 1337, 1248, 1178, 1080, 1046, 860, 837, 742, 698 cm−1; HRMS[ESI] m/z calcd for C30H41N3O7NaSSi [M+Na]+ 638.2327, found 638.2330. (E)-9a: [α]23D −3.5 (c 0.89, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.01 (m, 1H), 7.14–7.32 (m, 10H), 6.99 (dd, 1H, J = 13.7, 6.6 Hz), 5.59 (d, 1H, J = 13.7 Hz), 5.42–5.44 (m, 2H), 5.05 (s, 2H), 4.48 (dd, 1H, J = 5.5, 2.7 Hz), 4.40–4.42 (m, 1H), 4.13–4.17 (m, 2H), 3.64 (s, 3H), 3.34–3.38 (m, 1H), 3.06 (d, 2H, J = 6.8 Hz), 1.29 (d, 3H, J = 7.5 Hz), 1.00–1.01 (m, 2H), 0.03 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.2, 168.3, 156.8, 156.3, 136.0, 135.9, 129.4, 129.3, 129.0, 128.7, 128.5, 128.2, 127.4, 104.1, 67.5, 63.9, 57.9, 56.4, 52.5, 45.3, 38.2, 18.6, 17.8, −1.4; IR (neat) 3303, 2953, 1725, 1688, 1669, 1629, 1505, 1454, 1341, 1288, 1261, 1246, 1209, 1176, 1079, 1046, 938, 862, 835, 750, 697 cm−1; HRMS[ESI] m/z calcd for C30H41N3O7NaSSi [M+Na]+ 638.2327, found 638.2334.
3.4.2. The β-Thioenamides (Z)-9b and (E)-9b
Compounds (Z)-9b and (E)-9b were prepared from the carboxylic acid 6b (63.7 mg, 101 μmol) according to the procedure above described for 9a, and purified by preparative TLC (eluted with hexane/EtOAc = 7:2, hexane/IPA = 20:1) to be obtained in 32% yield (18.6 mg, 32.0 μmol) as a white amorphous solid and 32% yield (18.9 mg, 32.5 μmol) as a white amorphous solid, respectively. (Z)-9b: [α]24D −49 (c 1.2, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.42 (d, 1H, J = 10.9 Hz), 7.28–7.30 (m, 2H), 7.22–7.24 (m, 3H), 7.12–7.15 (m, 1H), 5.45–5.47 (m, 1H), 5.28–5.29 (m, 1H), 5.14–5.16 (m, 1H), 4.52 (d, 2H, J = 10.3 Hz), 4.14–4.17 (m, 2H), 3.70 (s, 3H), 3.34–3.36 (m, 1H), 3.18 (dd, 1H, J = 14.0, 5.8 Hz), 3.04–3.06 (m, 1H), 1.39 (s, 9H), 1.32 (d, 3H, J = 6.8 Hz), 0.98–0.99 (m, 2H), 0.01 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.3, 169.3, 156.8, 155.6, 136.5, 129.5, 129.3, 128.8, 127.1, 99.6, 80.6, 63.9, 58.7, 55.8, 52.6, 45.8, 37.9, 28.3, 18.4, 17.8, −1.4; IR (neat) 3324, 2953, 1690, 1627, 1499, 1366, 1338, 1285, 1249, 1171, 1080, 1047, 860, 837, 754, 699 cm−1; HRMS [ESI] m/z calcd for C27H43N3NaO7SSi [M+Na]+ 604.2483, found 604.2490. (E)-9b: [α]24D −6.8 (c 1.1, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.04–8.05 (m, 1H), 7.28 (t, 2H, J = 7.5 Hz), 7.22 (t, 1H, J = 7.5 Hz), 7.15 (d, 2H, J = 7.5 Hz), 7.00 (dd, 1H, J = 13.7, 10.9 Hz), 5.59 (d, 1H, J = 13.7 Hz), 5.42 (d, 1H, J = 6.0 Hz), 5.06 (d, 1H, J = 8.2 Hz), 4.47 (dd, 1H, J = 9.2, 3.1 Hz), 4.33 (s, 1H), 4.15–4.16 (m, 2H), 3.65 (s, 3H), 3.37–3.38 (m, 1H), 3.05–3.06 (m, 1H), 2.99–3.00 (m, 1H), 1.37 (s, 9H), 1.28 (d, 3H, J = 6.8 Hz), 0.98–1.00 (m, 2H), 0.04 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.2, 168.7, 156.8, 155.8, 136.3, 129.6, 129.3, 128.8, 127.2, 103.6, 80.8, 63.5, 57.9, 56.0, 52.5, 45.3, 38.0, 28.3, 18.5, 17.8, −1.4; IR (neat) 3310, 2954, 1723, 1681, 1628, 1513, 1454, 1391, 1367, 1339, 1289, 1248, 1211, 1169, 1079, 1047, 941, 860, 837, 754, 698 cm−1; HRMS [ESI] m/z calcd for C27H43N3NaO7SSi [M+Na]+ 604.2483, found 604.2487.
3.4.3. The β-Thioenamides (Z)-9c and (E)-9c
To a solution of the carboxylic acid 6c (75.2 mg, 100 µmol, 1.0 equiv) and N-hydroxyphthalimide (18.0 mg, 110 µmol, 1.1 equiv) in dry CH2Cl2 (1.0 mL) was added DIC (17.3 μL, 110 µmol, 1.1 equiv) at room temperature under an argon atmosphere, and the mixture was stirred at the same temperature for 5 h. After complete consumption of 5c (monitored by TLC analysis), the reaction mixture was cooled to −40 °C. A solution of Eosin Y (696 μg, 1.00 μmol, 1 mol%), Hantzsch ester (25.4 mg, 100 µmol, 1.0 equiv) and diphenyl diselenide (62.6 mg, 200 µmol, 2.0 equiv) in dry DMF (1.5 mL, used immediately after Freeze-Pump-Thaw cycling) was then added to the above mixture at −40 °C. After being stirred at the same temperature for 30 min under irradiated Blue LEDs, Et3N (41.9 μL, 300 μmol, 3.0 equiv) was added to the solution at −40 °C. After being stirred at room temperature for 3 h, the reaction mixture was quenched with saturated aqueous NaHCO3, diluted with Et2O, and stirred for 12 h. The organic layer was separated, and the aqueous layer was extracted three times with Et2O. The combined organic layers were dried over Na2SO4, and filtered. The filtrate was concentrated in vacuo, and the resulting residue was purified by preparative TLC (eluted with hexane/EtOAc = 7:2, hexane/IPA = 10:1) to afford the β-thioenamide (Z)-9c (14.6 mg, 20.7 µmol, 21%) as a white amorphous solid and the β-thioenamide (E)-9c (11.1 mg, 15.8 µmol, 16%) as a white amorphous solid. (Z)-9c: [α]24D −25 (c 0.83, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.41–8.42 (m, 1H), 7.75 (d, 2H, J = 7.5 Hz), 7.49–7.52 (m, 2H), 7.39 (t, 2H, J = 7.5 Hz), 7.25–7.28 (m, 7H), 7.17 (dd, 1H, J = 10.8, 7.6 Hz) 5.53–5.55 (m, 1H), 5.38–5.40 (m, 1H), 5.33 (d, 1H, J = 4.6 Hz), 4.59–4.61 (m, 1H), 4.50–4.52 (m, 1H), 4.42 (dd, 1H, J = 10.8, 7.5 Hz), 4.34–4.35 (m, 1H), 4.12–4.18 (m, 3H), 3.68 (s, 3H), 3.31–3.33 (m, 1H), 3.12–3.19 (m, 2H), 1.32 (d, 3H, J = 6.8 Hz), 0.93–0.95 (m, 2H), 0.01 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.4, 168.9, 156.8, 156.2, 143.83, 143.80, 141.4, 136.2, 129.8, 129.4, 129.0, 127.9, 127.4, 127.2, 125.2, 120.1, 99.8, 67.4, 64.0, 59.0, 56.4, 52.7, 47.2, 45.9, 38.3, 18.3, 17.8, −1.4; IR (neat) 3308, 3064, 3028, 2952, 1696, 1628, 1499, 1451, 1336, 1248, 1178, 1080, 1045, 859, 837, 757, 740, 699 cm−1; HRMS [ESI] m/z calcd for C37H45N3O7NaSSi [M+Na]+ 726.2640, found 726.2649. (E)-9c: [α]24D −16 (c 0.59, CHCl3); 1H NMR (600 MHz, CDCl3) δ 7.76 (d, 2H, J = 7.5 Hz), 7.50 (t, 2H, J = 7.5 Hz), 7.40 (t, 2H, J = 7.5 Hz), 7.25–7.29 (m, 8H), 6.95–6.99 (m, 1H), 5.58 (d, 1H, J = 13.7 Hz), 5.39 (d, 1H, J = 9.6 Hz), 5.26–5.28 (m, 1H), 4.45–4.47 (m, 2H), 4.34–4.36 (m, 2H), 4.15–4.17 (m, 3H), 3.63 (s, 3H), 3.36–3.37 (m, 1H), 3.06–3.07 (m, 2H), 1.28 (d, 3H, J = 6.8 Hz), 0.99–1.00 (m, 2H), 0.04 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.1, 168.0, 156.7, 156.3, 143.6, 141.4, 136.0, 129.3, 129.0, 127.9, 127.4, 127.2, 125.0, 120.1, 104.2, 67.3, 63.8, 57.9, 56.3, 52.5, 47.1, 45.2, 38.0, 18.5, 17.8, −1.4; IR (neat) 3316, 3064, 3027, 2950, 1691, 1626, 1509, 1450, 1338, 1289, 1247, 1216, 1079, 1045, 937, 859, 837, 757, 739, 700 cm−1; HRMS [ESI] m/z calcd for C37H45N3O7NaSSi [M+Na]+ 726.2640, found 726.2643.
3.4.4. The β-Thioenamides (Z)-9d and (E)-9d
Compounds (Z)-9d and (E)-9d were prepared from the carboxylic acid 6d (57.1 mg, 99.9 μmol) according to the procedure above described for 9a, and purified by preparative TLC (eluted with hexane/EtOAc = 7:2, toluene/acetone = 8:1, hexane/IPA = 10:1) to be obtained in 39% yield (20.2 mg, 38.4 μmol) as a white amorphous solid and 19% yield (9.9 mg, 19 μmol) as a white amorphous solid, respectively. (Z)-9d: [α]23D +3.0 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.57–8.59 (m, 1H), 7.25–7.34 (m, 5H), 7.14 (dd, 1H, J = 10.9, 7.5 Hz), 5.75–5.77 (m, 1H), 5.59–5.61 (m, 1H), 5.32 (d, 1H, J = 7.5 Hz), 5.18 (d, 1H, J = 12.3 Hz), 5.14 (d, 1H, J = 12.3 Hz), 4.54 (dd, 1H, J = 8.9, 2.7 Hz), 4.12–4.14 (m, 2H), 3.98 (d, 2H, J = 5.5 Hz), 3.64 (s, 3H), 3.35–3.37 (m, 1H), 1.34 (d, 3H, J = 6.8 Hz), 0.93–0.95 (m, 2H), −0.01 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.3, 167.1, 156.9, 156.8, 136.1, 129.3, 128.6, 128.4, 128.2, 100.1, 67.5, 63.9, 58.7, 52.5, 46.4, 44.9, 18.5, 17.8, −1.4; IR (neat) 3320, 2952, 1700, 1629, 1517, 1338, 1248, 1176, 1081, 1046, 860, 837, 738, 697 cm−1; HRMS [ESI] m/z calcd for C23H35N3O7NaSSi [M+Na]+ 548.1857, found 548.1864. (E)-9d: [α]23D +5.0 (c 0.50, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.31–8.32 (m, 1H), 7.31–7.36 (m, 5H), 7.05 (dd, 1H, J = 13.7, 10.9 Hz), 5.72 (d, 1H, J = 13.7 Hz), 5.55–5.56 (m, 1H), 5.45 (d, 1H, J = 8.9 Hz), 5.12 (s, 2H), 4.48 (dd, 1H, J = 8.9, 3.4 Hz), 4.15–4.16 (m, 2H), 3.87 (d, 2H, J = 5.5 Hz), 3.70 (s, 3H), 3.37–3.39 (m, 1H), 1.29 (d, 3H, J = 6.8 Hz), 0.98–1.00 (m, 2H), 0.03, (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.3, 166.4, 156.9, 156.7, 135.9, 129.5, 128.7, 128.5, 128.2, 103.7, 67.6, 63.8, 57.9, 52.6, 45.3, 44.8, 18.4, 17.8, −1.4; IR (neat) 3311, 2953, 1696, 1627, 1512, 1454, 1338, 1248, 1174, 1080, 1047, 860, 837, 697 cm−1; HRMS [ESI] m/z calcd for C23H35N3O7NaSSi [M+Na]+ 548.1857, found 548.1866.
3.4.5. The β-Thioenamides (Z)-9e and (E)-9e
Compounds (Z)-9e and (E)-9e were prepared from the carboxylic acid 6e (62.2 mg, 99.1 μmol) according to the procedure above described for 9a, and purified by preparative TLC (eluted with hexane/EtOAc = 7:2) to be obtained in 36% yield (20.5 mg, 35.2 μmol) as a white amorphous solid and 32% yield (18.3 mg, 31.5 μmol) as a white amorphous solid, respectively. (Z)-9e: [α]24D −45 (c 1.1, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.52 (d, 1H, J = 10.6 Hz), 7.30–7.33 (m, 5H), 7.19 (dd, 1H, J = 10.6, 7.5 Hz), 5.66 (d, 1H, J = 8.2 Hz), 5.41 (d, 1H, J = 10.0 Hz), 5.34 (d, 1H, J = 7.5 Hz), 5.15 (d, 1H, J = 12.3 Hz), 5.10 (d, 1H, J = 12.3 Hz), 4.55 (dd, 1H, J = 10.0, 5.0 Hz), 4.26–4.27 (m, 1H), 4.11–4.15 (m, 2H), 3.61 (s, 3H), 3.33–3.34 (m, 1H), 2.03–2.04 (m, 1H), 1.48–1.49 (m, 1H), 1.37 (d, 3H, J = 6.8 Hz), 1.18–1.24 (m, 1H), 0.97 (d, 3H, J = 7.6 Hz), 0.94–0.97 (m, 2H), 0.91 (t, 3H, J = 7.6 Hz), 0.01 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.5, 169.3, 156.8, 156.5, 136.2, 130.0, 128.6, 128.34, 128.26, 99.1, 67.4, 63.9, 60.1, 59.2, 52.6, 45.6, 37.3, 24.6, 18.2, 17.8, 15.7, 11.6, −1.4; IR (neat) 3314, 3065, 3033, 2959, 1720, 1695, 1628, 1512, 1381, 1337, 1283, 1248, 1178, 1127, 1080, 1043, 938, 860, 837, 773, 738, 696 cm−1; HRMS [ESI] m/z calcd for C27H43N3NaO7SSi [M+Na]+ 604.2483, found 604.2487. (E)-9e: [α]24D +8.8 (c 0.78, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.35–8.37 (m, 1H), 7.30–7.35 (m, 5H), 7.04 (dd, 1H, J = 13.7, 10.3 Hz), 5.71 (d, 1H, J = 13.7 Hz), 5.46–5.47 (m, 2H), 5.10 (d, 1H, J = 12.0 Hz), 5.05 (d, 1H, J = 12.0 Hz), 4.48 (dd, 1H, J = 8.9, 3.4 Hz), 4.14–4.16 (m, 2H), 4.01 (t, 1H, J = 7.9 Hz), 3.65 (s, 3H), 3.40–3.41 (m, 1H), 1.85–1.93 (m, 1H), 1.48–1.50 (m, 1H), 1.29 (d, 3H, J = 6.8 Hz), 1.08–1.11 (m, 1H), 0.98–1.00 (m, 2H), 0.91 (d, 3H, J = 6.8 Hz), 0.87 (t, 3H, J = 7.5 Hz), 0.02 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.3, 168.8, 156.8, 156.7, 136.0, 129.5, 128.7, 128.4, 128.1, 103.9, 67.4, 63.8, 59.9, 57.9, 52.5, 45.4, 37.1, 24.8, 18.6, 17.8, 15.6, 11.3, −1.4; IR (neat) 3297, 2960, 2929, 1747, 1714, 1692, 1664, 1630, 1515, 1455, 1380, 1335, 1283, 1242, 1211, 1176, 1081, 1041, 862, 836, 697 cm−1; HRMS [ESI] m/z calcd for C27H43N3NaO7SSi [M+Na]+ 604.2483, found 604.2485.
3.4.6. The β-Thioenamides (Z)-9f and (E)-9f
Compounds (Z)-9f and (E)-9f were prepared from the carboxylic acid 6f (74.9 mg, 101 μmol) according to the procedure above described for 9a, and purified by preparative TLC (eluted with hexane/EtOAc = 3.5:1 to 2:1, hexane/IPA = 20:1, toluene/acetone = 20:1, hexane/tBuOH = 9:1) to be obtained in 28% yield (19.9 mg, 28.6 μmol) as a white amorphous solid and 16% yield (11.5 mg, 16.5 μmol) as a white amorphous solid, respectively. (Z)-9f: [α]23D −7.6 (c 1.0, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.58 (d, 1H, J = 10.3 Hz), 7.30–7.34 (m, 5H), 7.14 (dd, 1H, J = 10.3, 6.8 Hz), 5.63–5.65 (m, 2H), 5.31 (d, 1H, J = 6.8 Hz), 5.15 (d, 1H, J = 12.3 Hz), 5.10 (d, 1H, J = 12.3 Hz), 4.62–4.61 (m, 1H), 4.55 (dd, 1H, J = 9.6, 3.4 Hz), 4.24–4.25 (m, 1H), 4.12–4.14 (m, 2H), 3.63 (s, 3H), 3.38–3.40 (m, 1H), 3.16–3.18 (m, 1H), 3.07–3.08 (m, 1H), 1.96–1.97 (m, 1H), 1.82–1.83 (m, 1H), 1.69–1.70 (m, 1H), 1.41–1.48 (m, 12H), 1.35 (d, 3H, J = 7.5 Hz), 0.95–0.96 (m, 2H), 0.03 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.4, 169.7, 156.9, 156.6, 156.4, 136.2, 129.6, 128.6, 128.3, 99.9, 79.3, 67.4, 63.8, 58.8, 55.3, 52.5, 46.1, 39.7, 31.4, 29.7, 28.5, 22.5, 18.5, 17.8, −1.4; IR (neat) 3325, 2952, 1698, 1628, 1522, 1365, 1337, 1249, 1172, 1080, 1045, 860, 837, 754, 697 cm−1; HRMS [ESI] m/z calcd for C32H52N4O9NaSSi [M+Na]+ 719.3116, found 719.3137. (E)-9f: [α]23D +0.1 (c 0.58, CHCl3); 1H NMR (600 MHz, CDCl3) δ 8.57–8.58 (m, 1H), 7.30–7.35 (m, 5H), 7.04 (dd, 1H, J = 13.3, 10.6 Hz), 5.71 (d, 1H, J = 13.3 Hz), 5.60–5.62 (m, 1H), 5.44 (d, 1H, J = 10.6 Hz), 5.08 (s, 2H), 4.68–4.70 (m, 1H), 4.47 (dd, 1H, J = 8.9, 3.4 Hz), 4.14–4.16 (m, 3H), 3.66 (s, 3H), 3.38–3.40 (m, 1H), 3.06–3.08 (m, 2H), 1.96–1.98 (m, 1H), 1.83–1.85 (m, 1H), 1.63–1.65 (m, 1H), 1.28–1.48 (m, 3H), 1.40 (s, 9H), 1.29 (d, 3H, J = 6.8 Hz), 0.98–1.00 (m, 2H), 0.02 (s, 9H); 13C{1H} NMR (150 MHz, CDCl3) δ 171.3, 169.3, 156.8, 156.7, 156.5, 136.0, 129.9, 128.6, 128.4, 128.2, 103.5, 79.5, 67.4, 63.8, 57.9, 54.8, 52.5, 45.4, 39.4, 31.3, 29.5, 28.5, 22.3, 18.6, 17.8, −1.4; IR (neat) 3313, 2952, 1694, 1628, 1513, 1454, 1365, 1337, 1248, 1172, 1081, 1045, 860, 837, 754 cm−1; HRMS [ESI] m/z calcd for C32H52N4O9NaSSi [M+Na]+ 719.3116, found 719.3125.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

