
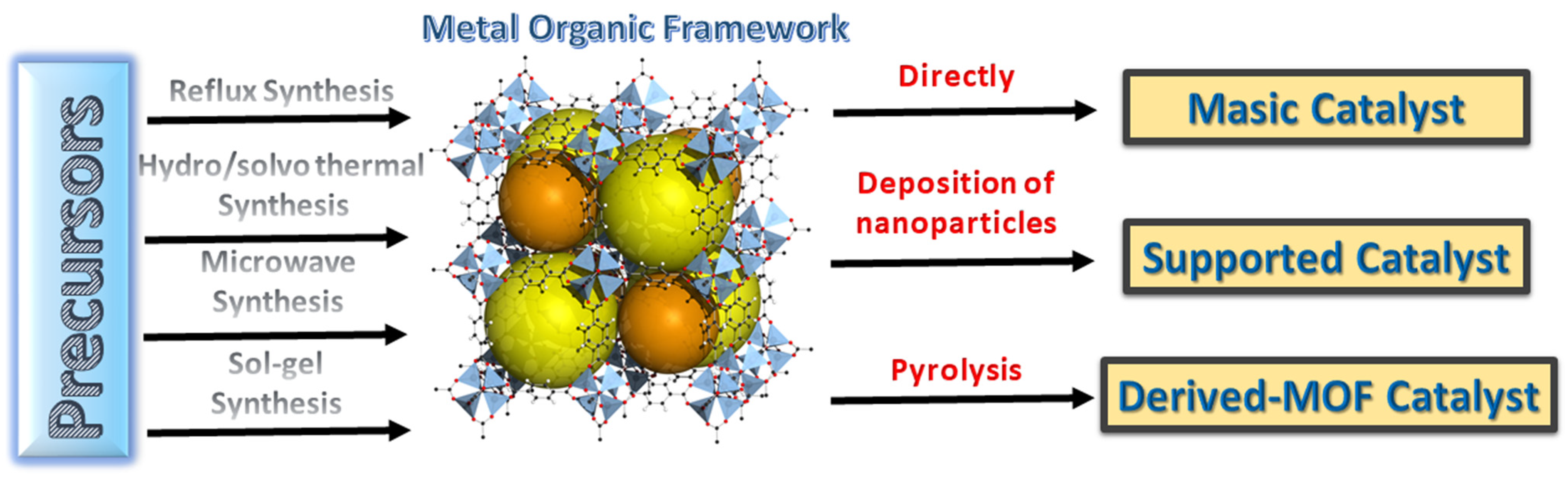
Metal–Organic Frameworks (MOFs) and Materials Derived from MOFs as Catalysts for the Development of Green Processes


Abstract
:1. Introduction
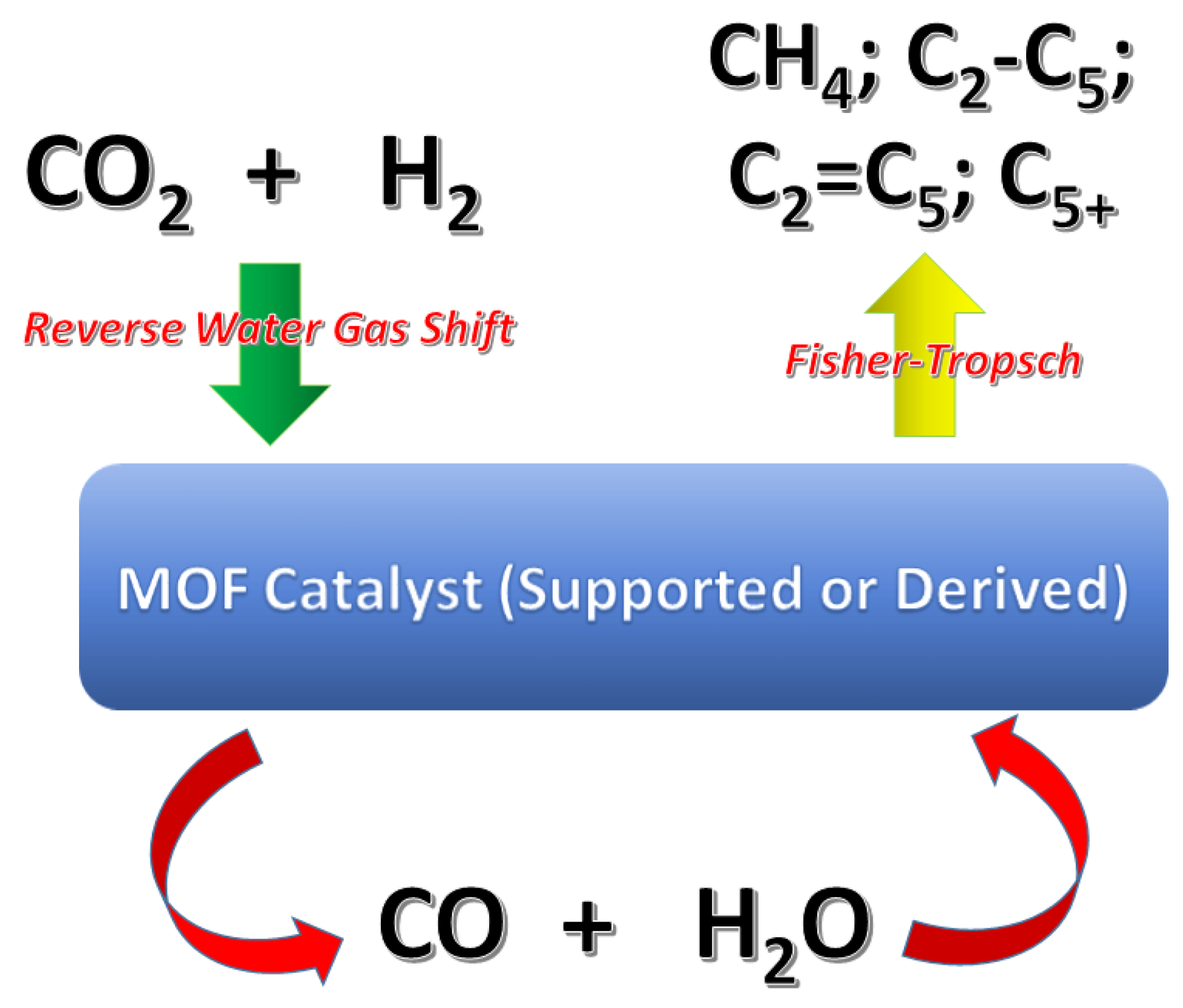
2. MOFs for Conversion of CO2 into Light Hydrocarbons Fuels
2.1. MOFs Used as Supporting Materials for Catalysts
2.2. Catalysts Obtained by Pyrolysis of MOFs
2.3. Perspectives and Conclusions
3. MOFs and Their Application in Hydrogenation and Oxidation Reactions for Biomass Valorisation
3.1. Hydrogenation
3.1.1. Surface Area and Pore Size
3.1.2. Acidity
3.1.3. Metal Nodes
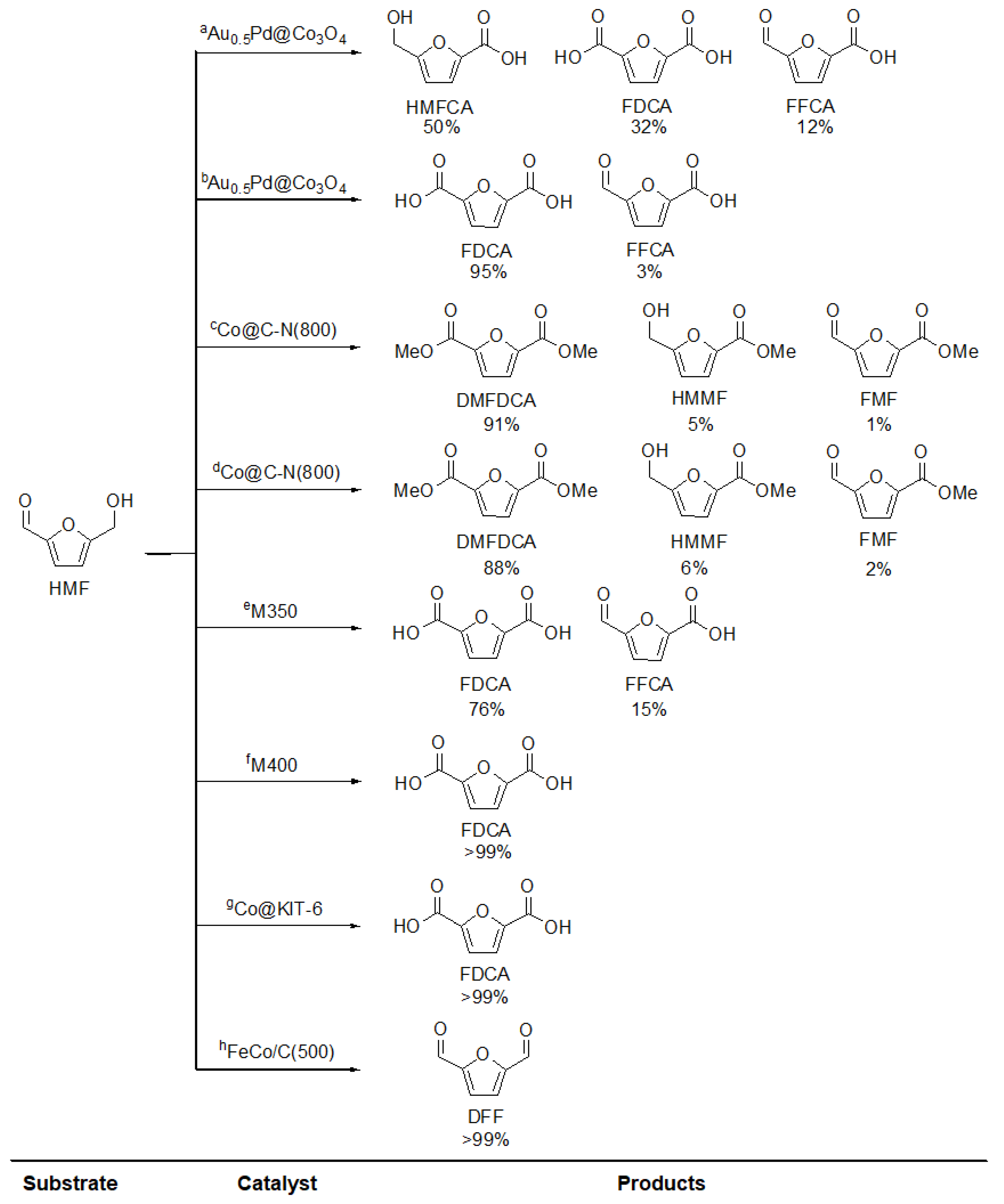
3.2. Oxidation
MOFs as Sacrifice Templates (MOF Derived Catalysts)
3.3. Perspectives and Conclusions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yu, D.; He, L. Introduction to CO2 utilization. Green Chem. 2021, 23, 3499–3501. [Google Scholar] [CrossRef]
- Garba, M.; Usman, M.; Khan, S. CO2 towards fuels: A review of catalytic conversion of carbon dioxide to hydrocarbons. J. Environ. Chem. Eng. 2021, 9, 104756. [Google Scholar] [CrossRef]
- Chen, Z.; Du, S.; Zhang, J.; Wu, X. From ‘Gift’ to gift: Producing organic solvents from CO2. Green Chem. 2020, 22, 8169–8182. [Google Scholar] [CrossRef]
- Hwang, S.-M.; Zhang, C.; Han, J.H.; Park, H.-G.; Kim, Y.T.; Yang, S.; Jun, K.-W.; Kim, S.K. Mesoporous carbon as an effective support for Fe catalyst for CO2 hydrogenation to liquid hydrocarbons. J. CO2 Util. 2020, 37, 65–73. [Google Scholar] [CrossRef]
- Wang, J.; Al Qahtani, M.; Wang, X. One-step plasma-enabled catalytic carbon dioxide hydrogenation to higher hydrocarbons: Significance of catalyst-bed configuration. Green Chem. 2021, 23, 1642–1647. [Google Scholar] [CrossRef]
- Wang, Y.; Winter, L.; Chen, J.; Yan, B. CO2 hydrogenation over heterogeneous catalysts at atmospheric pressure: From electronic properties to product selectivity. Green Chem. 2021, 23, 249–267. [Google Scholar] [CrossRef]
- Li, X.; Luo, X.; Jin, Y. Heterogeneous sulfur-free hydrodeoxygenation catalysts for selectively upgrading the renewable bio-oils to second generation biofuels. Renew. Sustain. Energy Rev. 2018, 82, 3762–3797. [Google Scholar] [CrossRef]
- Santillan-Jimenez, E.; Crocker, M. Catalytic deoxygenation of fatty acids and their derivatives to hydrocarbon fuels via decarboxylation/decarbonylation. J. Chem. Technol. Biotechnol. 2012, 87, 1041–1050. [Google Scholar] [CrossRef]
- Davis, K.; Yoo, S.; Shuler, E.; Sherman, B.; Lee, S.; Leem, G. Photocatalytic hydrogen evolution from biomass conversion. Nano Converg. 2021, 8, 6. [Google Scholar] [CrossRef]
- Rajeswari, G.; Jacob, S.; Chandel, A.; Kumar, V. Unlocking the potential of insect and ruminant host symbionts for recycling of lignocellulosic carbon with a biorefinery approach: A review. Microb. Cell. Fact 2021, 20, 107. [Google Scholar] [CrossRef]
- Serrano-Ruiz, J.C.; West, R.M.; Dumesic, J.A. Catalytic Conversion of Renewable Biomass Resources to Fuels and Chemicals. Annu. Rev. Chem. Biomelecular Eng. 2010, 1, 79–100. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Padrón, D.; Zhao, D.; Carrillo-Carrion, C. Exploring the potential of biomass-templated Nb/ZnO nanocatalysts for the sustainable synthesis of N-heterocycles. Catal. Today 2021, 368, 243–249. [Google Scholar] [CrossRef]
- Galarza, E.; Fermanelli, C.; Pierella, L.; Saux, C.; Renzini, M. Influence of the Sn incorporation method in ZSM-11 zeolites in the distribution of bio-oil products obtained from biomass pyrolysis. J. Anal. Appl. Pyrolysis 2021, 156, 105116. [Google Scholar] [CrossRef]
- Liang, J.; Shan, G.; Sun, Y. Catalytic fast pyrolysis of lignocellulosic biomass: Critical role of zeolite catalysts. Renew. Sustain. Energy Rev. 2021, 139, 110707. [Google Scholar] [CrossRef]
- Ribeiro, L.; de Melo Órfão, J.; Ribeiro Pereira, M. An overview of the hydrolytic hydrogenation of lignocellulosic biomass using carbon-supported metal catalysts. Mater. Today Sustain. 2020, 11, 100058. [Google Scholar] [CrossRef]
- Safaei, M.; Foroughi, M.; Ebrahimpoor, N.; Jahani, S.; Omidi, A.; Khatami, M. A review on metal–organic frameworks: Synthesis and applications. TrAC Trends Anal. Chem. 2019, 118, 401–425. [Google Scholar] [CrossRef]
- Zhang, L.; Li, S.; Xin, J. A non-enzymatic voltammetric xanthine sensor based on the use of platinum nanoparticles loaded with a metal–organic framework of type MIL-101(Cr). Application to simultaneous detection of dopamine, uric acid, xanthine and hypoxanthine. Microchim. Acta 2019, 186. [Google Scholar] [CrossRef]
- Mirkovic, I.; Lei, L.; Ljubic, D.; Zhu, S. Crystal Growth of Mmetal-Organic Framework-5 around Cellulose-Based Fibers Having a Necklace Morphology. ACS Omega 2019, 4, 169–175. [Google Scholar] [CrossRef]
- Zhang, L.; Jiang, K.; Zhang, J. Low-Cost and High-Performance Microporous Mmetal-Organic Framework for Separation of Acetylene from Carbon Dioxide. ACS Sustain. Chem. Eng. 2018, 7, 1667–1672. [Google Scholar] [CrossRef]
- Huan, W.; Xing, M.; Cheng, C.; Li, J. Facile Fabrication of Magnetic Mmetal-Organic Framework Nanofibers for Specific Capture of Phosphorylated Peptides. ACS Sustain. Chem. Eng. 2018, 7, 2245–2254. [Google Scholar] [CrossRef]
- Wei, X.; Wang, Y.; Chen, J. Poly(deep eutectic solvent)-functionalized magnetic metal organic framework composites coupled with solid-phase extraction for the selective separation of cationic dyes. Anal. Chim. Acta 2019, 1056, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; He, X.; Yin, F.; Chen, B.; Li, G.; Yin, H. Co-CoO-Co3O4/N-doped carbon derived from metal organic framework: The addition of carbon black for boosting oxygen electrocatalysis and Zn-Air battery. Electrochim. Acta 2019, 295, 966–977. [Google Scholar] [CrossRef]
- Cao, A.; Zhang, L.; Wang, Y. 2D–2D Heterostructured UNiMOF/g-C3N4 for Enhanced Photocatalytic H2 Production under Visible-Light Irradiation. ACS Sustain. Chem. Eng. 2018, 7, 2492–2499. [Google Scholar] [CrossRef]
- Baek, J.; Rungtaweevoranit, B.; Pei, X. Bioinspired Mmetal-Organic Framework Catalysts for Selective Methane Oxidation to Methanol. J. Am. Chem. Soc. 2018, 140, 18208–18216. [Google Scholar] [CrossRef] [PubMed]
- Andrew, R.M. Global CO2 emissions from cement production. Earth Syst. Sci. Data Discuss. 2018, 10, 195–217. [Google Scholar] [CrossRef] [Green Version]
- Aresta, M. Carbon dioxide reduction to C1 or Cn molecules. In Carbon Dioxide Recovery and Utilization; Aresta, M., Ed.; Springer: Dordrecht, The Netherlands, 2003; pp. 293–312. [Google Scholar]
- Whang, H.S.; Lim, J.; Choi, M.S.; Lee, J.; Lee, H. Heterogeneous catalysts for catalytic CO2 conversion into value-added chemicals. BMC Chem. Eng. 2019, 1, 9. [Google Scholar] [CrossRef]
- Wei, J.; Ge, Q.; Yao, R.; Wen, Z.; Fang, C.; Guo, L.; Xu, H.; Sun, J. Directly converting CO2 into a gasoline fuel. Nat. Commun. 2017, 8, 15174. [Google Scholar] [CrossRef] [Green Version]
- Porosoff, M.D.; Yan, B.; Chen, J.G. Catalytic reduction of CO2 by H2 for synthesis of CO, methanol and hydrocarbons: Challenges and opportunities. Energy Environ. Sci. 2016, 9, 62–73. [Google Scholar] [CrossRef]
- Li, W.; Wang, H.; Jiang, X.; Zhu, J.; Liu, Z.; Guo, X.; Song, C. A short review of recent advances in CO2 hydrogenation to hydrocarbons over heterogeneous catalysts. RSC Adv. 2018, 8, 7651–7669. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Dang, S.; Li, S.; Bu, X.; Liu, Z.; Qiu, M.; Yang, C.; Wang, H.; Zhong, L.; Han, Y.; et al. Direct production of lower olefins from CO2 conversion via bifunctional catalysis. ACS Catal. 2018, 8, 571–578. [Google Scholar] [CrossRef]
- Fujiwara, M.; Ando, H.; Tanaka, M.; Souma, Y. Hydrogenation of carbon dioxide over Cu Zn-chro mate/zeolite composite catalyst: The effects of reaction behavior of alkenes on hydrocarbon synthesis. Appl. Catal. A Gen. 1995, 130, 105–116. [Google Scholar] [CrossRef]
- Dang, S.; Gao, P.; Liu, Z.; Chen, X.; Yang, C.; Wang, H.; Zhong, L.; Li, S.; Sun, Y. Role of zirconium in direct CO2 hydrogenation to lower olefins on oxide/zeolite bifunctional catalysts. J. Catal. 2018, 364, 382–393. [Google Scholar] [CrossRef]
- Gao, J.; Jia, C.; Liu, B. Direct and selective hydrogenation of CO2 to ethylene and propene by bifunctional catalysts. Catal. Sci. Technol. 2017, 7, 5602–5607. [Google Scholar] [CrossRef]
- Fujiwara, M.; Kieffer, R.; Ando, H.; Xu, Q.; Souma, Y. Change of catalytic properties of FeZnO/zeolite composite catalyst in the hydrogenation of carbon dioxide. Appl. Catal. A Gen. 1997, 154, 87–101. [Google Scholar] [CrossRef]
- Helal, A.; Usman, M.; Arafat, M.E.; Abdelnaby, M.M. Allyl functionalized UiO-66 metal–organic framework as a catalyst for the synthesis of cyclic carbonates by CO2 cycloaddition. J. Ind. Eng. Chem. 2020, 89, 104–110. [Google Scholar] [CrossRef]
- Helal, A.; Cordova, K.E.; Arafat, M.E.; Usman, M.; Yamani, Z.H. Defect-engineeringa metal–organic framework for CO2 fixation in the synthesis of bioactive oxazolidinones. Inorg. Chem. Front. 2020, 7, 3571–3577. [Google Scholar] [CrossRef]
- Santibañez, L.; Escalona, N.; Torres, J.; Kremer, C.; Cancino, P.; Spodine, E. CuII- and CoII-Based MOFs: {[La2Cu3(μ-H2O)(ODA)6(H2O)3]·3H2O}n and {[La2Co3(ODA)6(H2O)6]·12H2O}n. The Relevance of Physicochemical Properties on the Catalytic Aerobic Oxidation of Cyclohexene. Catalysts 2020, 10, 589. [Google Scholar] [CrossRef]
- Cancino, P.; Santibañez, L.; Stevens, C.; Fuentealba, P.; Audebrand, N.; Aravena, D.; Torres, J.; Martinez, S.; Kremer, C.; Spodine, E. Influence of the channel size of isostructural 3d–4f MOFs on the catalytic aerobic oxidation of cycloalkenes. New J. Chem. 2019, 43, 11057–11064. [Google Scholar] [CrossRef]
- Han, Y.; Xu, H.; Su, Y.; Xu, Z.-l.; Wang, K.; Wang, W. Noble metal (Pt, Au@Pd) nanoparticles supported on metal organic framework (MOF-74) nanoshuttles as high-selectivity CO2 conversion catalysts. J. Catal. 2019, 370, 70–78. [Google Scholar] [CrossRef]
- Wang, T.; Shi, L.; Tang, J.; Malgras, V.; Asahina, S.; Liu, G.; Zhang, H.; Meng, X.; Chang, K.; He, J.; et al. A Co3O4-embedded porous ZnO rhombic dodecahedron prepared using zeolitic imidazolate frameworks as precursors for CO2 photoreduction. Nanoscale 2016, 8, 6712–6720. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, T.; Wang, J.; Liu, H.D.; Dao, T.; Li, M.; Liu, G.; Meng, X.; Chang, K.; Shi, L.; et al. Surface-Plasmon-Enhanced Photodriven CO2 Reduction Catalyzed by Mmetal-Organic-Framework-Derived Iron Nanoparticles Encapsulated by Ultrathin Carbon Layers. Adv. Mater. 2016, 28, 3703–3710. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Guan, B.Y.; Lu, Y.; Lou, X.W.D. Formation of Hierarchical In2S3–CdIn2S4 Heterostructured Nanotubes for Efficient and Stable Visible Light CO2 Reduction. J. Am. Chem. Soc. 2017, 139, 17305–17308. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Liu, M.; Ding, F.; Song, C.; Zhang, G.; Guo, X. Hydrothermally stable MOFs for CO2 hydrogenation over iron-based catalyst to light olefins. J. CO2 Util. 2016, 15, 89–95. [Google Scholar] [CrossRef]
- Tarasov, A.L.; Isaeva, V.I.; Tkachenko, O.P.; Chernyshev, V.V.; Kustov, L.M. Conversion of CO2 into liquid hydrocarbons in the presence of a Cocontaining catalyst based on the microporous metal–organic framework MIL-53(Al)Fuel. Proc. Tech. 2018, 176, 101–106. [Google Scholar] [CrossRef]
- Zhao, Z.-W.; Zhou, X.; Liu, Y.-N.; Shen, C.-C.; Yuan, C.-Z.; Jiang, Y.-F.; Zhao, S.-J.; Ma, L.-B.; Cheang, T.-Y.; Xu, A.-W. Ultrasmall Ni nanoparticles embedded in Zr-based MOFs provide high selectivity for CO2 hydrogenation to methane at low temperatures. Catal. Sci. Technol. 2018, 8, 3160–3165. [Google Scholar] [CrossRef]
- Zhen, W.; Gao, F.; Tian, B.; Ding, B.; Deng, Y.; Li, Z.; Gao, H.; Lu, G. Enhancing activity for carbon dioxide methanation by encapsulating (111) facet Ni particle in metal–organic frameworks at low temperature. J. Catal. 2017, 348, 200–211. [Google Scholar] [CrossRef]
- Zhen, W.; Li, B.; Lu, G.; Ma, J. Enhancing catalytic activity and stability for CO2 methanation on Ni@MOF-5 via control of active species dispersion. Chem. Commun. 2015, 51, 1728–1731. [Google Scholar] [CrossRef]
- Rungtaweevoranit, B.; Baek, J.; Araujo, J.R.; Archanjo, B.S.; Yaghi, O.M.; Somorjai, G.A. Copper Nanocrystals Encapsulated in Zr-based Mmetal-Organic Frameworks for Highly Selective CO2 Hydrogenation to Methanol. Nano Lett. 2016, 16, 7645–7649. [Google Scholar] [CrossRef]
- An, B.; Zhang, J.; Cheng, K.; Ji, P.; Wang, C.; Lin, W. Confinement of Ultrasmall Cu/ZnOx Nanoparticles in Mmetal-Organic Frameworks for Selective Methanol Synthesis from Catalytic Hydrogenation of CO2. J. Am. Chem. Soc. 2017, 139, 3834–3840. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, A.; Liu, M.; Hu, S.; Ding, F.; Song, C.; Guo, X. Fe-MOF-derived highly active catalysts for carbon dioxide hydrogenation to valuable hydrocarbons. J. CO2 Util. 2017, 21, 100–107. [Google Scholar] [CrossRef]
- Wang, Y.; Kazumi, S.; Gao, W.; Gao, X.; Li, H.; Guo, X.; Yoneyama, Y.; Yang, G.; Tsubaki, N. Direct conversion of CO2 to aromatics with high yield via a modified Fischer-Tropsch synthesis pathway. Appl. Catal. B Env. 2020, 269, 118792–118801. [Google Scholar] [CrossRef]
- Martín, N.; Portillo, A.; Ateka, A.; Cirujano, F.G.; Oar-Arteta, L.; Aguayo, A.T.; Dusselier, M. MOF-derived/zeolite hybrid catalyst for the production of light olefins from CO2. ChemCatChem 2020, 12, 5750–5758. [Google Scholar] [CrossRef]
- Ramirez, A.; Gevers, L.; Bavykina, A.; Ould-Chikh, S.; Gascon, J. Metal Organic Framework-Derived Iron Catalysts for the Direct Hydrogenation of CO2 to Short Chain Olefins. ACS Catal. 2018, 8, 9174–9182. [Google Scholar] [CrossRef]
- Chen, C.S.; Cheng, W.H.; Lin, S. Mechanism of CO formation in reverse water–gas shift reaction over Cu/Al2O3 catalyst. Catal. Lett. 2000, 68, 45–48. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Baldwin, J.; Peng, X.; Mpourmpakis, G.; Willauer, H.D. Potassium Promoted Molybdenum Carbide as a Highly Active and Selective Catalyst for CO2 Conversion to CO. ChemSusChem 2017, 10, 2408–2415. [Google Scholar] [CrossRef] [PubMed]
- Matsubu, J.C.; Yang, V.N.; Christopher, P. Isolated Metal Active Site Concentration and Stability Control Catalytic CO2 Reduction Selectivity. J. Am. Chem. Soc. 2015, 137, 3076–3084. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.S.; Lee, H.H.; Hong, S.C. A study on the effect of support’s reducibility on the reverse water-gas shift reaction over Pt catalysts. Appl. Catal. A 2012, 423, 100–107. [Google Scholar] [CrossRef]
- Lu, B.; Kawamoto, K. Preparation of mesoporous CeO2 and monodispersed NiO particles in CeO2, and enhanced selectivity of NiO/CeO2 for reverse water gas shift reaction. Mater. Res. Bull. 2014, 53, 70–78. [Google Scholar] [CrossRef]
- Loiland, J.A.; Wulfers, M.J.; Marinkovic, N.S.; Lobo, R.F. Fe/γ-Al2O3 and Fe−K/γ-Al2O3 as reverse water-gas shift catalysts. Catal. Sci. Technol. 2016, 6, 5267. [Google Scholar] [CrossRef] [Green Version]
- Pour, A.N.; Shahri, S.M.K.; Bozorgzadeh, H.R.; Zamani, Y.; Tavasoli, A.; Marvast, M.A. Effect of Mg, La and Ca promoters on the structure and catalytic behavior of iron-based catalysts in Fischer–Tropsch synthesis. Appl. Catal. A 2008, 348, 201–208. [Google Scholar] [CrossRef]
- Li, J.; Cheng, X.; Zhang, C.; Wang, J.; Dong, W.; Yang, Y.; Li, Y. Alkalis in iron-based Fischer–Tropsch synthesis catalysts; distribution, migration and promotion. J. Chem. Technol. Biotechnol. 2017, 92, 1472–1480. [Google Scholar] [CrossRef]
- Dorner, R.W.; Hardy, D.R.; Williams, F.W.; Willauer, H.D. K and Mn doped iron-based CO2 hydrogenation catalysts; Detection of KAlH4 as part of the catalyst’s active phase. Appl. Catal. A 2010, 373, 112–121. [Google Scholar] [CrossRef]
- Rodemerck, U.; Holenă, M.; Wagner, E.; Smejkal, Q.; Barkschat, A.; Baerns, M. Catalyst Development for CO2 Hydrogenation to Fuels. ChemCatChem 2013, 5, 1948–1955. [Google Scholar] [CrossRef]
- Numpilai, T.; Witoon, T.; Chanlek, N.; Limphirat, W.; Bonura, G.; Chareonpanich, M.; Limtrakul, L. Structure–activity relationships of Fe-Co/K-Al2O3 catalysts calcined at different temperatures for CO2 hydrogenation to light olefins. Appl. Catal. A 2017, 547, 219–229. [Google Scholar] [CrossRef]
- Zhao, T.; Hui, Y.; Li, Z. Controllable preparation of ZIF-67 derived catalyst for CO2 methanation. Mol. Catal. 2019, 474, 110421–110430. [Google Scholar] [CrossRef]
- Lin, X.; Wang, S.; Tu, W.; Hu, Z.; Ding, Z.; Hou, Y.; Xu, R.; Dai, W. MOF-derived hierarchical hollow spheres composed of carbon-confined Ni nanoparticles for efficient CO2 methanation. Catal. Sci. Technol. 2019, 9, 731–738. [Google Scholar] [CrossRef]
- Fang, R.; Chen, L.; Shen, Z.; Li, Y. Efficient hydrogenation of furfural to fufuryl alcohol over hierarchical MOF immobilized metal catalysts. Catal. Today 2021, 368, 217–223. [Google Scholar] [CrossRef]
- Song, Y.; Feng, X.; Chen, J.S.; Brzezinski, C.; Xu, Z.; Lin, W. Multistep Engineering of Synergistic Catalysts in a Metal–Organic Framework for Tandem C-O Bond Cleavage. J. Am. Chem. Soc. 2020, 142, 4872–4882. [Google Scholar] [CrossRef]
- Phan, D.P.; Lee, E.Y. Phosphoric acid enhancement in a Pt-encapsulated Metal–Organic Framework (MOF) bifunctional catalyst for efficient hydro-deoxygenation of oleic acid from biomass. J. Catal. 2020, 386, 19–29. [Google Scholar] [CrossRef]
- Yuan, Q.; Zhang, D.; Van Haandel, L.; Ye, F.; Xue, T.; Hensen, E.J.M.; Guan, Y. Selective liquid phase hydrogenation of furfural to furfuryl alcohol by Ru/Zr-MOFs. J. Mol. Catal. A Chem. 2015, 406, 58–64. [Google Scholar] [CrossRef]
- Fulajtárova, K.; Soták, T.; Hronec, M.; Vávra, I.; Dobročka, E.; Omastová, M. Aqueous phase hydrogenation of furfural to furfuryl alcohol over Pd-Cu catalysts. Appl. Catal. A Gen. 2015, 502, 78–85. [Google Scholar] [CrossRef]
- Feng, J.; Li, M.; Zhong, Y.; Xu, Y.; Meng, X.; Zhao, Z.; Feng, C. Hydrogenation of levulinic acid to γ-valerolactone over Pd@UiO-66-NH2 with high metal dispersion and excellent reusability. Microporous Mesoporous Mater. 2020, 294, 109858–109867. [Google Scholar] [CrossRef]
- Aijaz, A.; Karkamkar, A.; Choi, Y.J.; Tsumori, N.; Rönnebro, E.; Autrey, T.H.; Shioyama, H.; Xu, Q. Immobilizing Highly Catalytically Active Pt Nanoparticles inside the Pores of Metal–Organic Framework. J. Am. Chem. Soc. 2012, 134, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Kumar Reddy, D.H.; Bediako, J.K.; Song, M.H.; Wei, W.; Kim, J.A.; Yun, Y.S. Effective adsorption of Pd(II), Pt(IV) and Au(III) by Zr(IV)-based metal–organic frameworks from strongly acidic solutions. J. Mater. Chem. A 2017, 5, 13557–13564. [Google Scholar] [CrossRef]
- Li, X.; Guo, Z.; Xiao, C.; Goh, T.W.; Tesfagaber, D.; Huang, W. Tandem catalysis by palladium nanoclusters encapsulated in metal–organic frameworks. ACS Catal. 2014, 4, 3490–3497. [Google Scholar] [CrossRef] [Green Version]
- Klet, R.C.; Liu, Y.; Wang, T.C.; Hupp, J.T.; Farha, O.K. Evaluation of Brønsted acidity and proton topology in Zr- and Hf-based metal–organic frameworks using potentiometric acid-base titration. J. Mater. Chem. A 2016, 4, 1479–1485. [Google Scholar] [CrossRef]
- Cirujano, F.G.; Corma, A.; Llabrés, I.; Xamena, F.X. Zirconium-containing metal organic frameworks as solid acid catalysts for the esterification of free fatty acids: Synthesis of biodiesel and other compounds of interest. Catal. Today 2015, 257, 213–220. [Google Scholar] [CrossRef]
- Li, X.; Deng, Q.; Yu, L.; Gao, R.; Tong, Z.; Lu, C.; Wang, J.; Zeng, Z.; Zou, J.J.; Deng, S. Double-metal cyanide as an acid and hydrogenation catalyst for the highly selective ring-rearrangement of biomass-derived furfuryl alcohol to cyclopentenone compounds. Green Chem. 2020, 22, 2549–2557. [Google Scholar] [CrossRef]
- Kim, D.W.; Kim, H.G.; Cho, D.H. Catalytic performance of MIL-100 (Fe, Cr) and MIL-101 (Fe, Cr) in the isomerization of endo- to exo-dicyclopentadiene. Catal. Commun. 2016, 73, 69–73. [Google Scholar] [CrossRef]
- Rinaldi, R.; Schüth, F. Design of solid catalysts for the conversion of biomass. Energy Environ. Sci. 2009, 2, 610–626. [Google Scholar] [CrossRef]
- Geilen, F.M.A.; Engendahl, B.; Harwardt, A.; Marquardt, W.; Klankermayer, J.; Leitner, W. Selective and flexible transformation of biomass-derived platform chemicals by a multifunctional catalytic system. Angew. Chem. Int. Ed. 2010, 49, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ye, F.; Guan, Y.; Wang, Y.; Hensen, E.J.M. Hydrogenation of γ-valerolactone in ethanol over Pd nanoparticles supported on sulfonic acid functionalized MIL-101. RSC Adv. 2014, 4, 39558–39564. [Google Scholar] [CrossRef]
- Valekar, A.H.; Lee, M.; Yoon, J.W.; Kwak, J.; Hong, D.Y.; Oh, K.R.; Cha, G.Y.; Kwon, Y.U.; Jung, J.; Chang, J.S.; et al. Catalytic Transfer Hydrogenation of Furfural to Furfuryl Alcohol under Mild Conditions over Zr-MOFs: Exploring the Role of Metal Node Coordination and Modification. ACS Catal. 2020, 10, 3720–3732. [Google Scholar] [CrossRef]
- Bai, Y.; Dou, Y.; Xie, L.H.; Rutledge, W.; Li, J.R.; Zhou, H.C. Zr-based metal–organic frameworks: Design, synthesis, structure, and applications. Chem. Soc. Rev. 2016, 45, 2327–2367. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; He, J.; Riisager, A.; Saravanamurugan, S.; Song, B.; Yang, S. Acid-base bifunctional zirconium N-alkyltriphosphate nanohybrid for hydrogen transfer of biomass-derived carboxides. ACS Catal. 2016, 6, 7722–7727. [Google Scholar] [CrossRef]
- Li, H.; Liu, X.; Yang, T.; Zhao, W.; Saravanamurugan, S.; Yang, S. Porous Zirconium–Furandicarboxylate Microspheres for Efficient Redox Conversion of Biofuranics. ChemSusChem 2017, 10, 1761–1770. [Google Scholar] [CrossRef]
- Yang, D.; Bernales, V.; Islamoglu, T.; Farha, O.K.; Hupp, J.T.; Cramer, C.J.; Gagliardi, L.; Gates, B.C. Tuning the Surface Chemistry of Metal Organic Framework Nodes: Proton Topology of the Metal-Oxide-Like Zr6 Nodes of UiO-66 and NU-1000. J. Am. Chem. Soc. 2016, 138, 15189–15196. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Gaggioli, C.A.; Ray, D.; Babucci, M.; Gagliardi, L.; Gates, B.C. Tuning Catalytic Sites on Zr6O8 Metal–Organic Framework Nodes via Ligand and Defect Chemistry Probed with tert-Butyl Alcohol Dehydration to Isobutylene. J. Am. Chem. Soc. 2020, 142, 8044–8056. [Google Scholar] [CrossRef]
- Jung, K.D.; Bell, A.T. Role of Hydrogen Spillover in Methanol Synthesis over Cu/ZrO2. J. Catal. 2000, 193, 207–223. [Google Scholar] [CrossRef]
- Li, D.; Xu, H.Q.; Jiao, L.; Jiang, H.L. Metal-organic frameworks for catalysis: State of the art, challenges, and opportunities. EnergyChem 2019, 1, 100005. [Google Scholar] [CrossRef]
- Han, A.; Wang, B.; Kumar, A.; Qin, Y.; Jin, J.; Wang, X.; Yang, C.; Dong, B.; Jia, Y.; Liu, J.; et al. Recent Advances for MOF-Derived Carbon-Supported Single-Atom Catalysts. Small Methods 2019, 3, 1800471–1800492. [Google Scholar] [CrossRef]
- Salunkhe, R.R.; Kaneti, Y.V.; Kim, J.; Kim, J.H.; Yamauchi, Y. Nanoarchitectures for Metal–Organic Framework-Derived Nanoporous Carbons toward Supercapacitor Applications. Acc. Chem. Res. 2016, 49, 2796–2806. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Astruc, D. State of the Art and Prospects in Metal–Organic Framework (MOF)-Based and MOF-Derived Nanocatalysis. Chem. Rev. 2020, 120, 1438–1511. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Peng, L.; Bulut, S.; Queen, W.L. Recent Advances of MOFs and MOF-Derived Materials in Thermally Driven Organic Transformations. Chem. Eur. J. 2019, 25, 2161–2178. [Google Scholar] [CrossRef]
- Chaikittisilp, W.; Hu, M.; Wang, H.; Huang, H.S.; Fujita, T.; Wu, K.C.W.; Chen, L.C.; Yamauchi, Y.; Ariga, K. Nanoporous carbons through direct carbonization of a zeolitic imidazolate framework for supercapacitor electrodes. Chem. Commun. 2012, 48, 7259–7261. [Google Scholar] [CrossRef]
- Fang, R.; Dhakshinamoorthy, A.; Li, Y.; Garcia, H. Metal organic frameworks for biomass conversion. Chem. Soc. Rev. 2020, 49, 3638–3687. [Google Scholar] [CrossRef]
- Liao, Y.; Van Chi, N.; Ishiguro, N.; Young, A.P.; Tsung, C.; Wu, K.C. Environmental Engineering a homogeneous alloy-oxide interface derived from metal- organic frameworks for selective oxidation of 5-hydroxymethylfurfural to 2, 5-furandicarboxylic acid. Applied Catalysis B 2020, 270, 118805–118817. [Google Scholar] [CrossRef]
- Liao, Y.; Chen, J.E.; Isida, Y.; Yonezawa, T.; Chang, W. De Novo Synthesis of Gold-Nanoparticle-Embedded, Nitrogen-Doped Nanoporous Carbon Nanoparticles (Au@NC) with Enhanced Reduction Ability. ChemCatChem 2016, 44, 502–509. [Google Scholar] [CrossRef]
- Liao, Y.; Huang, Y.; Chen, H.M.; Komaguchi, K.; Hou, H.; Henzie, J.; Yamauchi, Y.; Ide, Y.; Wu, K.C.; Liao, Y.; et al. Mesoporous TiO2 Embedded with a Uniform Distribution of CuO Exhibit Enhanced Charge Separation and Photocatalytic Efficiency Mesoporous TiO2 Embedded with a Uniform Distribution of CuO Exhibit Enhanced Charge Separation and Photocatalytic Efficiency. ACS Appl. Mater. Interfaces 2017, 9, 42425–42429. [Google Scholar] [CrossRef]
- Abad, A.; Concepción, P.; Corma, A. A Collaborative Effect between Gold and a Support Induces the Selective Oxidation of Alcohols. Angew. Chem. Int. Ed. 2005, 44, 4066–4069. [Google Scholar] [CrossRef]
- Kim, B.Y.; Shim, I.B.; Araci, Z.O.; Scott Saavedra, S.; Monti, O.L.A.; Armstrong, N.R.; Sahoo, R.; Srivastava, D.N.; Pyun, J. Synthesis and colloidal polymerization of ferromagnetic Au-Co nanoparticles into Au-Co3O4 nanowires. J. Am. Chem. Soc. 2010, 132, 3234–3235. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Deng, J.; Liu, Y.; Xie, S.; Jiang, Y.; Zhao, X.; Yang, J.; Arandiyan, H.; Guo, G.; Dai, H. Three-dimensionally ordered mesoporous Co3O4-supported Au—Pd alloy nanoparticles: High-performance catalysts for methane combustion. J. Catal. 2015, 332, 13–24. [Google Scholar] [CrossRef]
- Casanova, O.; Iborra, S.; Corma, A. Biomass into Chemicals: Aerobic Oxidation of 5-Hydroxy-methyl-2-furfural into 2, 5-Furandicarboxylic Acid with Gold Nanoparticle Catalysts. ChemSusChem 2009, 2, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Ardemani, L.; Cibin, G.; Dent, A.J.; Isaacs, M.A.; Kyriakou, G.; Lee, A.F.; Parlett, C.M.A.; Parry, S.A.; Wilson, K. Solid base catalysed 5-HMF oxidation to 2,5-FDCA over Au/hydrotalcites: Fact or fiction? Chem. Sci. 2015, 6, 4940–4945. [Google Scholar] [CrossRef] [Green Version]
- Van Nguyen, C.; Liao, Y.T.; Kang, T.C.; Chen, J.E.; Yoshikawa, T.; Nakasaka, Y.; Masuda, T.; Wu, K.C.W. A Metal-Free, High Nitrogen-Doped Nanoporous Graphitic Carbon Catalyst for an Effective Aerobic HMF-to-FDCA Conversion. Green Chem. 2016, 18, 5957–5961. [Google Scholar] [CrossRef]
- Zhou, C.; Deng, W.; Wan, X.; Zhang, Q.; Yang, Y. Functionalized Carbon Nanotubes for Biomass Conversion: The Base-Free Aerobic Oxidation of 5-Hydroxymethyl- furfural to 2,5-Furandicarboxylic Acid over Platinum Supported on a Carbon Nanotube Catalyst. ChemCatChem 2015, 7, 2853–2863. [Google Scholar] [CrossRef]
- Feng, Y.; Jia, W.; Yan, G.; Zeng, X.; Sperry, J.; Xu, B.; Sun, Y.; Tang, X.; Lei, T.; Lin, L. Insights into the active sites and catalytic mechanism of oxidative esterification of 5-hydroxymethylfurfural by metal–organic frameworks-derived N-doped carbon. J. Catal. 2020, 381, 570–578. [Google Scholar] [CrossRef]
- Zhang, M.; Townend, I.; Cai, H.; He, J.; Mei, X. The influence of seasonal climate on the morphology of the mouth-bar in the Yangtze Estuary, China. Cont. Shelf Res. 2018, 153, 30–49. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; Liu, H.; Bai, C.; Liao, S.; Li, Y. Base-free oxidation of alcohols to esters at room temperature and atmospheric conditions using nanoscale Co-based catalysts. ACS Catal. 2015, 5, 1850–1856. [Google Scholar] [CrossRef]
- Wu, G.; More, K.L.; Johnston, C.M.; Zelenay, P. High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt. Science 2011, 332, 443–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, D.; Yu, L.; Chen, X.; Wang, G.; Jin, L.; Pan, X.; Deng, J.; Sun, G.; Bao, X. Iron Encapsulated within Pod-like Carbon Nanotubes for Oxygen Reduction Reaction. Angew. Chem. Int. Ed. 2013, 52, 371–375. [Google Scholar] [CrossRef]
- Bao, L.; Sun, F.; Zhang, G.; Hu, T.L. High-efficient Aerobic Oxidation of Biomass-derived 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic Acid over Holey 2D Mn2O3 Nanoflakes from a Mn-based MOF. ChemSusChem 2019, 13, 548–555. [Google Scholar] [CrossRef]
- Fang, R.; Tian, P.; Yang, X.; Luque, R.; Li, Y. Encapsulation of ultra fi ne metal-oxide nanoparticles within mesopores for biomass-derived catalytic applications. Chem. Sci. 2018, 9, 1854–1859. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Niu, C.; Xu, L.; Li, J.; Liu, X.; Wang, X.; Wu, Y.; Xu, X.; Chen, W.; Li, Q.; et al. General Oriented Formation of Carbon Nanotubes from Metal–Organic Frameworks. J. Am. Chem. Soc. 2017, 139, 8212–8221. [Google Scholar] [CrossRef]
- Fang, R.; Luque, R.; Li, Y. Selective aerobic oxidation of biomass-derived HMF to 2,5-diformylfuran using a MOF-derived magnetic hollow Fe–Co nanocatalyst. Green Chem. 2016, 18, 3152–3157. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | CO2 Conversion (%) | Mass of Catalysts (g) | Temperature (°C) | Selectivity (%) | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|
| CO | CH4 | C2–C4− | C2–C4= | C5+ | |||||
| MIL-53(Al)/Fe2O3 | 20 | 1 | 300 | 20 | 41 | 34 | 0 | 5 | [44] |
| ZIF-8(a)(Zn)/Fe2O3 | 23 | 1 | 300 | 23 | 21 | 23 | 21 | 12 | [44] |
| ZIF-8(b)(Zn)/Fe2O3 | 25 | 1 | 300 | 21 | 22 | 30 | 15 | 12 | [44] |
| ZIF-8(c)(Zn)/Fe2O3 | 30 | 1 | 300 | 17 | 25 | 40 | 8 | 10 | [44] |
| 10%Co/MIL-53(Al) | 25 | 0.8 | 260 | 26 | 26 | 22 | 0 | 26 | [45] |
| 10%Co/MIL-53(Al) | 30 | 0.8 | 300 | 19 | 28 | 24 | 0 | 28 | [45] |
| 10%Co/MIL-53(Al) | 38 | 0.8 | 340 | 9 | 32 | 27 | 0 | 32 | [45] |
| 5Ni@UiO-66 | 8 | 0.1 | 300 | # | # | # | # | # | [46] |
| 10Ni@UiO-66 | 13 | 0.1 | 300 | # | # | # | # | # | [46] |
| 15Ni@UiO-66 | 25 | 0.1 | 300 | # | # | # | # | # | [46] |
| 20Ni@UiO-66 | 58 | 0.1 | 300 | 100 | 0 | 0 | 0 | 0 | [46] |
| 25Ni@UiO-66 | 50 | 0.1 | 300 | # | # | # | # | # | [46] |
| 30Ni@UiO-66 | 41 | 0.1 | 300 | # | # | # | # | # | [46] |
| 20Ni@MIL-101 | 100 | 0.2 | 300 | 100 | 0 | 0 | 0 | 0 | [47] |
| 10Ni@MOF-5 | 57 | 0.2 | 300 | 100 | 0 | 0 | 0 | 0 | [48] |
| Catalyst | Conv. (%) | Selectivity (%) | ||||
|---|---|---|---|---|---|---|
| CO | C1 | C2–C6 | C2=C6 | C7+ | ||
| Fe/C | 24 | 39 | 39 | 12 | 1 | 9 |
| Fe/C + Fe | 25 | 40 | 38 | 12 | 1 | 9 |
| Fe/C + Cu | 29 | 23 | 53 | 15 | 2 | 7 |
| Fe/C + Mo | 22 | 50 | 30 | 9 | 1 | 10 |
| Fe/C + Li | 26 | 38 | 40 | 12 | 2 | 8 |
| Fe/C + Na | 27 | 38 | 32 | 15 | 4 | 11 |
| Fe/C + K | 35 | 17 | 18 | 7 | 40 | 18 |
| Fe/C + Mg | 22 | 48 | 39 | 9 | 1 | 3 |
| Fe/C + Ca | 24 | 40 | 43 | 13 | 1 | 3 |
| Fe/C + Zn | 23 | 41 | 45 | 13 | 1 | 0 |
| Fe/C + Ni | 26 | 34 | 52 | 13 | 1 | 0 |
| Fe/C + Co | 26 | 32 | 51 | 14 | 2 | 1 |
| Fe/C + Mn | 23 | 45 | 38 | 8 | 1 | 8 |
| Fe/C + Pt | 30 | 22 | 51 | 19 | 3 | 5 |
| Fe/C + Rh | 25 | 17 | 63 | 19 | 1 | 0 |
| Catalyst | Catalyst (mg) | Sudstrate | Substrate (mmol) | Hydrogen Sours | T (°C) | t (h) | Conv. (%) | Selectivity (%) | Product | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a | Pd/H-UiO-66 | 0.3 mol% | FUR | 5.0 | 0.5 MPa H2 | 60 | 3 | 100 | >99 | FOL | [68] |
| b | Ru/UiO-66 | 100 | FUR | 0.1 mL FUR in 9.9 mL H2O | 0.5 MPa H2 | 20 | 4 | 94.9 | 100 | FOL | [71] |
| c | Pd@UiO-66–NH2 | 150 | LA | 5 | 2 MPa H2 | 140 | 2 | 98 | 100 | GVL | [73] |
| d | FeZn-P | 100 | FOL | 10.2 | - | 150 | 6 | 95 | 88.2 | HCP | [79] |
| FeZn-P | 100 | FOL | 10.2 | 4 MPa H2 | 150 | 6 | 95 | 61.5 | CPE | ||
| e | Pd/MIL-101–SO3H | 100 | GVL | 10 | 3 MPa H2 | 250 | 10 | 98 | 83 | Ethyl valerate | |
| f a | Pt/P@MIL | 50 | Oleic acid | 0.0047 (mmol/s·gcat) | 2 MPa H2 | 300 | 2 | 75 | - | HDO products | [70] |
| g | M-MOF-808 | 100 | FUR | 10 | IPA b 416 mmol | 40 | 24 | 96.5 | 88.6 | FOL | [84] |
| Catalyst (mg) | Substrate (mmol) | Base | Oxidant | Temperature (°C) | Time (h) | Conv. HMF (%) | Ref. | |
|---|---|---|---|---|---|---|---|---|
| a | 10 | 0.3 | NaOH | H2O2 | 90 | 1 | 100 | [98] |
| b | 10 | 0.3 | NaOH/Na2CO3 | H2O2 | 90 | 1 | 100 | [98] |
| c | 100 | 0.5 | Na2CO3 | O2 | 100 | 5 | 99 | [108] |
| d | 100 | 0.5 | Na2CO3 | O2 | 100 | 5 | 99 | [108] |
| e | 150 | 50 mM | NaHCO3 | O2 | 100 | 24 | >99 | [113] |
| f | 150 | 50 mM | NaHCO3 | O2 | 100 | 24 | >99 | [113] |
| g | 1 mol% | 0.1 | - | Air | 80 | 2 | 100 | [114] |
| h | 20 mol% | 1 | Na2CO3 | O2 | 100 °C | 6 | >99 | [116] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdebenito, G.; Gonzaléz-Carvajal, M.; Santibañez, L.; Cancino, P. Metal–Organic Frameworks (MOFs) and Materials Derived from MOFs as Catalysts for the Development of Green Processes. Catalysts 2022, 12, 136. https://doi.org/10.3390/catal12020136
Valdebenito G, Gonzaléz-Carvajal M, Santibañez L, Cancino P. Metal–Organic Frameworks (MOFs) and Materials Derived from MOFs as Catalysts for the Development of Green Processes. Catalysts. 2022; 12(2):136. https://doi.org/10.3390/catal12020136
Chicago/Turabian StyleValdebenito, Gonzalo, Marco Gonzaléz-Carvajal, Luis Santibañez, and Patricio Cancino. 2022. "Metal–Organic Frameworks (MOFs) and Materials Derived from MOFs as Catalysts for the Development of Green Processes" Catalysts 12, no. 2: 136. https://doi.org/10.3390/catal12020136