
Activation of Small Molecules and Hydrogenation of CO2 Catalyzed by Frustrated Lewis Pairs


Abstract
:1. Introduction
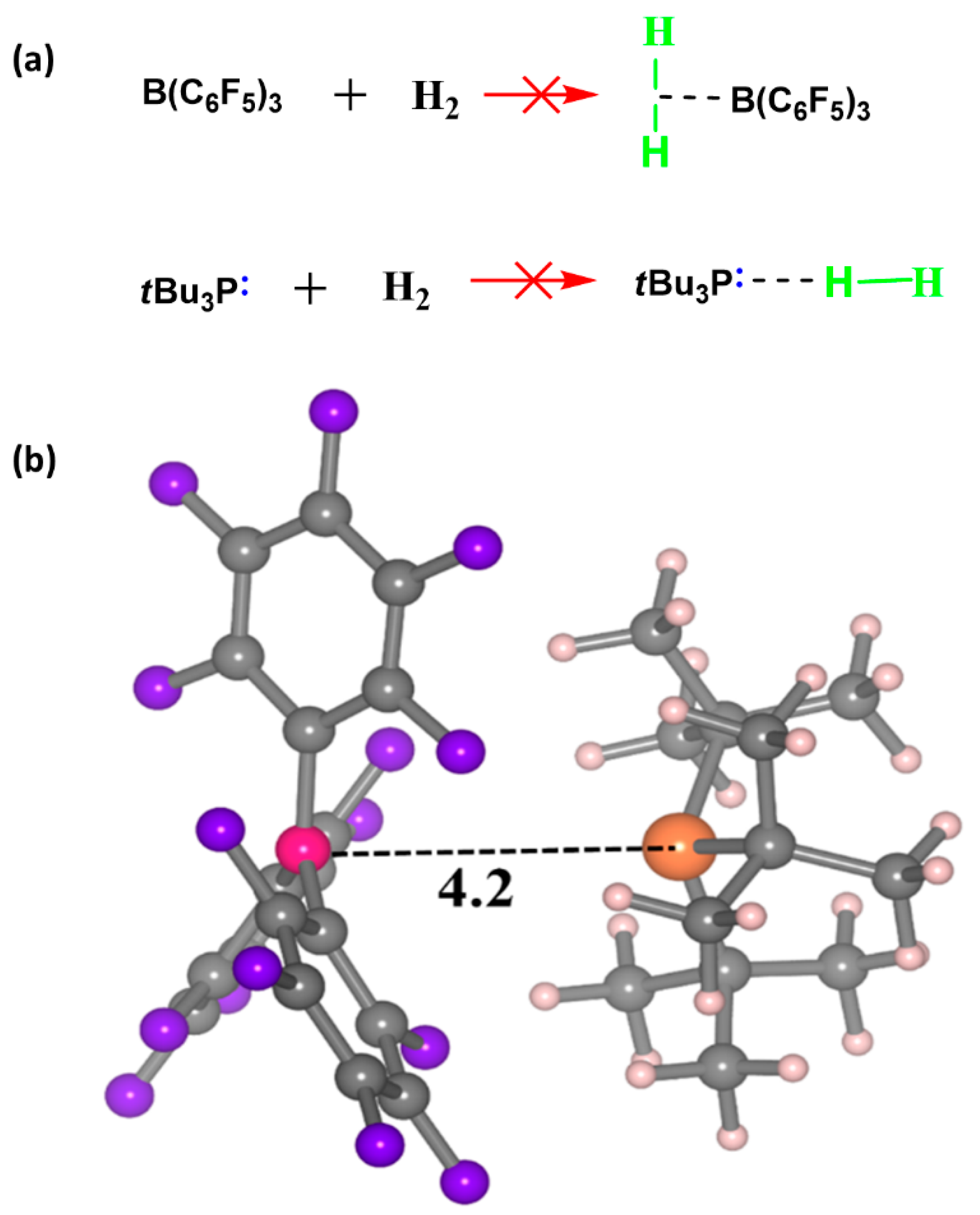
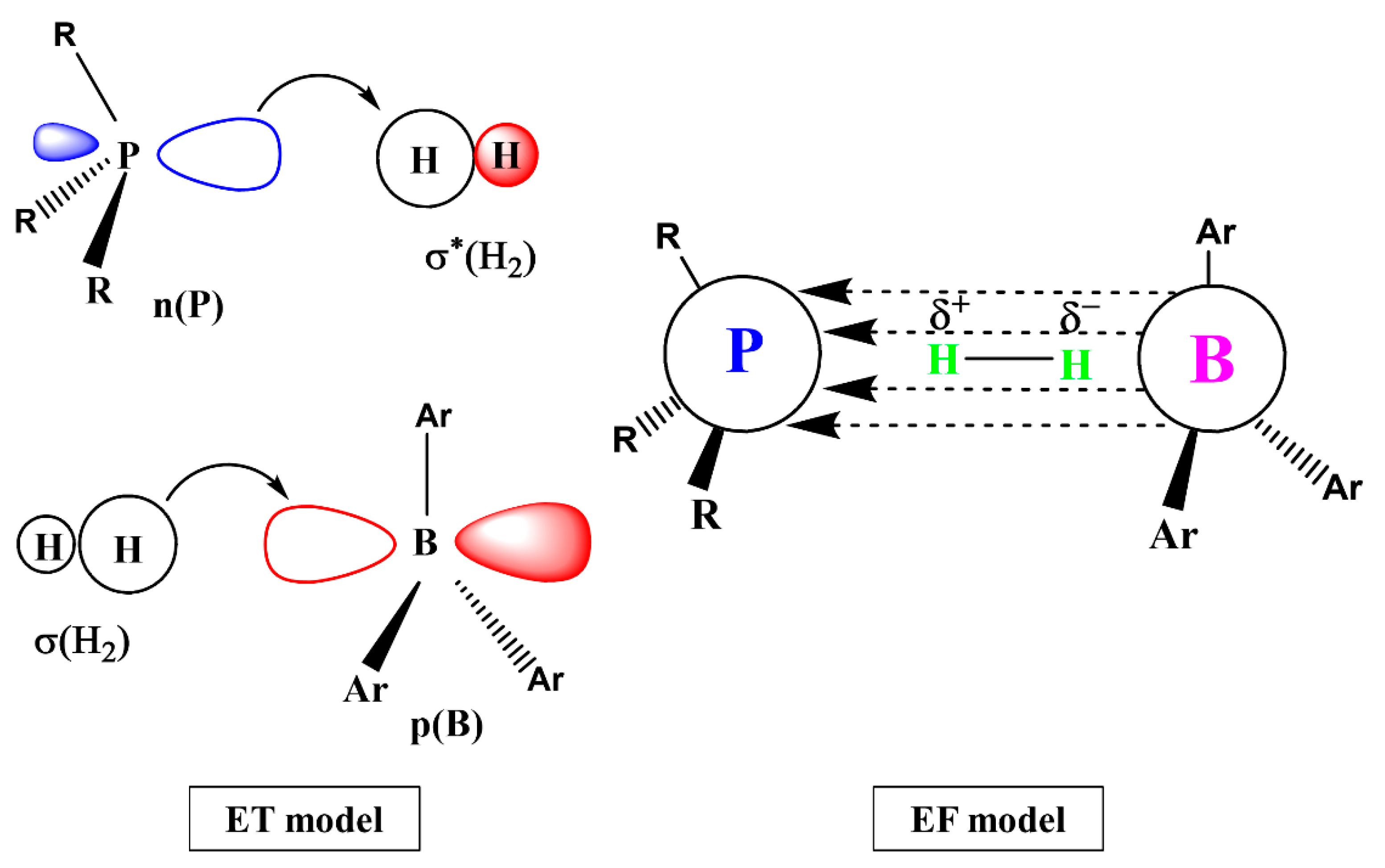
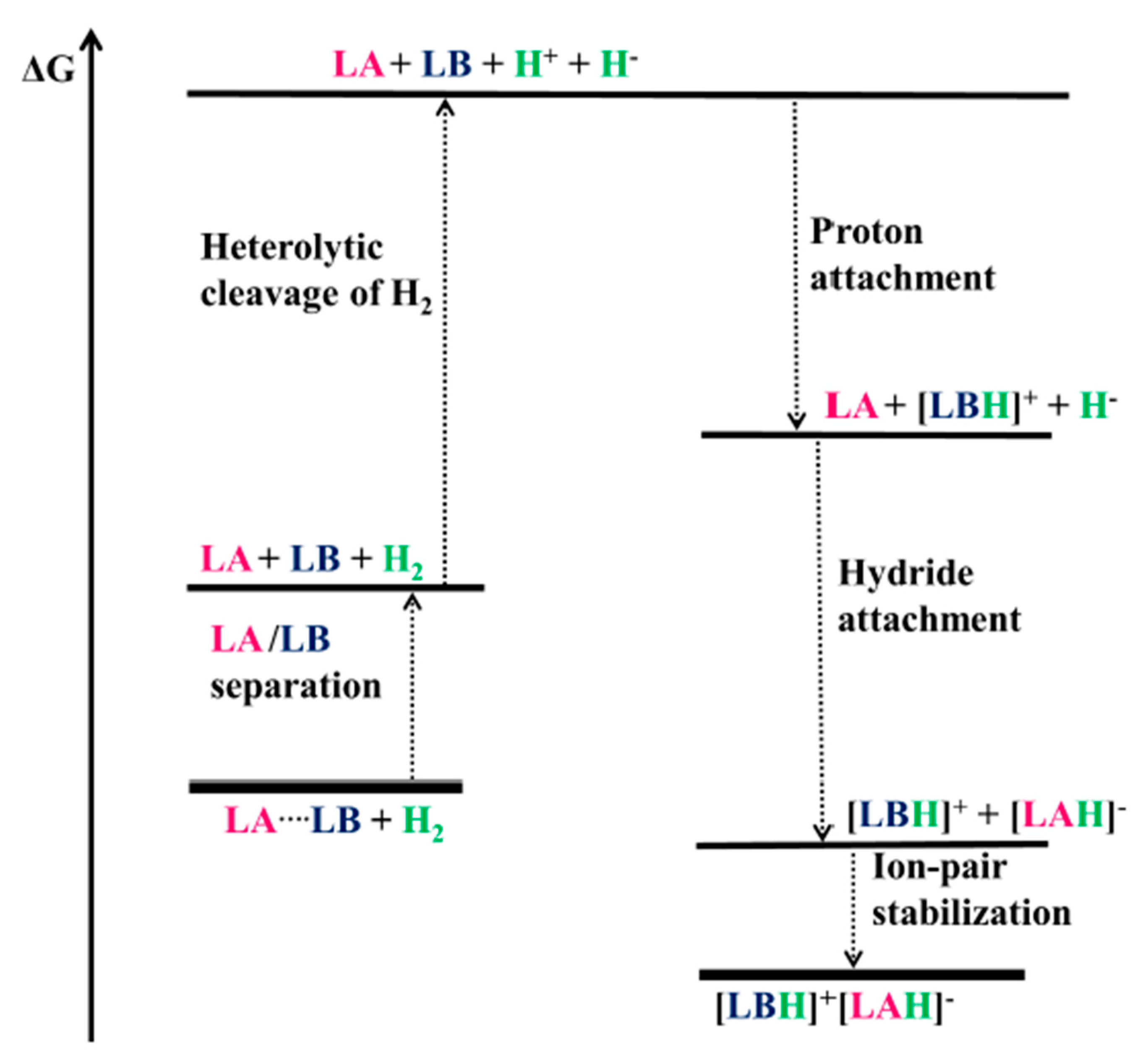
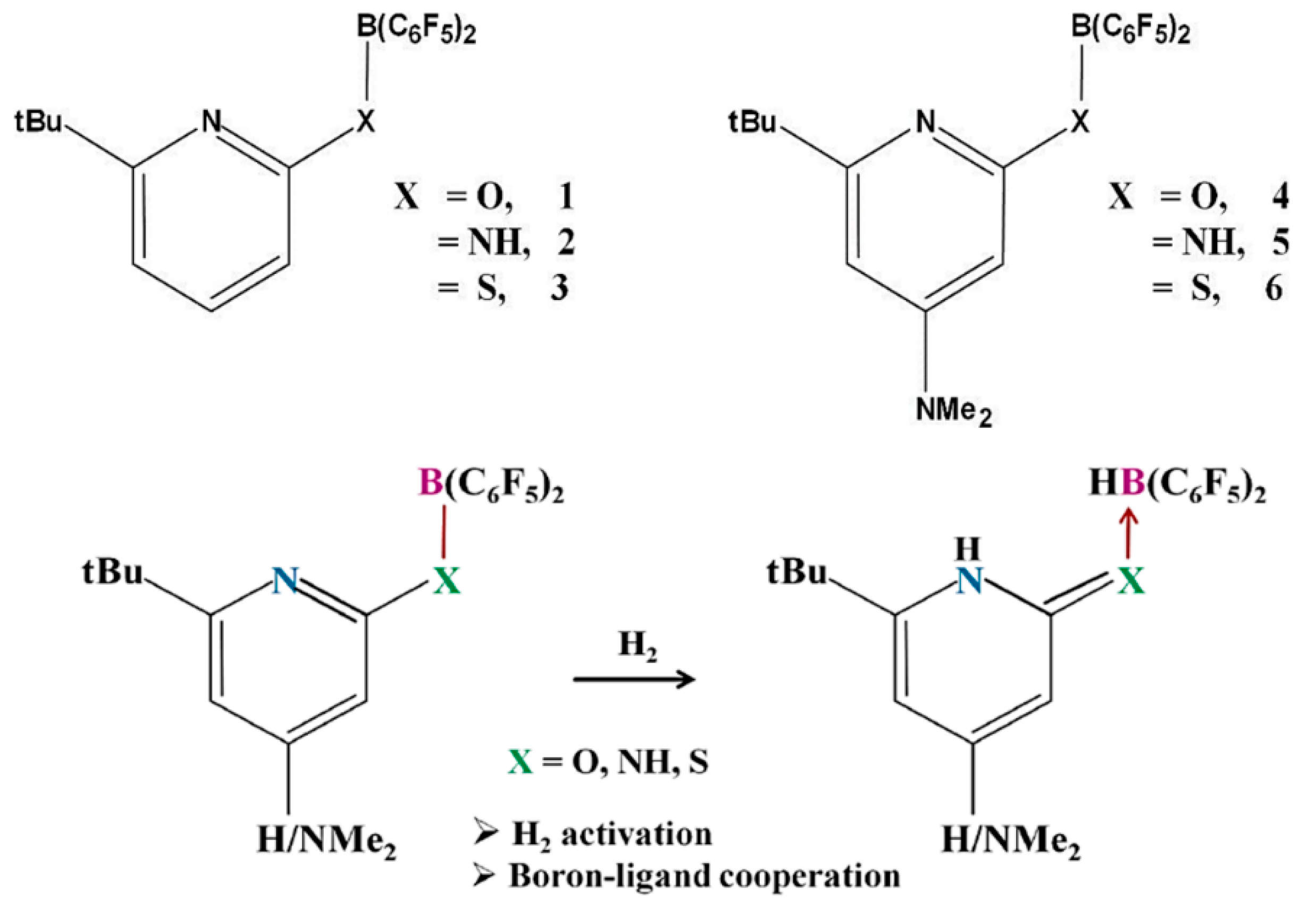
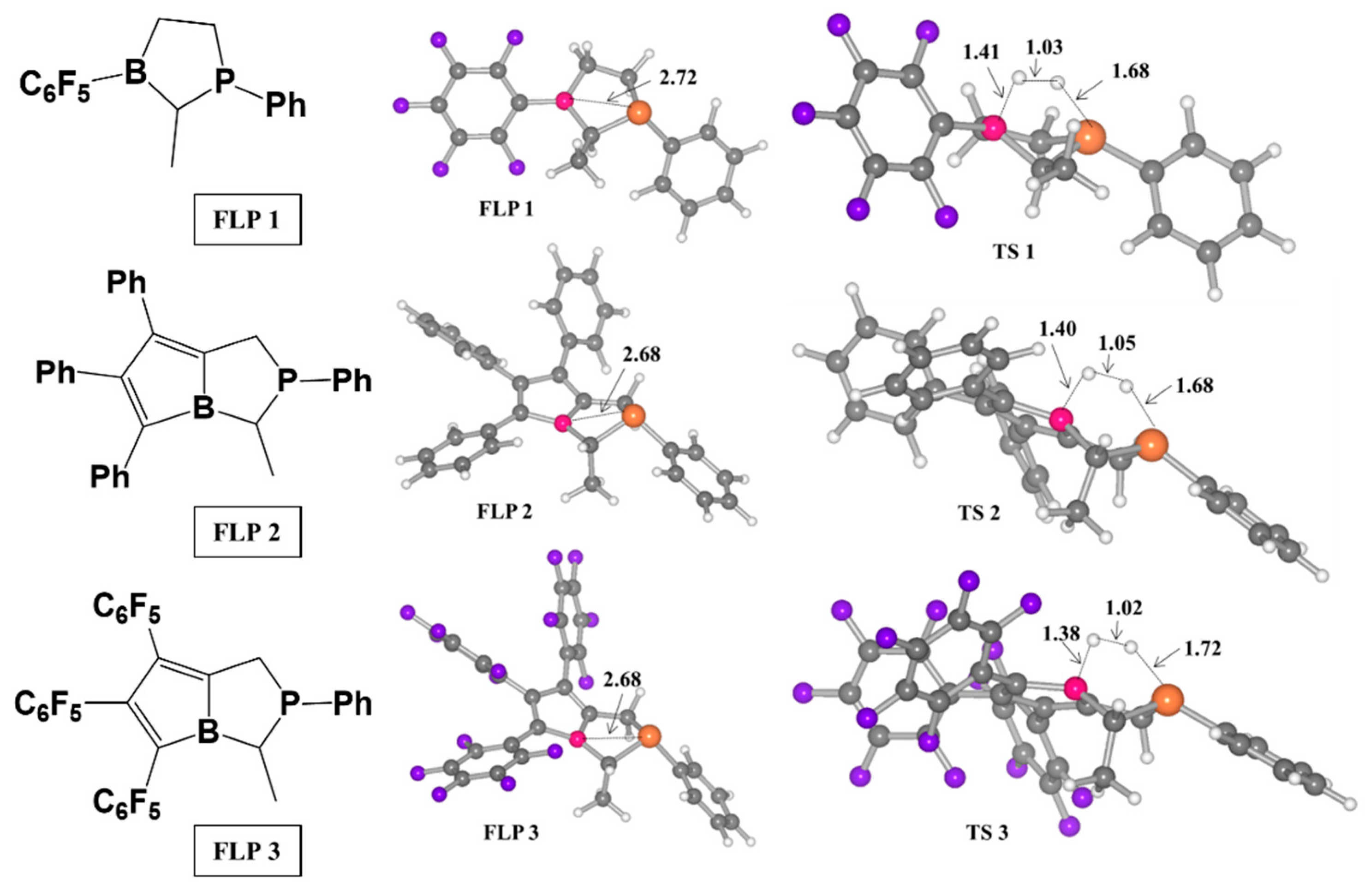
2. H2 Activation by FLPs
3. Catalytic Hydrogenation by FLPs
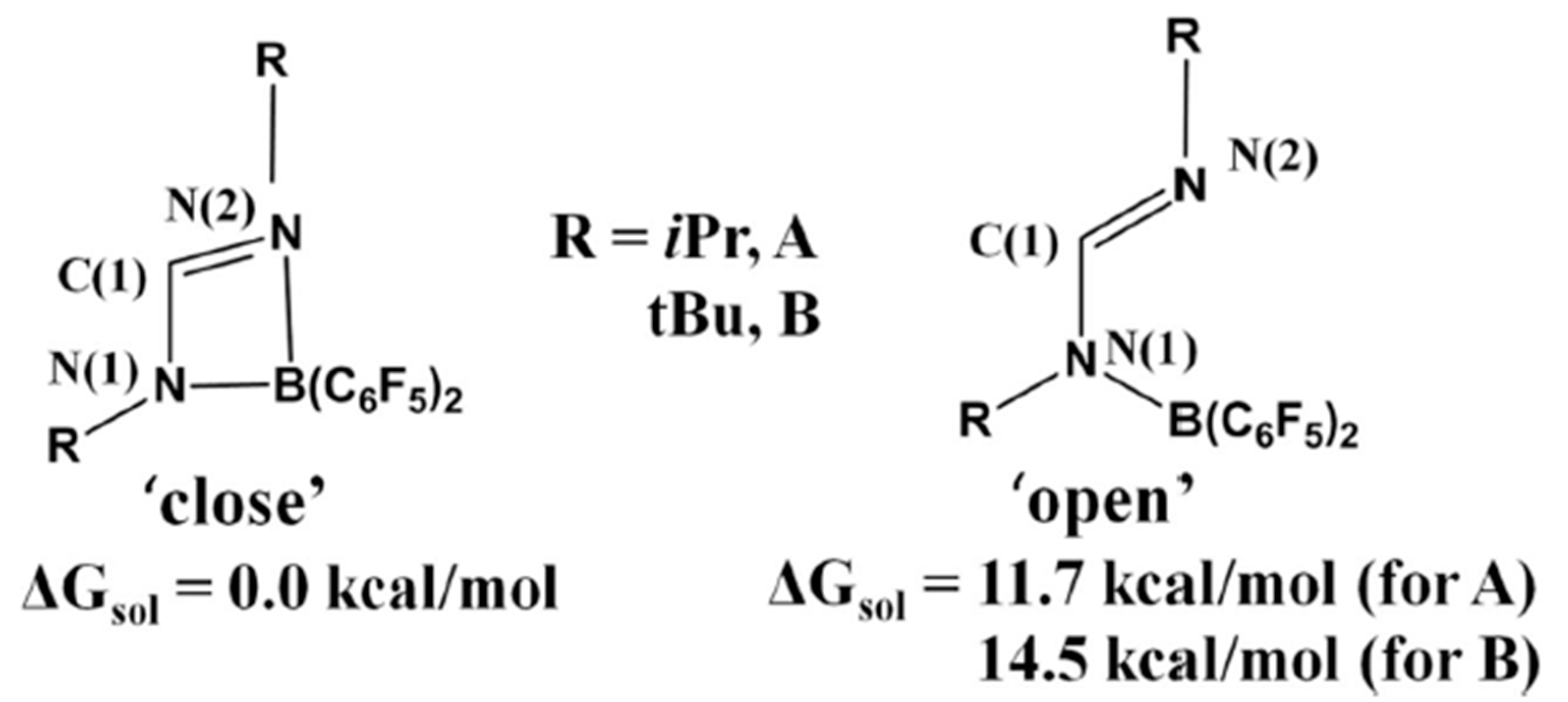
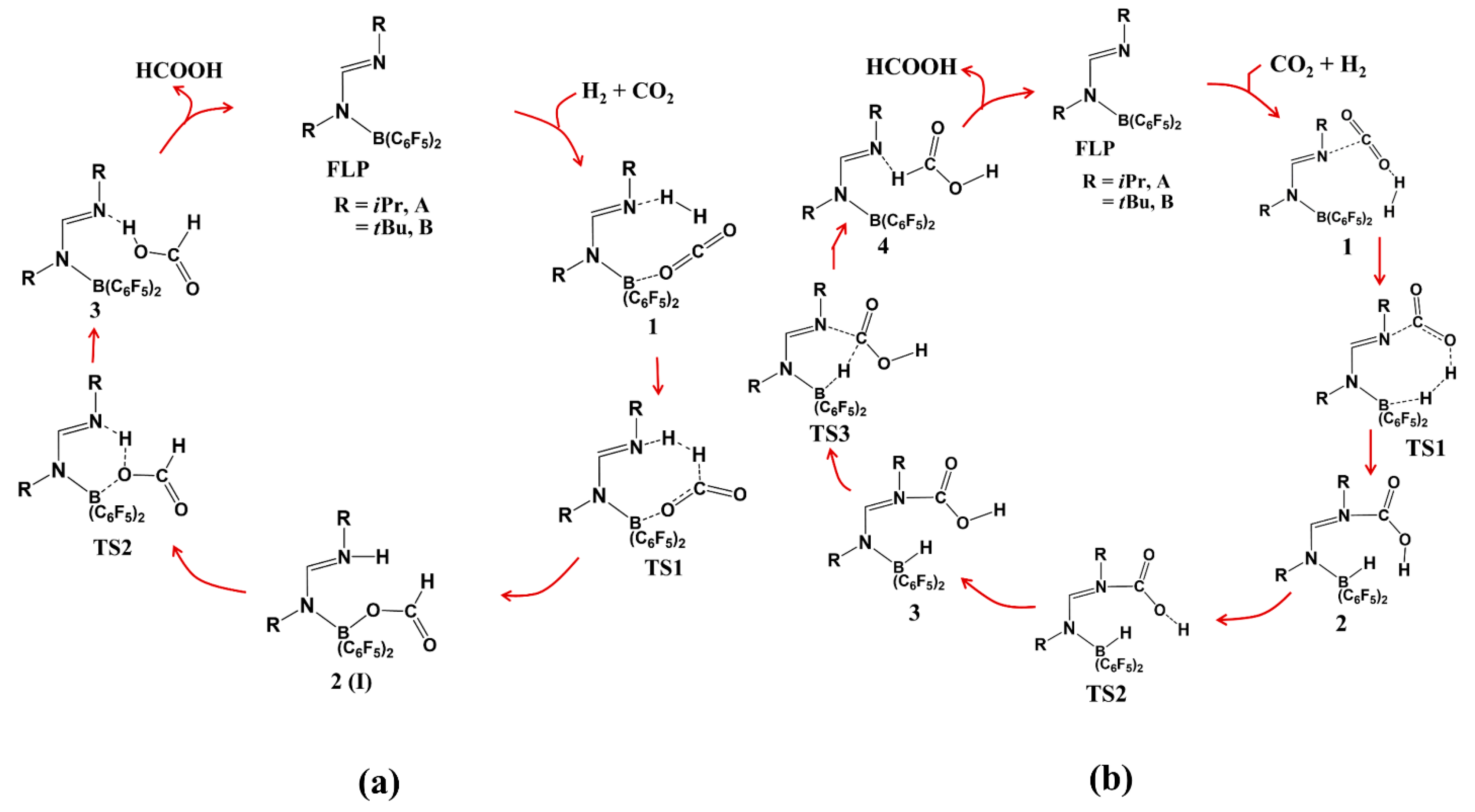
4. Catalytic Hydrogenation of CO2
5. Activation of Other Small Molecules
6. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Berke, H. Conceptual approach to the reactivity of dihydrogen. Chem. Phys. Chem. 2010, 11, 1837–1849. [Google Scholar] [CrossRef] [PubMed]
- de Vries, J.G.; Elsevier, C.J. Handbook of Homogeneous Hydrogenation; WeinheimWiley: Weinheim, Germany, 2007. [Google Scholar]
- Siegbahn, P.E.; Tye, J.W.; Hall, M.B. Computational studies of [NiFe] and [FeFe] hydrogenases. Chem. Rev. 2007, 107, 4414–4435. [Google Scholar] [CrossRef] [PubMed]
- Spielmann, J.; Buch, F.; Harder, S. Early Main-Group Metal Catalysts for the Hydrogenation of Alkenes with H2. Angew. Chem. 2008, 120, 9576–9580. [Google Scholar] [CrossRef]
- Ashley, A.E.; Thompson, A.L.; O’Hare, D. Non-metal-mediated homogeneous hydrogenation of CO2 to CH3OH. Angew. Chem. Int. Ed. 2009, 48, 9839–9843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Power, P.P. Main-group elements as transition metals. Nature 2010, 463, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Frey, G.D.; Lavallo, V.; Donnadieu, B.; Schoeller, W.W.; Bertrand, G. Facile splitting of hydrogen and ammonia by nucleophilic activation at a single carbon center. Science 2007, 316, 439–441. [Google Scholar] [CrossRef] [Green Version]
- Protchenko, A.V.; Birjkumar, K.H.; Dange, D.; Schwarz, A.D.; Vidovic, D.; Jones, C.; Kaltsoyannis, N.; Mountford, P.; Aldridge, S. A stable two-coordinate acyclic silylene. J. Am. Chem. Soc. 2012, 134, 6500–6503. [Google Scholar] [CrossRef]
- Peng, Y.; Guo, J.-D.; Ellis, B.D.; Zhu, Z.; Fettinger, J.C.; Nagase, S.; Power, P.P. Reaction of hydrogen or ammonia with unsaturated germanium or tin molecules under ambient conditions: Oxidative addition versus arene elimination. J. Am. Chem. Soc. 2009, 131, 16272–16282. [Google Scholar] [CrossRef]
- Peng, Y.; Ellis, B.D.; Wang, X.; Power, P.P. Diarylstannylene activation of hydrogen or ammonia with arene elimination. J. Am. Chem. Soc. 2008, 130, 12268–12269. [Google Scholar] [CrossRef]
- Spikes, G.H.; Fettinger, J.C.; Power, P.P. Facile activation of dihydrogen by an unsaturated heavier main group compound. J. Am. Chem. Soc. 2005, 127, 12232–12233. [Google Scholar] [CrossRef]
- Peng, Y.; Brynda, M.; Ellis, B.D.; Fettinger, J.C.; Rivard, E.; Power, P.P. Addition of H2 to distannynes under ambient conditions. Chem. Commun. 2008, 6042–6044. [Google Scholar] [CrossRef] [PubMed]
- Welch, G.C.; Cabrera, L.; Chase, P.A.; Hollink, E.; Masuda, J.D.; Wei, P.; Stephan, D.W. Tuning Lewis acidity using the reactivity of “frustrated Lewis pairs”: Facile formation of phosphine-boranes and cationic phosphonium-boranes. Dalton Trans. 2007, 3407–3414. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.M.; Hatnean, J.A.; Stephan, D.W. Activation of hydrogen and hydrogenation catalysis by a borenium cation. J. Am. Chem. Soc. 2012, 134, 15728–15731. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, A.; Reißmann, M.; Schäfer, A.; Schmidtmann, M.; Müller, T. Dihydrogen Activation by a Silylium Silylene Frustrated Lewis Pair and the Unexpected Isomerization Reaction of a Protonated Silylene. Chem.–A Eur. J. 2014, 20, 9381–9386. [Google Scholar] [CrossRef]
- Ménard, G.; Stephan, D.W. H2 activation and hydride transfer to olefins by Al(C6F5)3-based frustrated Lewis pairs. Angew. Chem. Int. Ed. Engl. 2012, 51, 8272–8275. [Google Scholar] [CrossRef]
- vom Stein, T.; Peréz, M.; Dobrovetsky, R.; Winkelhaus, D.; Caputo, C.B.; Stephan, D.W. Electrophilic Fluorophosphonium Cations in Frustrated Lewis Pair Hydrogen Activation and Catalytic Hydrogenation of Olefins. Angew. Chem. Int. Ed. 2015, 54, 10178–10182. [Google Scholar] [CrossRef]
- Clark, E.R.; Ingleson, M.J. N-Methylacridinium Salts: Carbon Lewis Acids in Frustrated Lewis Pairs for σ-Bond Activation and Catalytic Reductions. Angew. Chem. 2014, 126, 11488–11491. [Google Scholar] [CrossRef]
- Metters, O.J.; Forrest, S.J.; Sparkes, H.A.; Manners, I.; Wass, D.F. Small molecule activation by intermolecular Zr (IV)-phosphine frustrated Lewis pairs. J. Am. Chem. Soc. 2016, 138, 1994–2003. [Google Scholar] [CrossRef]
- Scott, D.J.; Phillips, N.A.; Sapsford, J.S.; Deacy, A.C.; Fuchter, M.J.; Ashley, A.E. Versatile catalytic hydrogenation using a simple tin (IV) Lewis acid. Angew. Chem. 2016, 128, 14958–14962. [Google Scholar] [CrossRef]
- Weicker, S.A.; Stephan, D.W. Main group Lewis acids in frustrated Lewis pair chemistry: Beyond electrophilic boranes. Bull. Chem. Soc. Jpn. 2015, 88, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, H.B.; King, A.M.; Sparkes, H.A.; Pridmore, N.E.; Wass, D.F. Zirconium–Nitrogen Intermolecular Frustrated Lewis Pairs. Inorg. Chem. 2019, 58, 6399–6409. [Google Scholar] [CrossRef] [PubMed]
- Sumerin, V.; Schulz, F.; Nieger, M.; Leskelä, M.; Repo, T.; Rieger, B. Facile heterolytic H2 activation by amines and B (C6F5) 3. Angew. Chem. Int. Ed. 2008, 47, 6001–6003. [Google Scholar] [CrossRef] [PubMed]
- Chase, P.A.; Stephan, D.W. Hydrogen and Amine Activation by a Frustrated Lewis Pair of a Bulky N-Heterocyclic Carbene and B (C6F5) 3. Angew. Chem. 2008, 120, 7543–7547. [Google Scholar] [CrossRef]
- Holschumacher, D.; Bannenberg, T.; Hrib, C.G.; Jones, P.G.; Tamm, M. Heterolytic dihydrogen activation by a frustrated carbene–borane Lewis pair. Angew. Chem. 2008, 120, 7538–7542. [Google Scholar] [CrossRef]
- Hounjet, L.J.; Bannwarth, C.; Garon, C.N.; Caputo, C.B.; Grimme, S.; Stephan, D.W. Combinations of ethers and B (C6F5) 3 function as hydrogenation catalysts. Angew. Chem. 2013, 125, 7640–7643. [Google Scholar] [CrossRef]
- Li, H.; Aquino, A.J.; Cordes, D.B.; Hung-Low, F.; Hase, W.L.; Krempner, C. A zwitterionic carbanion frustrated by boranes–Dihydrogen cleavage with weak Lewis acids via an “inverse” frustrated Lewis pair approach. J. Am. Chem. Soc. 2013, 135, 16066–16069. [Google Scholar] [CrossRef]
- Mummadi, S.; Unruh, D.K.; Zhao, J.; Li, S.; Krempner, C. “Inverse” frustrated Lewis pairs–activation of dihydrogen with organosuperbases and moderate to weak Lewis acids. J. Am. Chem. Soc. 2016, 138, 3286–3289. [Google Scholar] [CrossRef]
- Douglas, W.S. Diverse Uses of the Reaction of Frustrated Lewis Pair (FLP) with Hydrogen. J. Am. Chem. Soc. 2021, 143, 20002–20014. [Google Scholar]
- Sapsford, J.S.; Csókás, D.; Turnell-Ritson, R.C.; Parkin, L.A.; Crawford, A.D.; Pápai, I.; Ashley, A.E. Transition Metal-Free Direct Hydrogenation of Esters via a Frustrated Lewis Pair. ACS Catal. 2021, 11, 9143–9150. [Google Scholar] [CrossRef]
- Lambic, N.S.; Sommer, R.D.; Ison, E.A. Tuning Catalytic Activity in the Hydrogenation of Unactivated Olefins with Transition-Metal Oxos as the Lewis Base Component of Frustrated Lewis Pairs. ACS Catal. 2017, 7, 1170–1180. [Google Scholar] [CrossRef]
- Zhou, S.; Wan, Q.; Lin, S. Cu/O Frustrated Lewis Pairs on Cu Doped CeO2(111) for Acetylene Hydrogenation: A First-Principles Study. Catalysts 2022, 12, 74. [Google Scholar] [CrossRef]
- Liu, X.; Qiu, B.; Yang, X. Bioinspired Design and Computational Prediction of SCS Nickel Pincer Complexes for Hydrogenation of Carbon Dioxide. Catalysts 2020, 10, 319. [Google Scholar] [CrossRef] [Green Version]
- Dureen, M.A.; Stephan, D.W. Terminal alkyne activation by frustrated and classical Lewis acid/phosphine pairs. J. Am. Chem. Soc. 2009, 131, 8396–8397. [Google Scholar] [CrossRef]
- Welch, G.C.; Stephan, D.W. Facile heterolytic cleavage of dihydrogen by phosphines and boranes. J. Am. Chem. Soc. 2007, 129, 1880–1881. [Google Scholar] [CrossRef]
- Otten, E.; Neu, R.C.; Stephan, D.W. Complexation of nitrous oxide by frustrated Lewis pairs. J. Am. Chem. Soc. 2009, 131, 9918–9919. [Google Scholar] [CrossRef] [Green Version]
- McCahill, J.S.; Welch, G.C.; Stephan, D.W. Reactivity of "frustrated Lewis pairs": Three-component reactions of phosphines, a borane, and olefins. Angew. Chem. Int. Ed. Engl. 2007, 46, 4968–4971. [Google Scholar] [CrossRef]
- Ullrich, M.; Seto, K.S.H.; Lough, A.J.; Stephan, D.W. 1,4-Addition reactions of frustrated Lewis pairs to 1,3-dienes. Chem. Commun. 2009, 2335–2337. [Google Scholar] [CrossRef]
- Dureen, M.A.; Lough, A.; Gilbert, T.M.; Stephan, D.W. B–H Activation by frustrated Lewis pairs: Borenium or boryl phosphonium cation? Chem. Commun. 2008, 4303–4305. [Google Scholar] [CrossRef]
- Ghara, M.; Pan, S.; Chattaraj, P.K. A theoretical investigation on boron–ligand cooperation to activate molecular hydrogen by a frustrated Lewis pair and subsequent reduction of carbon dioxide. Phys. Chem. Chem. Phys. 2019, 21, 21267–21277. [Google Scholar] [CrossRef]
- Dureen, M.A.; Stephan, D.W. Reactions of boron amidinates with CO2 and CO and other small molecules. J. Am. Chem. Soc. 2010, 132, 13559–13568. [Google Scholar] [CrossRef]
- Sajid, M.; Klose, A.; Birkmann, B.; Liang, L.; Schirmer, B.; Wiegand, T.; Eckert, H.; Lough, A.J.; Froehlich, R.; Daniliuc, C.G. Reactions of phosphorus/boron frustrated Lewis pairs with SO2. Chem. Sci. 2013, 4, 213–219. [Google Scholar] [CrossRef]
- Cardenas, A.J.P.; Culotta, B.J.; Warren, T.H.; Grimme, S.; Stute, A.; Fröhlich, R.; Kehr, G.; Erker, G. Capture of NO by a Frustrated Lewis Pair: A New Type of Persistent N-Oxyl Radical. Angew. Chem. Int. Ed. 2011, 50, 7567–7571. [Google Scholar] [CrossRef]
- Mömming, C.M.; Frömel, S.; Kehr, G.; Fröhlich, R.; Grimme, S.; Erker, G. Reactions of an intramolecular frustrated Lewis pair with unsaturated substrates: Evidence for a concerted olefin addition reaction. J. Am. Chem. Soc. 2009, 131, 12280–12289. [Google Scholar] [CrossRef] [PubMed]
- Longobardi, L.E.; Wolter, V.; Stephan, D.W. Frustrated Lewis Pair Activation of an N-Sulfinylamine: A Source of Sulfur Monoxide. Angew. Chem. Int. Ed. 2015, 54, 809–812. [Google Scholar] [CrossRef] [PubMed]
- Sajid, M.; Lawzer, A.; Dong, W.; Rosorius, C.; Sander, W.; Schirmer, B.; Grimme, S.; Daniliuc, C.G.; Kehr, G.; Erker, G. Carbonylation Reactions of Intramolecular Vicinal Frustrated Phosphane/Borane Lewis Pairs. J. Am. Chem. Soc. 2013, 135, 18567–18574. [Google Scholar] [CrossRef] [PubMed]
- Ekkert, O.; Miera, G.G.; Wiegand, T.; Eckert, H.; Schirmer, B.; Petersen, J.L.; Daniliuc, C.G.; Fröhlich, R.; Grimme, S.; Kehr, G. Remarkable coordination behavior of alkyl isocyanides toward unsaturated vicinal frustrated P/B Lewis pairs. Chem. Sci. 2013, 4, 2657–2664. [Google Scholar] [CrossRef]
- Appelt, C.; Westenberg, H.; Bertini, F.; Ehlers, A.W.; Slootweg, J.C.; Lammertsma, K.; Uhl, W. Geminal Phosphorus/Aluminum-Based Frustrated Lewis Pairs: C–H versus C≡C Activation and CO2 Fixation. Angew. Chem. Int. Ed. 2011, 50, 3925–3928. [Google Scholar] [CrossRef]
- Welch, G.C.; San Juan, R.R.; Masuda, J.D.; Stephan, D.W. Reversible, metal-free hydrogen activation. Science 2006, 314, 1124–1126. [Google Scholar] [CrossRef] [Green Version]
- Nori, V.; Pesciaioli, F.; Sinibaldi, A.; Giorgianni, G.; Carlone, A. Boron-Based Lewis Acid Catalysis: Challenges and Perspectives. Catalysts 2022, 12, 5. [Google Scholar] [CrossRef]
- Schreiner, P.R.; Schaefer, H.F., III; von Ragué Schleyer, P. The structure and stability of BH5. Does correlation make it a stable molecule? Qualitative changes at high levels of theory. J. Chem. Phys. 1994, 101, 7625–7632. [Google Scholar] [CrossRef]
- Rokob, T.A.; Hamza, A.; Stirling, A.; Soós, T.; Pápai, I. Turning frustration into bond activation: A theoretical mechanistic study on heterolytic hydrogen splitting by frustrated Lewis pairs. Angew. Chem. Int. Ed. 2008, 47, 2435–2438. [Google Scholar] [CrossRef] [PubMed]
- Jupp, A.R.; Stephan, D.W. New directions for frustrated Lewis pair chemistry. Trends Chem. 2019, 1, 35–48. [Google Scholar] [CrossRef]
- Brown, L.C.; Hogg, J.M.; Gilmore, M.; Moura, L.; Imberti, S.; Gärtner, S.; Gunaratne, H.N.; O’Donnell, R.J.; Artioli, N.; Holbrey, J.D. Frustrated Lewis pairs in ionic liquids and molecular solvents–A neutron scattering and NMR study of encounter complexes. Chem. Commun. 2018, 54, 8689–8692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakó, I.; Stirling, A.; Bálint, S.; Pápai, I. Association of frustrated phosphine–borane pairs in toluene: Molecular dynamics simulations. Dalton Trans. 2012, 41, 9023–9025. [Google Scholar] [CrossRef]
- Hamza, A.; Stirling, A.; András Rokob, T.; Pápai, I. Mechanism of hydrogen activation by frustrated Lewis pairs: A molecular orbital approach. Int. J. Quant. Chem. 2009, 109, 2416–2425. [Google Scholar] [CrossRef]
- Grimme, S.; Kruse, H.; Goerigk, L.; Erker, G. The mechanism of dihydrogen activation by frustrated Lewis pairs revisited. Angew. Chem. Int. Ed. 2010, 49, 1402–1405. [Google Scholar] [CrossRef]
- Rokob, T.A.; Bako, I.; Stirling, A.; Hamza, A.; Papai, I. Reactivity models of hydrogen activation by frustrated Lewis pairs: Synergistic electron transfers or polarization by electric field? J. Am. Chem. Soc. 2013, 135, 4425–4437. [Google Scholar] [CrossRef]
- Skara, G.; De Vleeschouwer, F.; Geerlings, P.; De Proft, F.; Pinter, B. Heterolytic Splitting of Molecular Hydrogen by Frustrated and Classical Lewis Pairs: A Unified Reactivity Concept. Sci. Rep. 2017, 7, 16024. [Google Scholar] [CrossRef]
- Rokob, T.A.; Hamza, A.; Pápai, I. Rationalizing the reactivity of frustrated Lewis pairs: Thermodynamics of H2 activation and the role of acid–base properties. J. Am. Chem. Soc. 2009, 131, 10701–10710. [Google Scholar] [CrossRef]
- Bordwell, F.G. Equilibrium acidities in dimethyl sulfoxide solution. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar] [CrossRef]
- Beckett, M.A.; Strickland, G.C.; Holland, J.R.; Varma, K.S. A convenient nmr method for the measurement of Lewis acidity at boron centres: Correlation of reaction rates of Lewis acid initiated epoxide polymerizations with Lewis acidity. Polymer 1996, 37, 4629–4631. [Google Scholar] [CrossRef]
- Heiden, Z.M.; Lathem, A.P. Establishing the hydride donor abilities of main group hydrides. Organometallics 2015, 34, 1818–1827. [Google Scholar] [CrossRef]
- Stephan, D.W.; Erker, G. Frustrated Lewis pair chemistry: Development and perspectives. Angew. Chem. Int. Ed. 2015, 54, 6400–6441. [Google Scholar] [CrossRef]
- Stephan, D.W.; Erker, G. Frustrated Lewis pairs: Metal-free hydrogen activation and more. Angew. Chem. Int. Ed. 2010, 49, 46–76. [Google Scholar] [CrossRef]
- Chase, P.A.; Stephan, D.W. Lewis acid-catalyzed hydrogenation: B (C6F5) 3-mediated reduction of imines and nitriles with H2. Chem. Commun. 2008, 1701–1703. [Google Scholar] [CrossRef]
- Mahdi, T.; Stephan, D.W. Enabling catalytic ketone hydrogenation by frustrated Lewis pairs. J. Am. Chem. Soc. 2014, 136, 15809–15812. [Google Scholar] [CrossRef]
- Scott, D.J.; Fuchter, M.J.; Ashley, A.E. Nonmetal catalyzed hydrogenation of carbonyl compounds. J. Am. Chem. Soc. 2014, 136, 15813–15816. [Google Scholar] [CrossRef] [Green Version]
- Mahdi, T.; Stephan, D.W. Facile protocol for catalytic frustrated Lewis pair hydrogenation and reductive deoxygenation of ketones and aldehydes. Angew. Chem. Int. Ed. 2015, 54, 8511–8514. [Google Scholar] [CrossRef]
- Gao, B.; Feng, X.; Meng, W.; Du, H. Asymmetric hydrogenation of ketones and enones with chiral Lewis base derived frustrated Lewis pairs. Angew. Chem. Int. Ed. 2020, 59, 4498–4504. [Google Scholar] [CrossRef]
- Spies, P.; Schwendemann, S.; Lange, S.; Kehr, G.; Fröhlich, R.; Erker, G. Metal-Free Catalytic Hydrogenation of Enamines, Imines, and Conjugated Phosphinoalkenylboranes. Angew. Chem. Int. Ed. 2008, 47, 7543–7546. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Du, H. A highly enantioselective hydrogenation of silyl enol ethers catalyzed by chiral frustrated Lewis pairs. J. Am. Chem. Soc. 2014, 136, 12261–12264. [Google Scholar] [CrossRef] [PubMed]
- Greb, L.; Oña-Burgos, P.; Schirmer, B.; Grimme, S.; Stephan, D.W.; Paradies, J. Metal-free Catalytic Olefin Hydrogenation: Low-Temperature H2 Activation by Frustrated Lewis Pairs. Angew. Chem. Int. Ed. 2012, 51, 10164–10168. [Google Scholar] [CrossRef] [PubMed]
- Chernichenko, K.; Madarász, Á.; Pápai, I.; Nieger, M.; Leskelä, M.; Repo, T. A frustrated-Lewis-pair approach to catalytic reduction of alkynes to cis-alkenes. Nat. Chem. 2013, 5, 718–723. [Google Scholar] [CrossRef] [Green Version]
- Mahdi, T.; Heiden, Z.M.; Grimme, S.; Stephan, D.W. Metal-free aromatic hydrogenation: Aniline to cyclohexyl-amine derivatives. J. Am. Chem. Soc. 2012, 134, 4088–4091. [Google Scholar] [CrossRef] [PubMed]
- Segawa, Y.; Stephan, D.W. Metal-free hydrogenation catalysis of polycyclic aromatic hydrocarbons. Chem. Commun. 2012, 48, 11963–11965. [Google Scholar] [CrossRef]
- Geier, S.J.; Chase, P.A.; Stephan, D.W. Metal-free reductions of N-heterocycles via Lewis acid catalyzed hydrogenation. Chem. Commun. 2010, 46, 4884–4886. [Google Scholar] [CrossRef]
- Tussing, S.; Greb, L.; Tamke, S.; Schirmer, B.; Muhle-Goll, C.; Luy, B.; Paradies, J. Autoinduced catalysis and inverse equilibrium isotope effect in the frustrated Lewis pair catalyzed hydrogenation of imines. Chem.–A Eur. J. 2015, 21, 8056–8059. [Google Scholar] [CrossRef]
- Greb, L.; Tussing, S.; Schirmer, B.; Ona-Burgos, P.; Kaupmees, K.; Lokov, M.; Leito, I.; Grimme, S.; Paradies, J. Electronic effects of triarylphosphines in metal-free hydrogen activation: A kinetic and computational study. Chem. Sci. 2013, 4, 2788–2796. [Google Scholar] [CrossRef]
- Tussing, S.; Kaupmees, K.; Paradies, J. Structure–Reactivity Relationship in the Frustrated Lewis Pair (FLP)-Catalyzed Hydrogenation of Imines. Chem.–A Eur. J. 2016, 22, 7422–7426. [Google Scholar] [CrossRef]
- Courtemanche, M.-A.; Pulis, A.P.; Rochette, É.; Légaré, M.-A.; Stephan, D.W.; Fontaine, F.-G. Intramolecular B/N frustrated Lewis pairs and the hydrogenation of carbon dioxide. Chem. Commun. 2015, 51, 9797–9800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Vankova, N.; Heine, T. A kinetic study on the reduction of CO2 by frustrated Lewis pairs: From understanding to rational design. Phys. Chem. Chem. Phys. 2016, 18, 3567–3574. [Google Scholar] [CrossRef] [PubMed]
- Ménard, G.; Stephan, D.W. Room temperature reduction of CO2 to methanol by Al-based frustrated Lewis pairs and ammonia borane. J. Am. Chem. Soc. 2010, 132, 1796–1797. [Google Scholar] [CrossRef] [PubMed]
- Shinde, G.Y.; Mote, A.S.; Gawande, M.B. Recent Advances of Photocatalytic Hydrogenation of CO2 to Methanol. Catalysts 2022, 12, 94. [Google Scholar] [CrossRef]
- Bibi, M.; Ullah, R.; Sadiq, M.; Sadiq, S.; Khan, I.; Saeed, K.; Zia, M.A.; Iqbal, Z.; Ullah, I.; Ahmad, S.; et al. Catalytic Hydrogenation of Carbon Dioxide over Magnetic Nanoparticles: Modification in Fixed-Bed Reactor. Catalysts 2021, 11, 592. [Google Scholar] [CrossRef]
- Ghara, M.; Chattaraj, P.K. A computational study on hydrogenation of CO2, catalyzed by a bridged B/N frustrated Lewis pair. Struct. Chem. 2019, 30, 1067–1077. [Google Scholar] [CrossRef]
- Jiang, B.; Zhang, Q.; Dang, L. Theoretical studies on bridged frustrated Lewis pair (FLP) mediated H2 activation and CO2 hydrogenation. Org. Chem. Front. 2018, 5, 1905–1915. [Google Scholar] [CrossRef]
- Mentoor, K.; Twigge, L.; Hans Niemantsverdriet, J.W.; Swarts, J.C.; Erasmus, E. Silica Nanopowder Supported Frustrated Lewis Pairs for CO2 Capture and Conversion to Formic Acid. Inorg. Chem. 2021, 60, 55–69. [Google Scholar] [CrossRef]
- Liu, L.; Lukose, B.; Ensing, B. A free energy landscape of CO2 capture by frustrated Lewis pairs. ACS Catal. 2018, 8, 3376–3381. [Google Scholar] [CrossRef]
- Mömming, C.M.; Otten, E.; Kehr, G.; Fröhlich, R.; Grimme, S.; Stephan, D.W.; Erker, G. Reversible metal-free carbon dioxide binding by frustrated Lewis pairs. Angew. Chem. Int. Ed. 2009, 48, 6643–6646. [Google Scholar] [CrossRef] [Green Version]
- Roth, D.; Stirn, J.; Stephan, D.W.; Greb, L. Lewis Superacidic Catecholato Phosphonium Ions: Phosphorus–Ligand Cooperative C–H Bond Activation. J. Am. Chem. Soc. 2021, 143, 15845–15851. [Google Scholar] [CrossRef]
- Ghara, M.; Giri, S.; Chattaraj, P.K. Cycloaddition Reactions between H2C = CHR (R = H, CN, CH3) and a Cyclic P/B Frustrated Lewis Pair: A DFT Study. J. Phys. Chem. A 2020, 124, 4455–4462. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Trujillo, J.J.; Fernández, I. Aromaticity can enhance the reactivity of P-donor/borole frustrated Lewis pairs. Chem. Commun. 2019, 55, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Daniliuc, C.G.; Kehr, G.; Erker, G. Formation of Active Cyclic Five-membered Frustrated Phosphane/Borane Lewis Pairs and their Cycloaddition Reactions. Chem. Eur. J. 2020, 26, 745. [Google Scholar] [CrossRef] [PubMed]
- Ghara, M.; Chattaraj, P.K. Can a Decrease in Anti-aromaticity Increase the Dihydrogen Activation Ability of a Frustrated Phosphorous/Borane Lewis Pair?: A DFT Study. Theor. Chem. Acc. 2020, 139, 183. [Google Scholar] [CrossRef]
- Zhuang, D.; Rouf, A.M.; Li, Y.; Dai, C.; Zhu, J. Aromaticity-promoted CO2 Capture by P/N-Based Frustrated Lewis Pairs: A Theoretical Study. Chem. Asian J. 2020, 15, 266–272. [Google Scholar] [CrossRef]
- Rouf, A.M.; Huang, Y.; Dong, S.; Zhu, J. Systematic Design of a Frustrated Lewis Pair Containing Methyleneborane and Carbene for Dinitrogen Activation. Inorg. Chem. 2021, 60, 5598–5606. [Google Scholar] [CrossRef]
- Voss, T.; Chen, C.; Kehr, G.; Nauha, E.; Erker, G.; Stephan, D.W. Cyclizations via frustrated Lewis pairs: Lewis acid induced intramolecular additions of amines to olefins and alkynes. Chem.–A Eur. J. 2010, 16, 3005–3008. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, L.; Yao, C.; Geng, J.; Wu, Y.; Hu, X. Controlling the Lewis Acidity and Polymerizing Effectively Prevent Frustrated Lewis Pairs from Deactivation in the Hydrogenation of Terminal Alkynes. Org. Lett. 2021, 23, 3685–3690. [Google Scholar] [CrossRef]
- Bhattacharjee, I.; Bhunya, S.; Paul, A. Frustrated Lewis Acid–Base-Pair-Catalyzed Amine-Borane Dehydrogenation. Inorg. Chem. 2020, 59, 1046–1056. [Google Scholar] [CrossRef]
- Sitte, N.A.; Bursch, M.; Grimme, S.; Paradies, J. Frustrated Lewis Pair Catalyzed Hydrogenation of Amides: Halides as Active Lewis Base in the Metal-Free Hydrogen Activation. J. Am. Chem. Soc. 2019, 141, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Gupta, R.; Jaiswal, A.K.; Young, R.D. Frustrated Lewis-pair-meditated selective single fluoride substitution in trifluoromethyl Groups. J. Am. Chem. Soc. 2020, 142, 2572–2578. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Mandal, D.; Jaiswal, A.K.; Young, R.D. FLP-Catalyzed Monoselective C–F Functionalization in Polyfluorocarbons at Geminal or Distal Sites. Org. Lett. 2021, 23, 1915–1920. [Google Scholar] [CrossRef]
- Ghara, M.; Giri, S.; Das, P.; Chattaraj, P.K. Possible C-F Bond Activation by B(C6F5)3/Lutidine and Al(C6F5)3/Lutidine Frustrated Lewis Pair: An In Silico Study. J. Chem. Sci. 2022, 134, 1–12. [Google Scholar] [CrossRef]
- Ison, E.A.; Tubb, J.L. Energy Decomposition Analysis of Lewis Acid/Base Adducts and Frustrated Lewis Pairs: The Use of EOrb/ESteric Ratios as a Reaction Parameter. Inorg. Chem. 2021, 60, 13797–13805. [Google Scholar] [CrossRef] [PubMed]
- McGraw, M.L.; Chen, E.Y.X. Lewis Pair Polymerization: Perspective on a Ten-Year Journey. Macromolecules 2020, 53, 6102–6122. [Google Scholar] [CrossRef]
- McGraw, M.L.; Clarke, R.W.; Chen, E.Y. Compounded Sequence Control in Polymerization of One-Pot Mixtures of Highly Reactive Acrylates by Differentiating Lewis Pairs. J. Am. Chem. Soc. 2020, 142, 5969–5973. [Google Scholar] [CrossRef]
- Su, Y.; Zhao, Y.; Zhang, H.; Luo, Y.; Xu, X. Rare-Earth Aryloxide/Ylide-Functionalized Phosphine Frustrated Lewis Pairs for the Polymerization of 4-Vinylpyridine and Its Derivatives. Macromolecules 2021, 54, 7724–7731. [Google Scholar] [CrossRef]
- Yolsal, U.; Horton, T.A.R.; Wang, M.; Shaver, M.P. Cyclic Ether Triggers for Polymeric Frustrated Lewis Pair Gels. J. Am. Chem. Soc. 2021, 143, 12980–12984. [Google Scholar] [CrossRef] [PubMed]
- Yolsal, U.; Wang, M.; Royer, J.R.; Shaver, M.P. Rheological Characterization of Polymeric Frustrated Lewis Pair Networks. Macromolecules 2019, 52, 3417–3425. [Google Scholar] [CrossRef]
- Shi, Q.; Chen, Z.; Hu, J. Recent Advances in Organocatalyzed Asymmetric Hydrosilylations. Curr. Org. Chem. 2018, 22, 557580. [Google Scholar] [CrossRef]
- Arkhipenko, S.; Whiting, A. Broadening the synthetic organic applications of Frustrated Lewis Pairs. Arkivoc 2017, 2017, 26–40. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial No. | Intermolecular FLP | Small Molecules Activated | Reference Number | |
|---|---|---|---|---|
| Donor | Acceptor | |||
| 1 | PtBu3 |  | H2 | [15] |
| 2 |  | (Me5C6)3Si+ | H2 | [16] |
| 3 | PtBu3 | Al(C6F5)3 | H2, Alkyne | [17,35] |
| 4 |  | 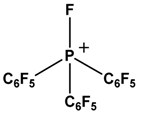 | H2 | [18] |
| 5 | 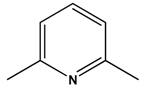 | 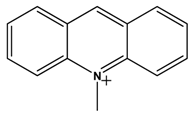 | H2 | [19] |
| 6 | PtBu3 |  | H2, CO2, THF, Phenylacetylene | [20] |
| 7 | 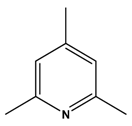 | 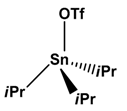 | H2 | [21] |
| 8 | 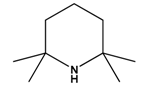 | B(C6F5)3 | H2 | [24] |
| 9 | 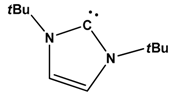 | B(C6F5)3 | H2 | [25] |
| 10 | 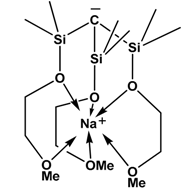 | BPh3 | H2 | [28] |
| 11 | PtBu3 | B(C6F5)3 | H2, N2O, Ethylene, Alkyne, 1,3-Diene, B-H bond | [35,36,37,38,39,40] |
| Serial No. | Intramolecular FLP | Small Molecules Activated | Reference Number |
|---|---|---|---|
| 1 |  | H2 | [14] |
| 2 | 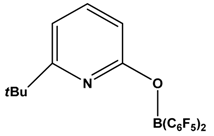 | H2 | [41] |
| 3 | 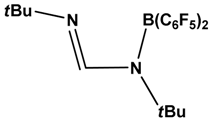 | CO, CO2, RNC, PhCCH, MeCN | [42] |
| 4 | 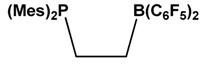 | SO2, NO, Olefin, N-Sulfinylamine | [43,44,45,46] |
| 5 | 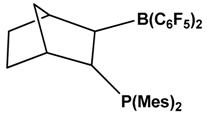 | CO | [47] |
| 6 | 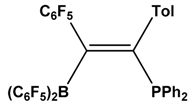 | RNC | [48] |
| 7 | 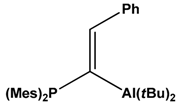 | Acetylene, CO2 | [49] |
| H2 Cleavage by Transition Metal | H2 Cleavage by FLP | ||
|---|---|---|---|
| Complex | Free Energy Barrier of H2 Activation | FLP | Free Energy Barrier of H2 Activation |
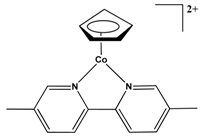 | 16.8 Kcal/mol at SMD(Water)-M06/6-311++G(2df,p)//M06/6-31+G(d,p) level |  | 21.7 Kcal/mol at SMD(Benzene)-ωB97XD/6-31++G(d,p) level |
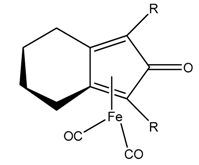 | 32.3 Kcal/mol at SMD(Methanol)-M06/6-31++G(d,p) level | 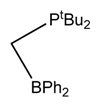 | 28.9 Kcal/mol at PCM(toluene)-M06-2X/def2-TZVP//M06-2X/def2-SVP level |
 | 5.9 Kcal/mol at SMD(Water)-M06/6-31++G(d,p) level | 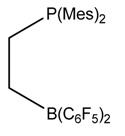 | 20.7 Kcal/mol at PCM(CH2Cl2)-B3LYP/6-311++G(d,p)//B3LYP/6-31G(d) level |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pal, R.; Ghara, M.; Chattaraj, P.K. Activation of Small Molecules and Hydrogenation of CO2 Catalyzed by Frustrated Lewis Pairs. Catalysts 2022, 12, 201. https://doi.org/10.3390/catal12020201
Pal R, Ghara M, Chattaraj PK. Activation of Small Molecules and Hydrogenation of CO2 Catalyzed by Frustrated Lewis Pairs. Catalysts. 2022; 12(2):201. https://doi.org/10.3390/catal12020201
Chicago/Turabian StylePal, Ranita, Manas Ghara, and Pratim Kumar Chattaraj. 2022. "Activation of Small Molecules and Hydrogenation of CO2 Catalyzed by Frustrated Lewis Pairs" Catalysts 12, no. 2: 201. https://doi.org/10.3390/catal12020201
APA StylePal, R., Ghara, M., & Chattaraj, P. K. (2022). Activation of Small Molecules and Hydrogenation of CO2 Catalyzed by Frustrated Lewis Pairs. Catalysts, 12(2), 201. https://doi.org/10.3390/catal12020201