Abstract
The results of recent studies on the mechanism of stereoinduction in asymmetric hydrogenation catalyzed by transition metal complexes suggest that hydrogen activation by metal atoms and the generation of enantioselectivity by organic ligands proceed independently. Hence, these reactions can be considered as variants of a cooperative organocatalytic reaction. This conclusion opens a broader view on rational catalyst design, suggesting that the structural ideas from different fields can be exploited reciprocally.
1. Introduction
The first homogeneous catalytic reaction (studied and realized as such) was the synthesis of ether from ethanol in the presence of catalytic amounts of sulfuric acid [1]. It was probably the case at that time that p-toluenesulfonic acid could not be found in Williamson’s laboratory, otherwise it could simultaneously have become the first organocatalytic reaction. On the other hand, had titanium tetrachloride been on the shelf, the first transition-metal-catalyzed reaction could have been discovered.
Although it is well-understood that the principles of catalysis are essentially the same for both types of asymmetric catalysis, the merits and drawbacks of these methods are usually discussed separately.
In my opinion, nowadays, considering that both areas are mature and each has been decorated with a Nobel Prize [2,3,4,5,6,7,8], it is important to look for ways to use our knowledge and experience accumulated for them in a collaborative way.
It is usually argued that asymmetric organocatalysis is a “greener” method compared to asymmetric transition metal catalysis. However, a recently published analysis of the environmental impacts showed that only very few asymmetric organocatalysts can really be considered as “green” ones due to the structural complexity and multistage syntheses necessary for their preparation [9].
Many organocatalytic reactions can be carried out without taking care of oxygen and/or moisture in the atmosphere (probably with the exception of aldol condensations), and the catalysts are more recyclable.
On the other hand, transition-metal-catalyzed reactions are usually characterized by significantly lower catalyst loadings.
Despite the evident success of organocatalysis in areas such as aldol reactions in addition to the Michael and Mannich additions, transformations such as catalytic hydrogenation or C-H bond activation normally require additional stimulation via the introduction of some extra activation center, either intra- (bifunctional catalysis [10], double-activation catalysis [11]) or intermolecularly (cascade catalysis [12], synergistic catalysis [13,14]). In the latter cases a combination of an organocatalyst (ligand) with a transition metal salt is used quite often [15]. The role of the metal is to weaken or break the chemical bonds, whereas the organocatalyst is responsible for creating the asymmetric environment essential for enantioselection during new bond formations. It is reasonably suggested that the metal salt and organic ligand can form a chelate complex in the course of the reaction [15]. As a result, the reactions set-ups appear exactly the same as those for transition-metal-catalyzed reactions carried out with in-situ-generated catalysts, e.g., [16].
The purpose of this essay is to illuminate experimental evidence that allows transition-metal-catalyzed asymmetric catalytic reactions to be considered as a kind of cooperation catalysis, where the functions of chemical bonds’ activation by transition metals and stereoinduction effectuated by the organic ligand are separated in space and time. This viewpoint gives reasons for a more productive exchange of ideas on rational catalyst design in a broad area of asymmetric catalysis.
2. Rh-Catalyzed Asymmetric Hydrogenation
Mechanistic studies on Rh-catalyzed asymmetric hydrogenation started almost immediately after its discovery. These efforts resulted in a decently grounded theory involving hydrogen activation after substrate coordination. It implied enantioselection during the oxidative addition step of hydrogen, i.e., the Rh atom coordinated to the double bond of the substrate was directly involved in the stage responsible for chirality generation [17,18].
However, more recent research actively carried out in the last 20 years brought extensive evidence that suggested instead that the role of transition metals in this process is restricted to the hydrogen activation, whereas the chiral ligand takes the responsibility for the stereoinduction [19,20]. This evidence is briefly summarized below.
2.1. Low-Temperature Reactions of Solvate Dihydrides with Prochiral Substrates
Numerous experiments with various catalysts and prochiral substrates demonstrated that reactions of solvate dihydrides 1 and activated olefins proceed instantly at −100 °C, yielding the corresponding monohydride intermediate; after the decomposition of the sample, hydrogenation products of the same handedness and level of optical purity as had been observed in the catalytic reaction were yielded (Scheme 1) [21,22,23,24].
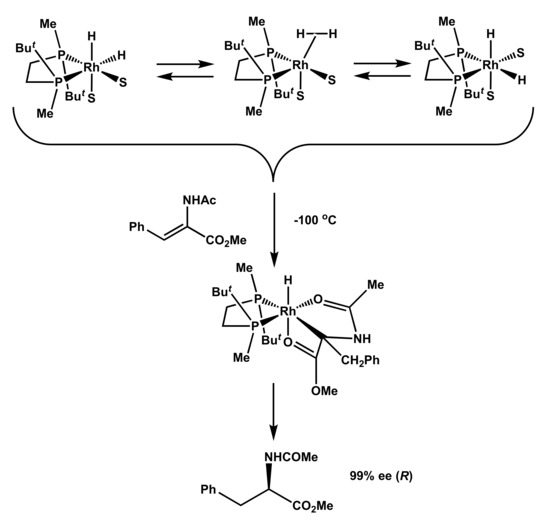
Scheme 1.
Practically perfect enantioselection in the reaction of solvate dihydrides 1 and 1′ with methyl (Z)-α-acetamidocinnamate (MAC) 3 at −100 °C [21].
These experiments testify the following:
- Stereoselection is possible after hydrogen activation and kinetically viable at significantly decreased temperatures.
- Substrate coordination and migratory insertion steps are extremely fast reactions.
2.2. Same Handedness and Level of Enantioselection in Hydrogenations of Differently Bound Catalyst–Substrate Complexes
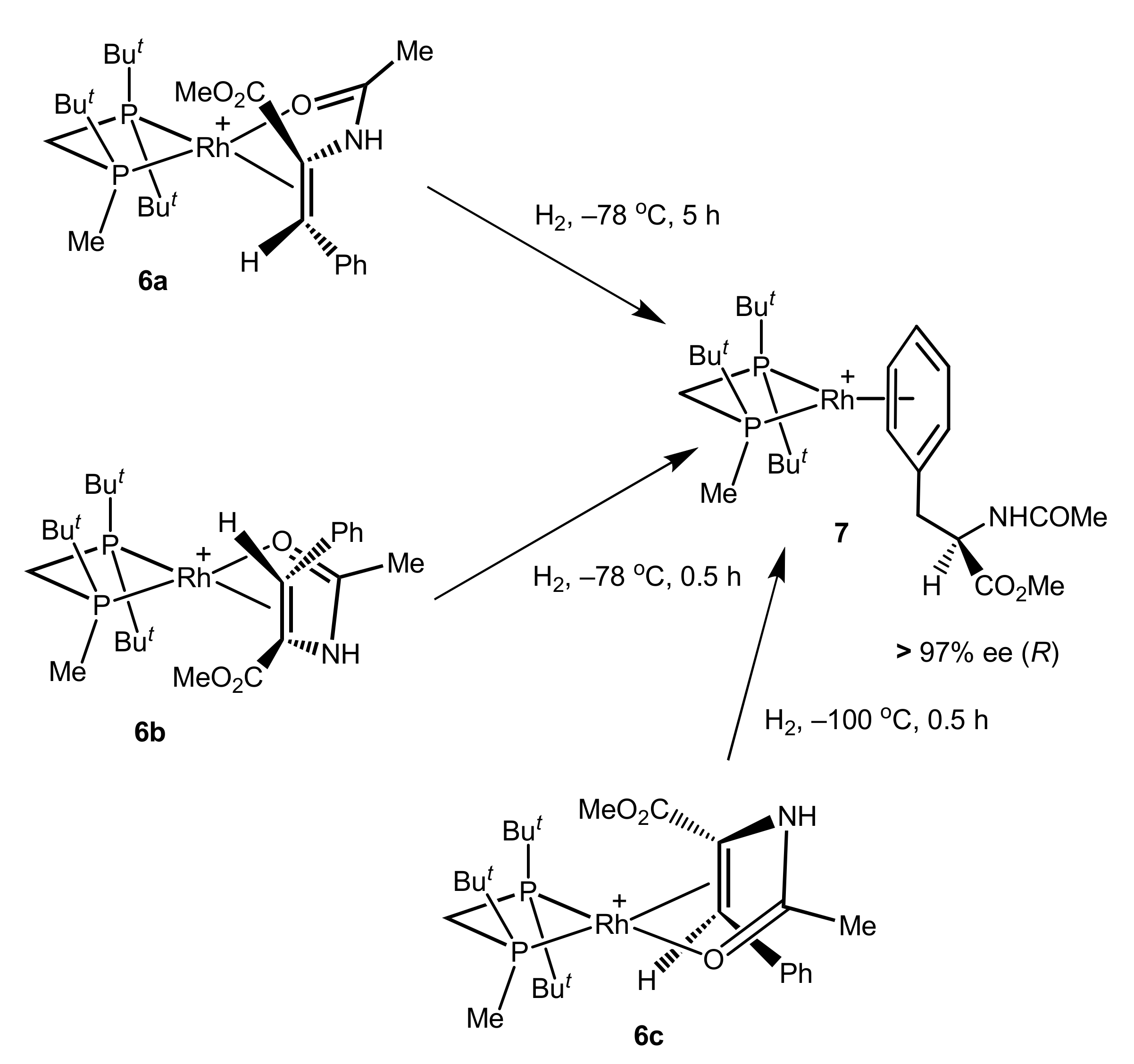
In the case of C1-symmetric catalyst TrichickenfootPHOS-Rh it was possible to perform low-temperature hydrogenations of three differently bound catalyst–substrate complexes with MAC: re-bound (6a) and si-bound (6b) from the side of the chiral phosphorus atom and si-bound on the side of the non-chiral phosphorus atom (6c) [25].
After the decomposition of samples, low-temperature hydrogenations of all three systems provided hydrogenation products with the same handedness and level of enantioselection as had been observed in the catalytic hydrogenation (Scheme 2).
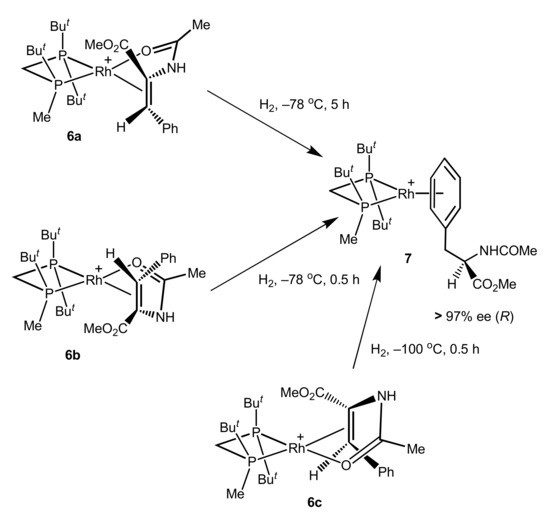
Scheme 2.
High enantioselection in the low-temperature hydrogenations of the catalyst–substrate complexes 6a–c.
These results clearly demonstrate that the stereochemical outcome of the hydrogenation does not depend on the mode of coordination in the Rh(I) square planar complex. In other words, the double bond can dissociate from this complex prior to the actual enantioselective step.
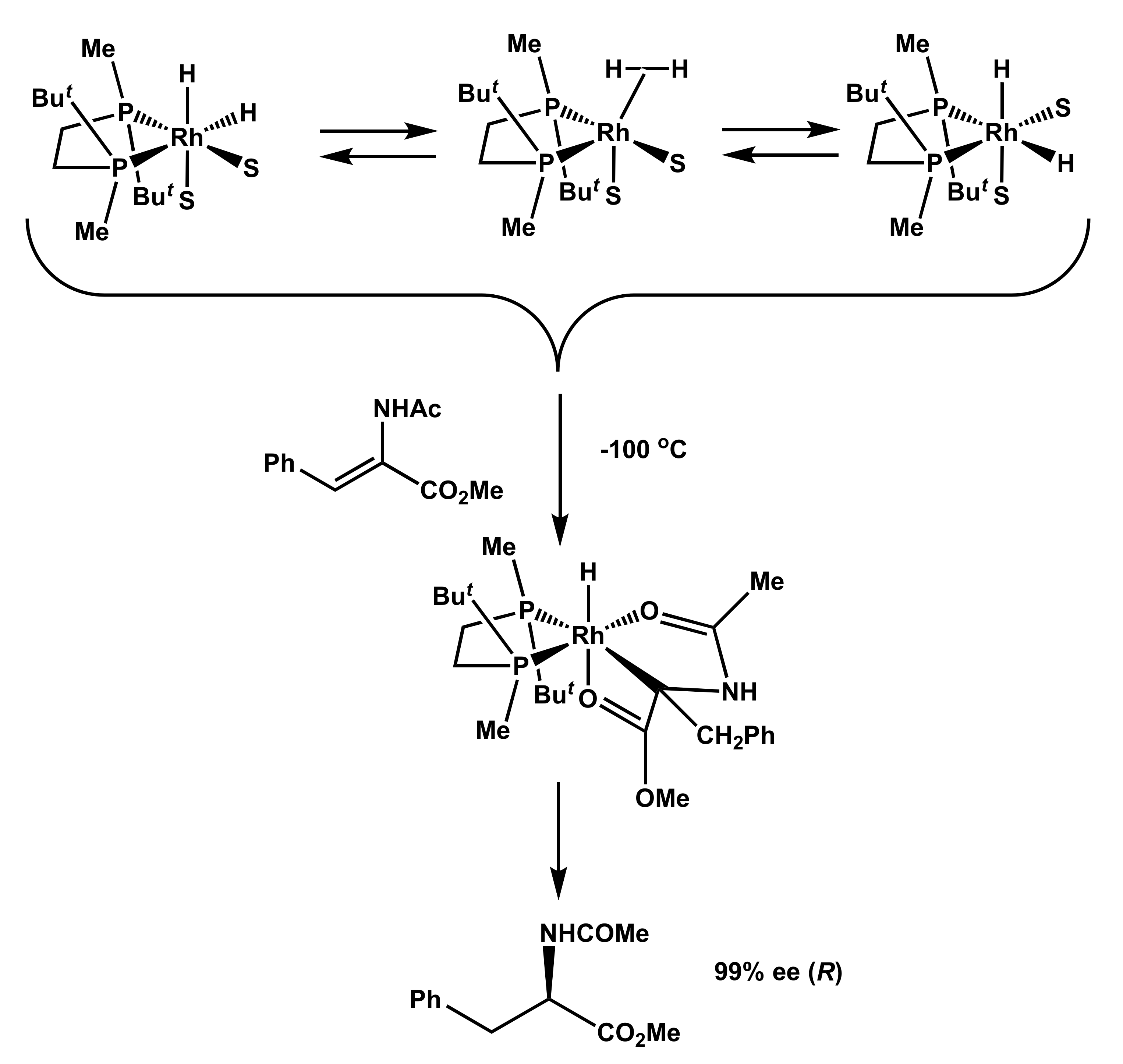
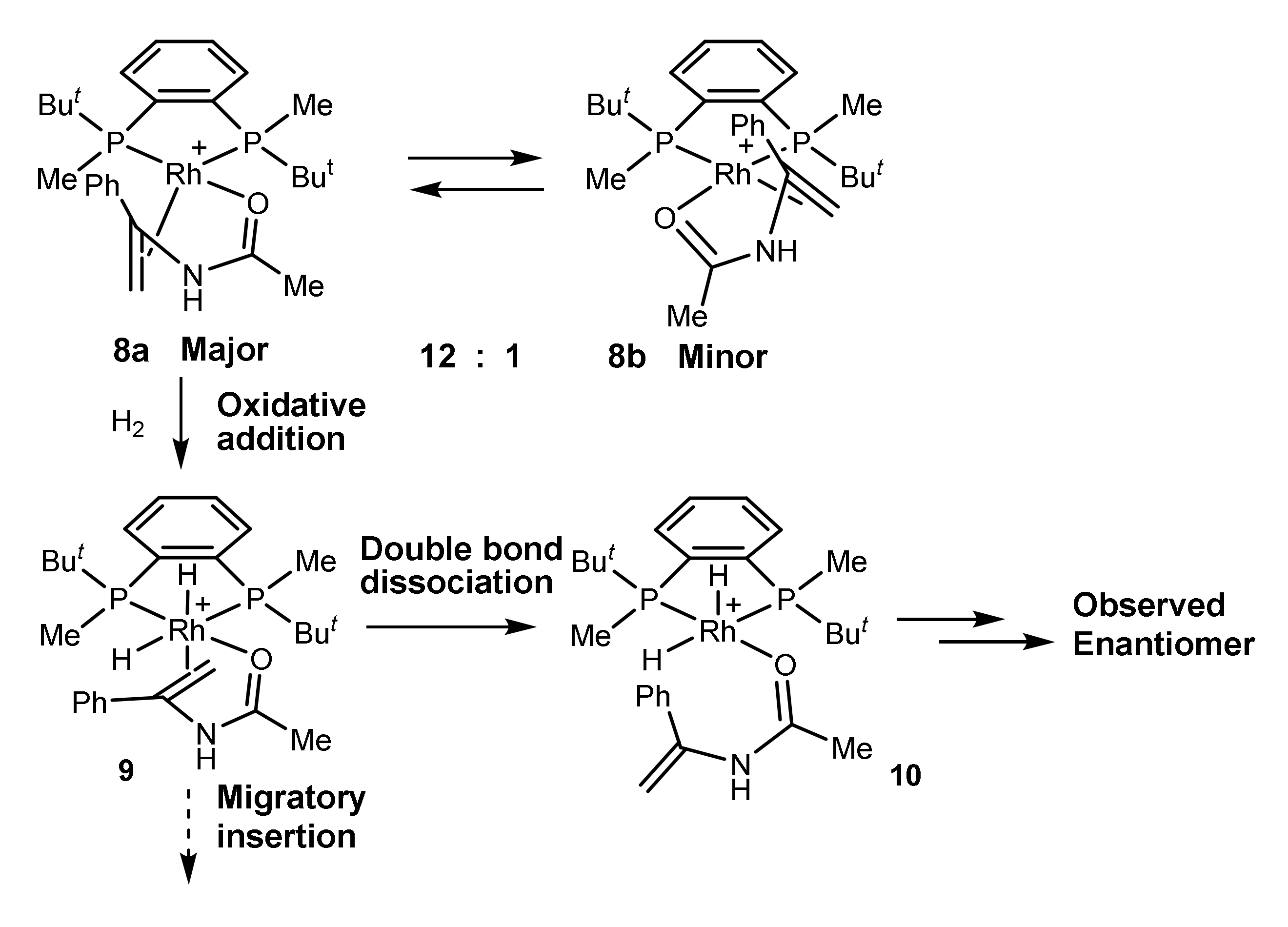
In the case of the system BenzP*-Rh and phenylenamide the latter conclusion has been developed further. Low-temperature hydrogenation of a 12:1 mixture of diastereomeric catalyst–substrate complexes 8a and 8b showed that the “wrong” isomer being more abundant is significantly more reactive, and bears most of the flux of catalysis. Nevertheless, the reaction product obtained with a high optical yield after the decomposition of the sample had the handedness opposite from the expected. Therefore, the double bond must have been dissociated (intermediate 10) prior to the enantioselective step (Scheme 3) [26].
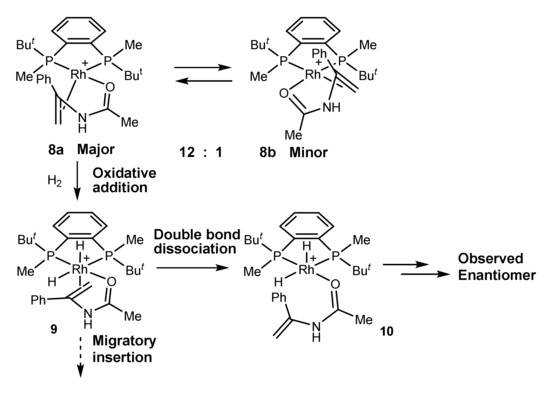
Scheme 3.
Evidence for the double bond dissociation prior to the enantioselective step [26].
2.3. Perfect Enantioselection without Substrate Binding in Rh(I) Square Complexes
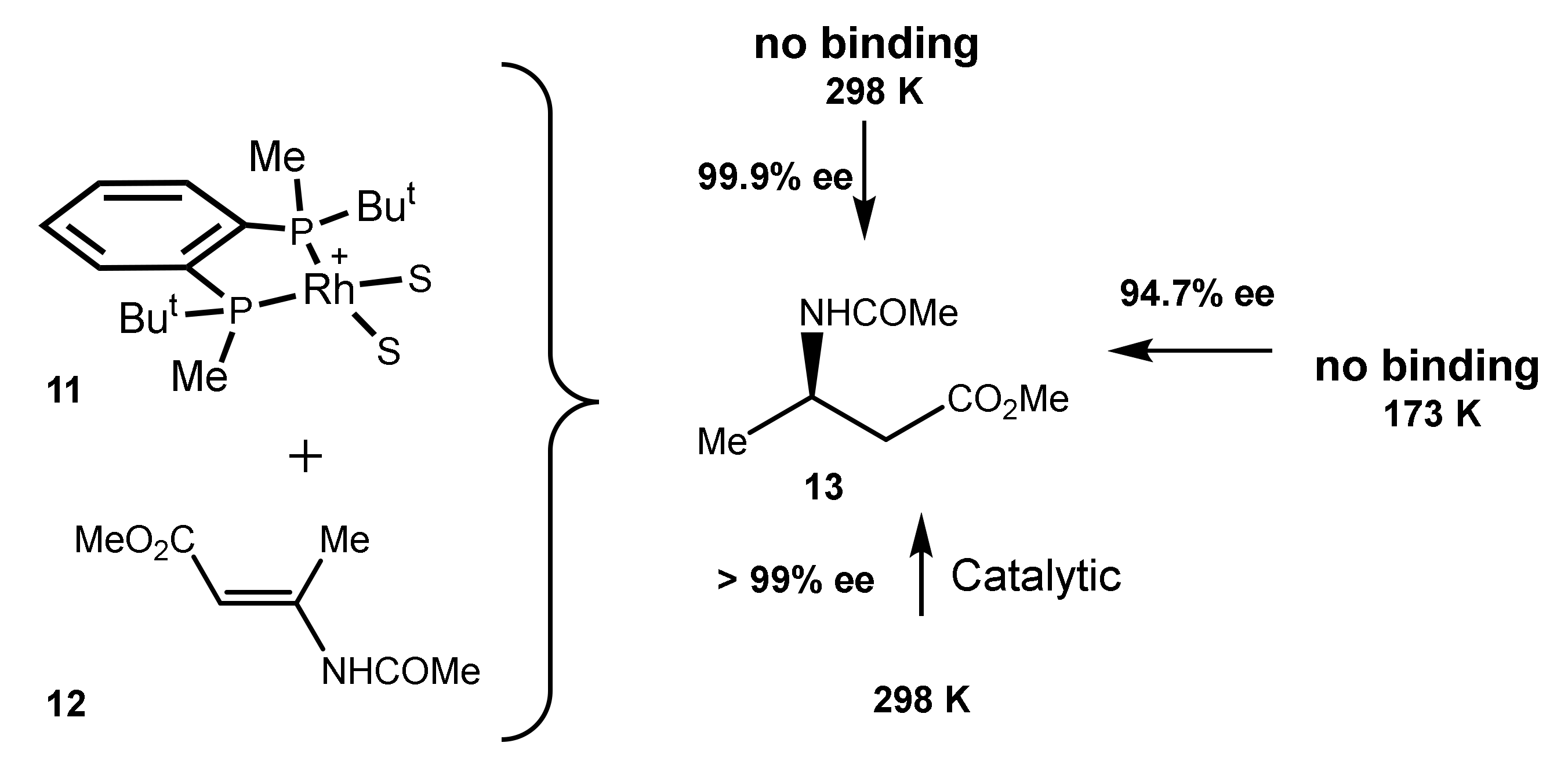
In contrast to numerous cases where the catalyst–substrate binding can be easily detected by NMR, in the case of the combination of the BenzP*-Rh catalyst and β-dehydroamino acid 12 no binding has been observed even at −100 °C. Nevertheless, either in catalytic conditions or in the low-temperature hydrogenations practically perfect enantioselection has been observed (Scheme 4) [27].
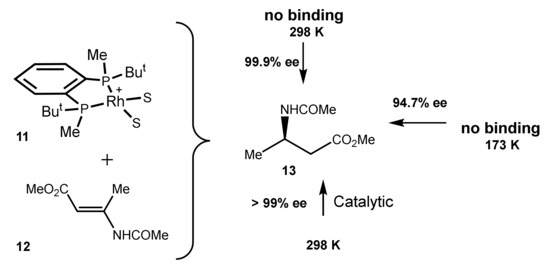
Scheme 4.
Practically perfect enantioselection in a system without the binding of the substrate 12 with the catalyst 11.
These results are also in line with the irrelevance of the binding strength of the metal for the enantioselection.
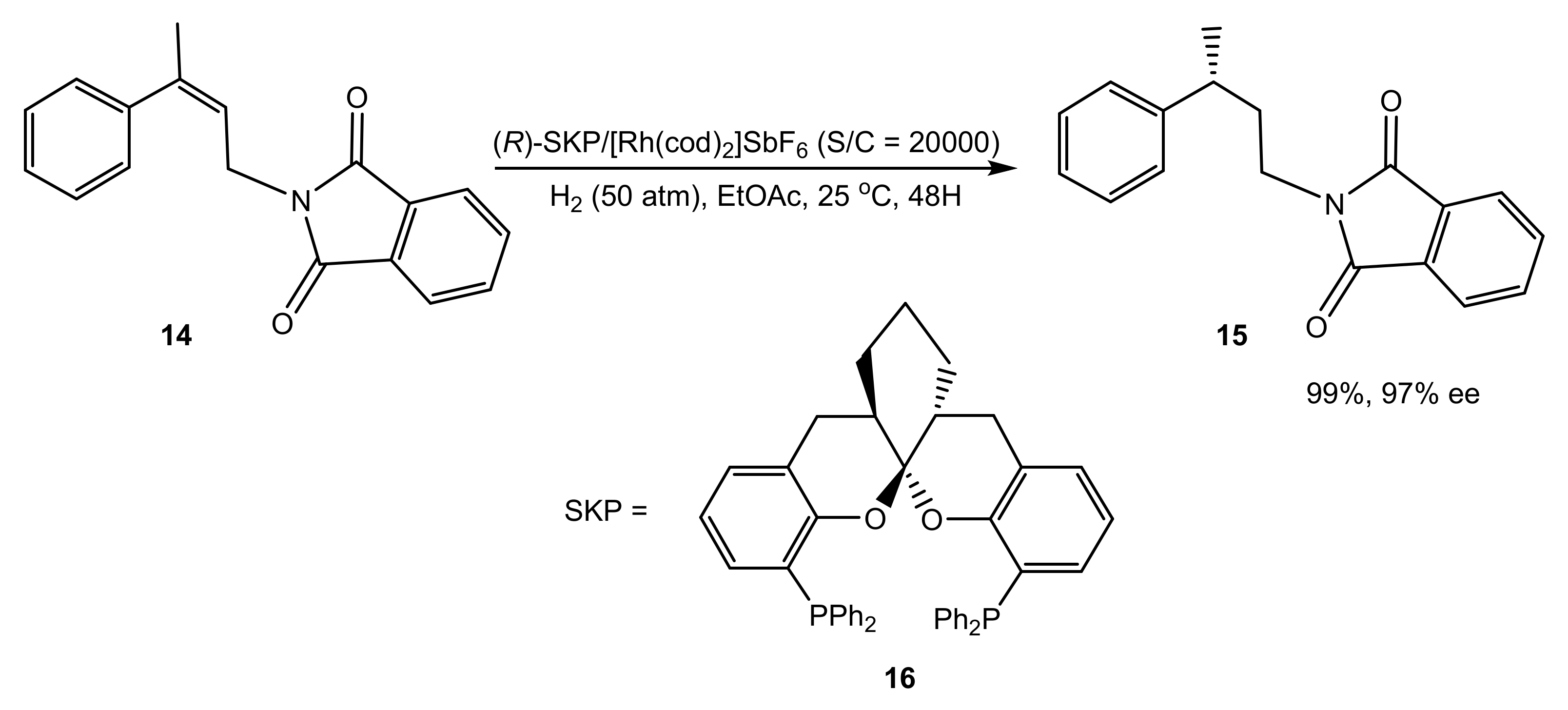
A still more convincing example of the metal excluded from the enantioselection process is the symmetric hydrogenation of γ-branched allylamines for the efficient synthesis of γ-chirogenic amines (Scheme 5) [28].
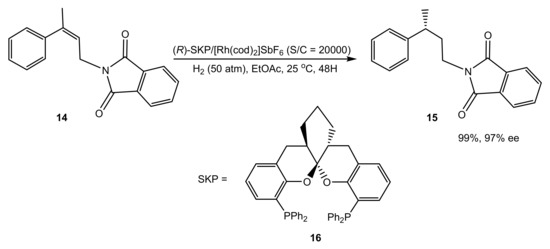
Scheme 5.
Multigram scale asymmetric hydrogenation with the Rh–SKP complex.
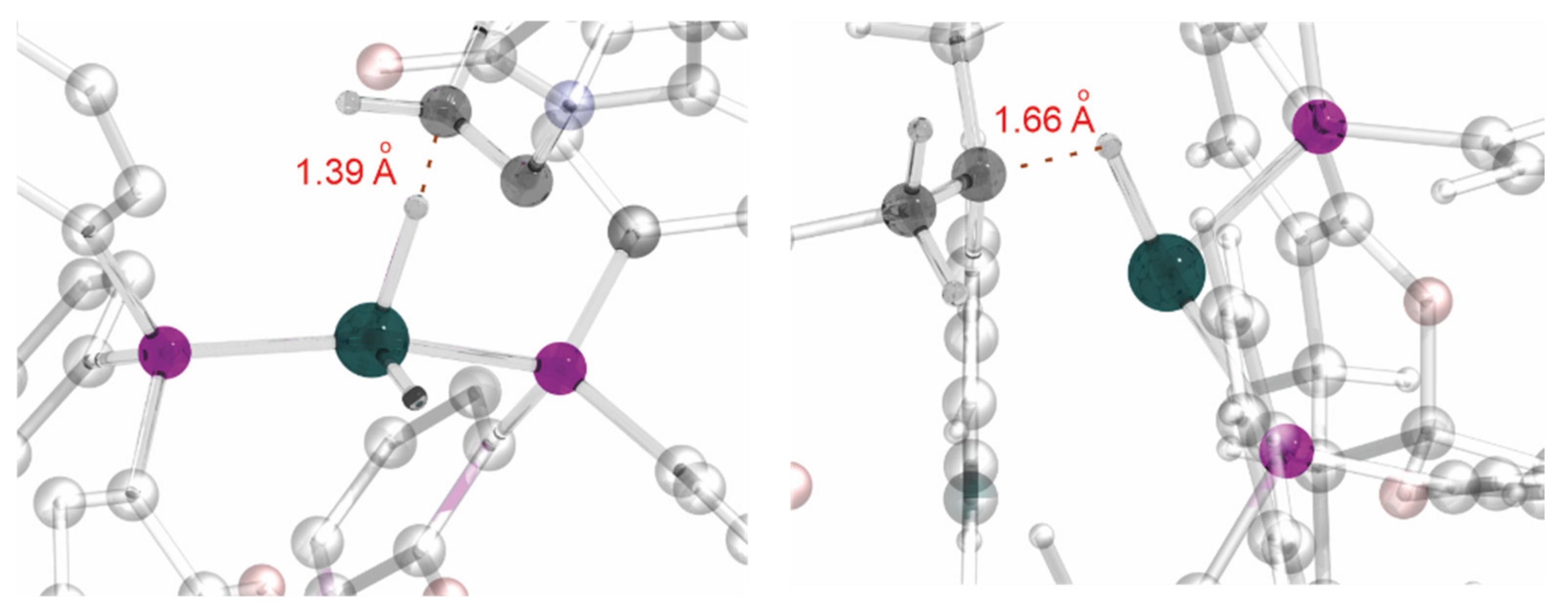
Due to the specific geometry of the Rh–SKP complex, with the phenyl groups effectively preventing any approach of the substrate to the metal atom, binding is not observed with the Rh(I) precursors. Moreover, computations suggested that even the migratory insertion stage takes place without coordination to Rh(III). The hydride from the metal is directly transferred to the double bond fixed in a proper position via numerous weak interactions between the catalyst and the substrate, i.e., exactly in an organocatalytic fashion (Figure 1).
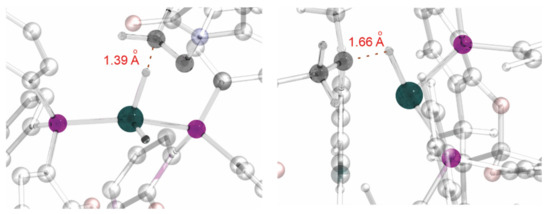
Figure 1.
Partial view of the computed structures of the TS for migratory insertion (left) and reductive elimination (right).
Thus, the presence of the metal in the catalyst helps again to activate hydrogen, but does not contribute to the process of stereoinduction.
2.4. Computational Evidence
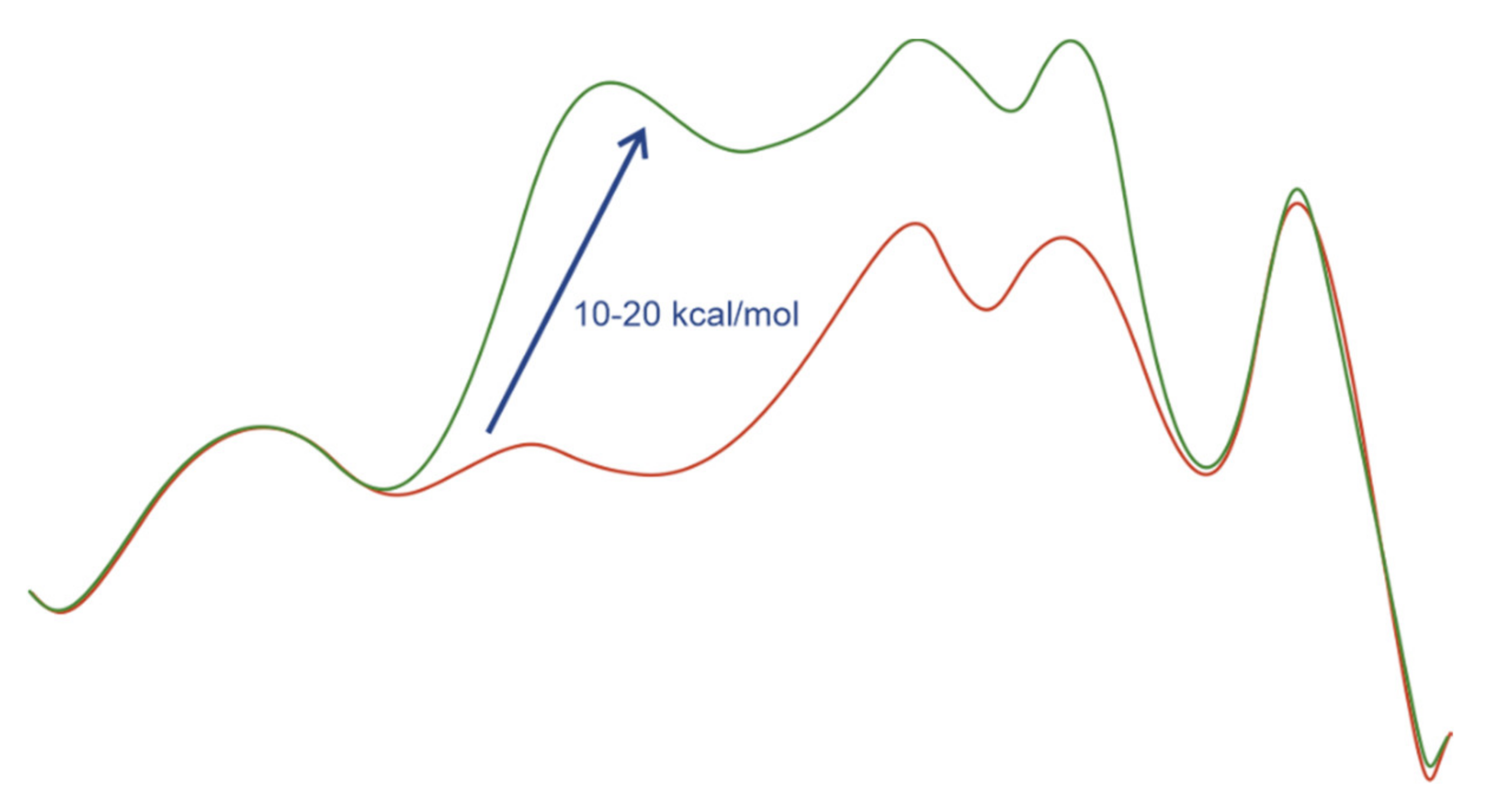
The latter conclusion was supported by computational studies. In several cases the hydrogenation pathways containing coordinated double bonds were 10–20 kcal/mol higher in energy than those with noncoordinated double bonds (Figure 2) [29,30,31].
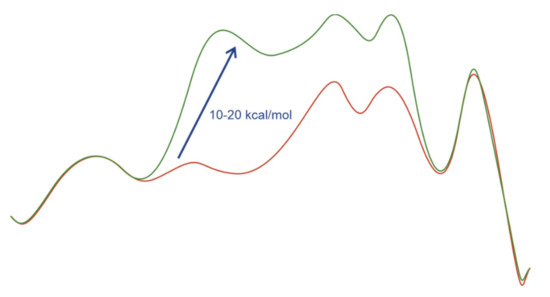
Figure 2.
Representative profiles of Gibbs free energies for the pathways with coordinated (green) and noncoordinated (red) double bonds in the case of electron-rich diphosphine ligands.
Even if the activation of hydrogen proceeded through the Rh(I) catalyst–substrate complex, the further dissociation of the double bond required lower energy than a direct migratory insertion–reductive elimination sequence [24].
Computational studies suggest that stereodiscrimination takes place during the secondary coordination of the double bond on the way to the perfect configuration for the barrierless (or characterized with very low barriers of 1–2 kcal/mol) migratory insertion. The main stereoregulating factors governing this process are noncovalent disperse interactions between the substituents on the ligand and the double bond, i.e., the process of stereoselection has intrinsically the same character as that in the organocatalytic reactions [20,29,30,31,32].
3. Asymmetric Hydrogenation Catalyzed by Co, Ni, and Pd Complexes
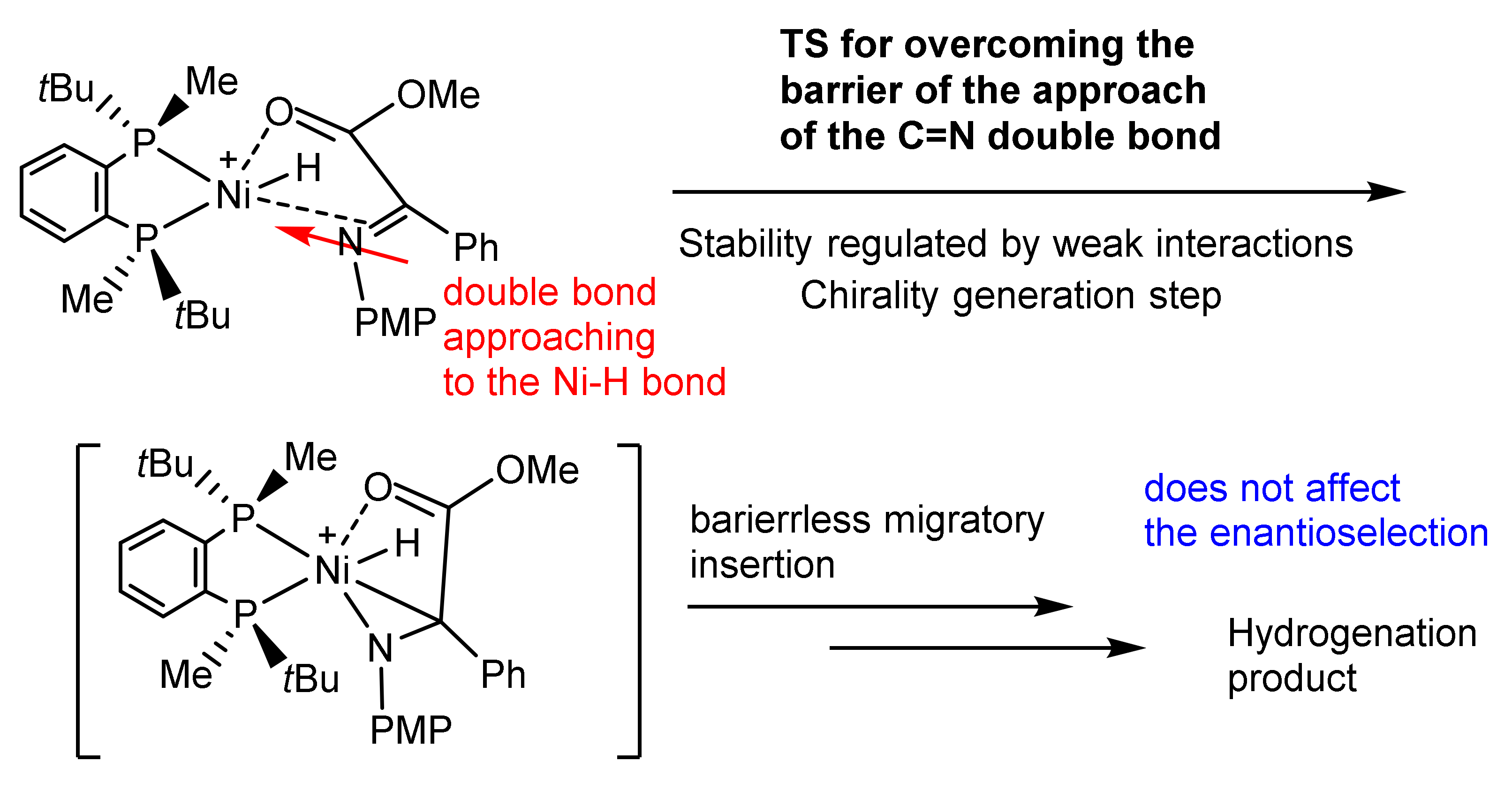
Despite absolutely different shapes of the energy profiles in these reactions preventing the detection of meaningful intermediates, computations suggest that in these cases the approach of the reagent to the configuration ideal for the barrierless migratory insertion (double bond coplanar to the M-H bond trans to the phosphorus) is the stage on which the chirality is generated [33,34,35,36,37].
In the diastereomeric transition states determining the handedness and level of enantioselection the double bond is distanced for approximately 3 Å from the metal atom, and their relative stabilities are determined by weak disperse interactions between the substituents of the ligand and those of the approaching catalyst (Scheme 6) [33,34,35,36,37].
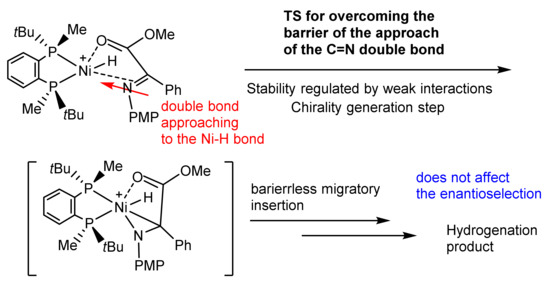
Scheme 6.
Mechanism of migratory insertion in Ni-catalyzed asymmetric hydrogenations.
4. Artificial Catalysis Mimics Enzymatic Behavior
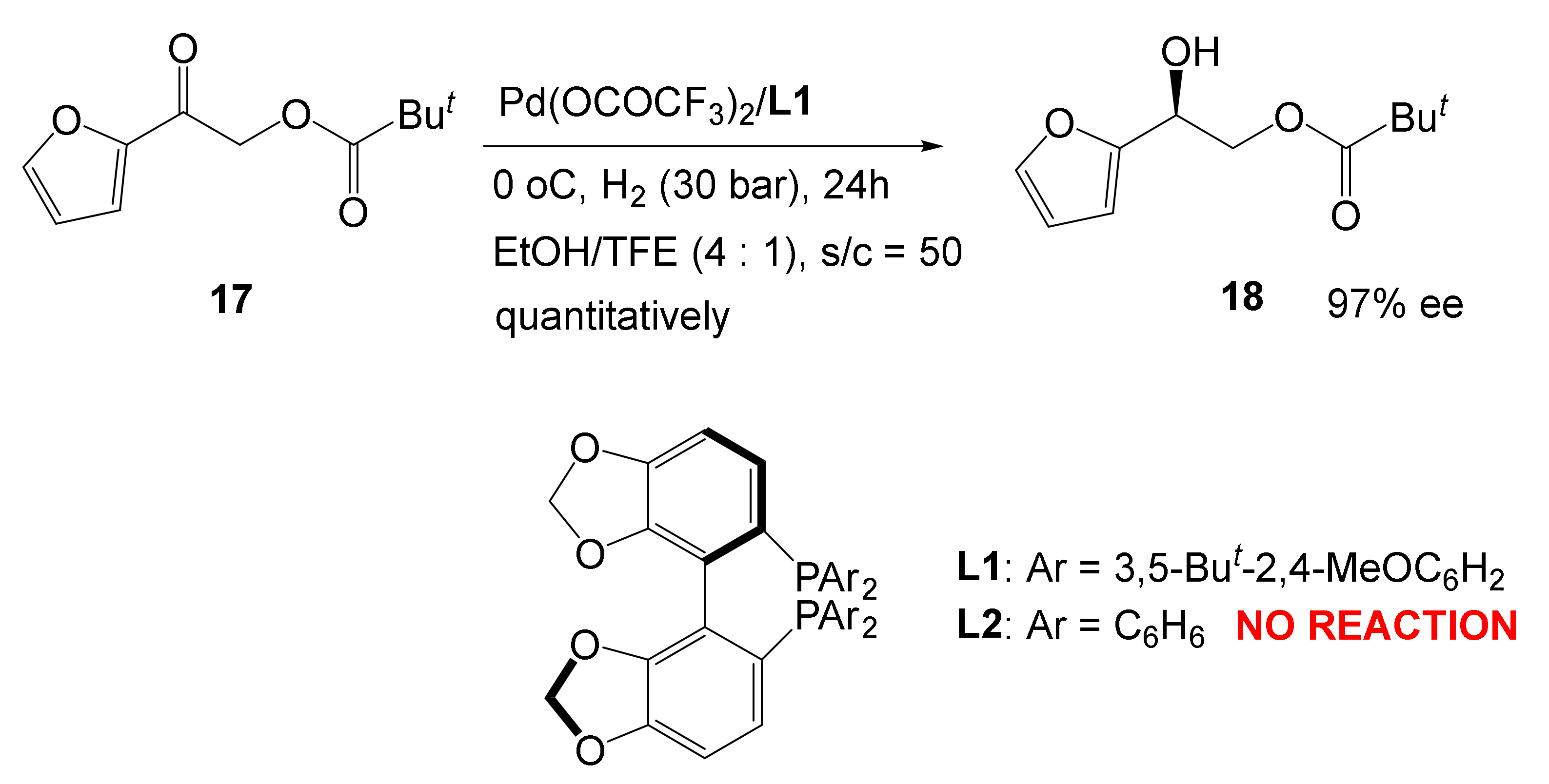
The most striking example demonstrating the actual closeness of transition metals and organocatalysis is the Pd-catalyzed hydrogenation of the C=O bond with a heavily substituted SEGPHOS catalyst [38].
Experimentally it was found with astonishment that only the heavily substituted ligand works, whereas normal SEGPHOS fails to effectuate the same transformation (Scheme 7).
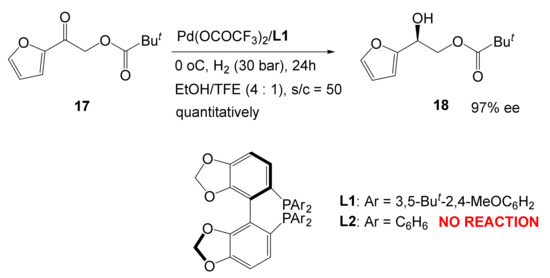
Scheme 7.
Only a heavily substituted ligand is an active catalyst.
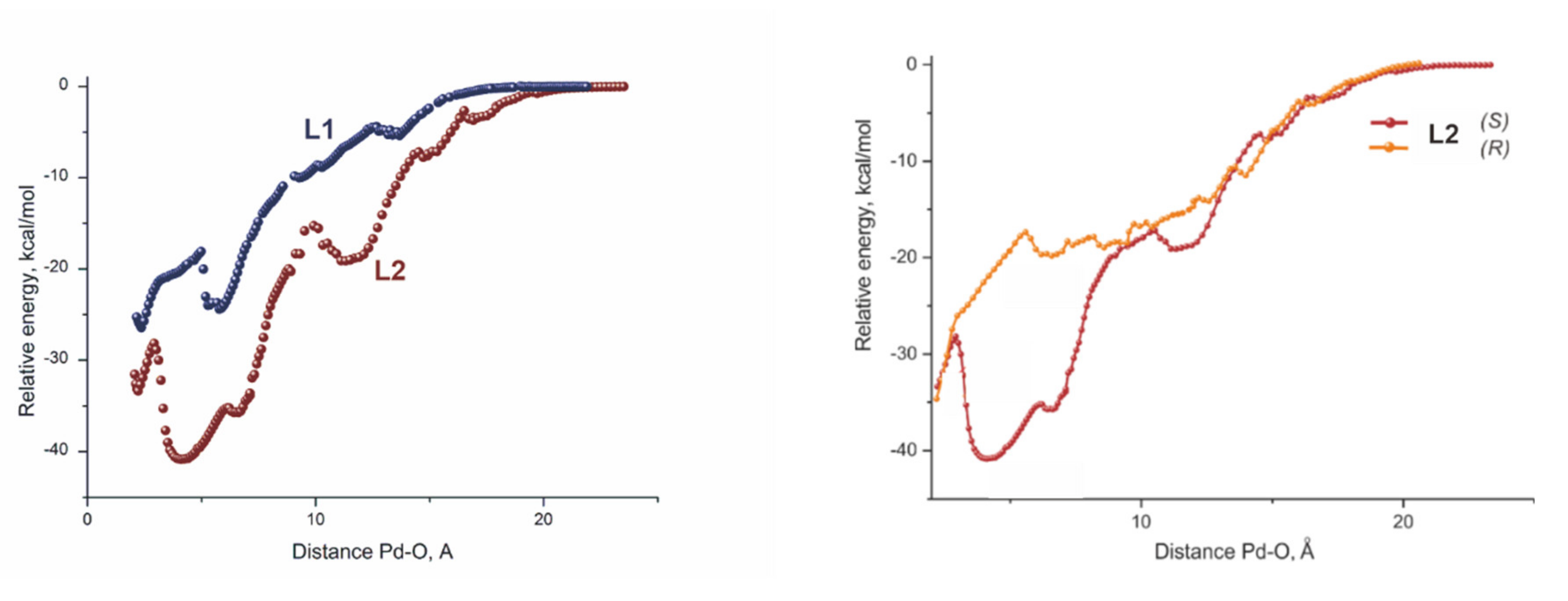
Computations of the catalytic cycles brought little understanding, since the activation barriers were very low and close in values for both cases. Nevertheless, the computations of the approach of the substrate to the catalytic site revealed a striking difference in the energetics of this process. A significant energy difference in favor of the larger catalyst was observed throughout the whole process of the substrate approach starting from almost 20 Å, separating the reactive centers when the first weak encounter complex was formed in the case of the heavily substituted SEGPHOS–Pd complex (Figure 3, left) [39].
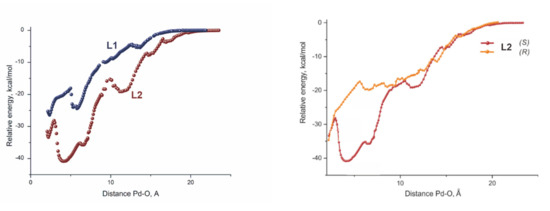
Figure 3.
Scans of the relative energy versus the interatomic distance Pd–O. The structures of the ligands L1 and L2 are shown on the Scheme 7.
In other words, the lack of numerous t-Bu groups, providing a possibility to form plenty of C-H⋯H-C interactions, does not allow the normal SEGPHOS catalyst to keep the substrate nearby for a time that is sufficient to approach the reactive center.
Interestingly, the same reason seems to be the origin of the enantioselectivity in this reaction. The curve of the energy scan of the substrate approaching the reactive site for the formation of an R-enantiomer (Figure 3, right) appeared strikingly similar to that for the unsubstituted SEGPHOS-Pd, featuring two reactions that do not occur.
The huge framework of the heavily substituted SEGPHOS ligand can be considered as a kind of a supramolecular catalyst [40], thus providing the first example of a fruitful (although innocent) exchange of ideas between different fields.
5. Conclusions
The above analysis of recent effective asymmetric hydrogenations demonstrates that metal atoms and chiral ligands work more or less independently in the processes of hydrogen activation and stereoinduction. This provides reasons to build an analogy with cooperative asymmetric organocatalysis, where the stages of nucleophile activation and stereoselective bond formation are similarly allocated for two different parts of the catalyst molecule or to two different components of the catalytic system. Noncovalent disperse interactions between the catalyst and the substrate play a key role in the stereoinduction process in both cases [32,41].
In my opinion, this conclusion opens the doors for a more conscious approach to the design of both transition metals and organocatalysts. Structural ideas that have proved their efficiency in one of the fields can find a broader application in another one. In particular, a significant number of effective C2-symmetric diphosphine ligands developed in the past 50 years could provide fruitful inspirations for the design of new chiral organocatalysts.
Funding
This research received no external funding.
Conflicts of Interest
The author declares no conflict of interest.
References
- Brock, W.H. The Fontana History of Chemistry; Fontana Press: London, UK, 1992; pp. 235–236. [Google Scholar]
- Sharpless, B. Searching for new reactivity (Nobel lecture). Angew. Chem. Int. Ed. 2002, 41, 2024–2032. [Google Scholar] [CrossRef]
- Knowles, W.S. Asymmetric hydrogenations (Nobel lecture). Angew. Chem. Int. Ed. 2002, 41, 1998–2007. [Google Scholar] [CrossRef]
- Noyori, R. Asymmetric catalysis: Science and opportunities (Nobel lecture). Angew. Chem. Int. Ed. 2002, 41, 2008–2022. [Google Scholar] [CrossRef]
- Suzuki, A. Cross-coupling reactions of organoboranes: An easy way to construct C–C bonds (Nobel lecture). Angew. Chem. Int. Ed. 2011, 50, 6722–6737. [Google Scholar] [CrossRef] [PubMed]
- Negishi, E.-I. Magical power of transition metals: Past, present, and future (Nobel lecture). Angew. Chem. Int. Ed. 2011, 50, 6738–6764. [Google Scholar] [CrossRef] [PubMed]
- List, B.; Lerner, R.A.; Barbas, C.F.J. Proline-catalyzed direct asymmetric aldol reactions. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Jen, W.S.; Wiener, J.J.M.; McMillan, D.W.C.J. New strategies for organic catalysis: The first enantioselective organocatalytic 1, 3-dipolar cycloaddition. Am. Chem. Soc. 2000, 122, 9874–9875. [Google Scholar] [CrossRef] [Green Version]
- Antenucci, A.; Dughera, S.; Renzi, P. Green chemistry meets asymmetric organocatalysis: A critical overview on catalysts synthesis. ChemSusChem 2021, 14, 2785–2853. [Google Scholar] [CrossRef]
- Shibasaki, M.; Kanai, M.; Matsunaga, S.; Kumagai, N. Recent progress in asymmetric bifunctional catalysis using multimetallic systems. Acc. Chem. Res. 2009, 42, 1117–1127. [Google Scholar] [CrossRef]
- Xu, H.; Zuend, S.J.; Woll, M.G.; Tao, Y.; Jacobsen, E.N. Asymmetric cooperative catalysis of strong Brønsted acid–promoted reactions using chiral ureas. Science 2010, 327, 986–990. [Google Scholar] [CrossRef] [Green Version]
- Simmons, B.; Walji, A.M.; MacMillan, D.W.C. Cycle-specific organocascade catalysis: Application to olefin hydroamination, hydro-oxidation, and amino-oxidation, and to natural product synthesis. Angew. Chem. Int. Ed. 2009, 48, 4349–4353. [Google Scholar] [CrossRef] [Green Version]
- Allen, A.E.; McMillan, D.W.C. Synergistic catalysis: A powerful synthetic strategy for new reaction development. Chem. Sci. 2012, 3, 633–658. [Google Scholar] [CrossRef]
- Qin, Y.; Zhu, L.; Luo, S. Organocatalysis in inert C–H bond functionalization. Chem. Rev. 2017, 117, 9433–9520. [Google Scholar] [CrossRef]
- Bo, L.; Dang, Y.; Houk, K.N. Dispersion and Steric Effects on Enantio-/Diastereoselectivities in Synergistic Dual Transition-Metal Catalysis. J. Am. Chem. Soc. 2022, 144, 1971–1985. [Google Scholar] [CrossRef]
- Liu, D.; Chen, J.; Gridnev, I.D.; Yan, D.; Zhang, W.B. Construction of chiral α-tert-amine scaffolds via amine-catalyzed asymmetric Mannich reactions of alkyl-substituted ketimines. Nat. Commun. 2020, 10, 1699. [Google Scholar]
- Halpern, J. Mechanism and stereoselectivity of asymmetric hydrogenation. Science 1982, 217, 401–407. [Google Scholar] [CrossRef]
- Brown, J.M. Hydrogenation of Functionalized Carbon-Carbon Double Bonds; Jacobsen, E.N., Pfalz, A., Yamamoto, H., Eds.; Springer: Berlin/Heidelberg, Germany, 1999; Volume 1, pp. 119–182. [Google Scholar]
- Gridnev, I.D.; Imamoto, T. On the mechanism of stereoselection in Rh-catalyzed asymmetric hydrogenation: A general approach for predicting the sense of enantioselectivity. Acc. Chem. Res. 2004, 37, 633–644. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Imamoto, T. Mechanism of enantioselection in Rh-catalyzed asymmetric hydrogenation. The origin of utmost catalytic performance. Chem. Commun. 2009, 48, 7447–7464. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Higashi, N.; Asakura, K.; Imamoto, T. Mechanism of Asymmetric Hydrogenation Catalyzed by a Rhodium Complex of (S,S)-1,2-Bis(tert-butylmethylphosphino)ethane. Dihydride Mechanism of Asymmetric Hydrogenation. J. Am. Chem. Soc. 2000, 122, 7183–7194. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Yasutake, M.; Higashi, N. Imamoto, T. Asymmetric hydrogenation of enamides with Rh-BisP* and Rh-MiniPHOS catalysts. Scope, limitations, and mechanism. J. Am. Chem. Soc. 2001, 123, 5268–5276. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Higashi, N.; Imamoto, T. Interconversion of monohydride intermediates in Rh (I)-catalyzed asymmetric hydrogenation of dimethyl 1-benzoyloxyethenephosphonate. J. Am. Chem. Soc. 2001, 123, 4631–4632. [Google Scholar] [CrossRef] [PubMed]
- Gridnev, I.D.; Yasutake, M.; Imamoto, T.; Beletskaya, I.P. Asymmetric Hydrogenation of α,β-umsaturated phosphonates with Rh-BisP* and Rh-MiniPHOS catalysts: Scope and mechanism of the reaction. Proc. Natl. Acad. Sci. USA 2004, 101, 5228–5235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gridnev, I.D.; Hoge, G.; Kouchi, T.M.; Takahashi, H.; Imamoto, T. Asymmetric Hydrogenation Catalyzed by a Rhodium Complex of (R)-(tert-Butylmethylphosphino)(di-tert-butylphosphino)methane: Scope of Enantioselectivity and Mechanistic Study. J. Am. Chem. Soc. 2008, 130, 2560–2572. [Google Scholar] [CrossRef] [PubMed]
- Gridnev, I.D.; Imamoto, T. Challenging the major/minor concept in Rh-catalyzed asymmetric hydrogenation. ACS Catal. 2015, 5, 2911–2915. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Liu, Y.; Imamoto, T. Mechanism of asymmetric hydrogenation of β-Dehydroamino acids catalyzed by rhodium complexes: Large-scale experimental and computational study. ACS Catal. 2014, 4, 203–219. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, T.; Wang, Y.; Zhou, F.; Zhang, Z.; Gridnev, I.D.; Zhang, W. Asymmetric hydrogenation for the synthesis of 2-substituted chiral morpholines. Nat. Sci. 2021, 1, e10021. [Google Scholar]
- Imamoto, T.; Tamura, K.; Zhang, Z.; Horiuchi, Y.; Sugiya, M.; Yoshida, K.; Yanagisawa, A.; Gridnev, I.D. Rigid P-Chiral Phosphine Ligands with tert-Butylmethylphosphino Groups for Rhodium-Catalyzed Asymmetric Hydrogenation of Functionalized Alkenes. J. Am. Chem. Soc. 2012, 134, 1754–1769. [Google Scholar] [CrossRef] [PubMed]
- Gridnev, I.D.; Imamoto, T. Enantioselection mechanism in Rh-catalyzed asymmetric hydrogenation. Russ. Chem. Bull. 2016, 65, 1514–1534. [Google Scholar] [CrossRef]
- Gridnev, I.D.; Dub, P.A. Enantioselection in Asymmetric Catalysis; CRC Press: Boca Raton, FL, USA, 2016; 234p. [Google Scholar]
- Gridnev, I.D. Attraction versus Repulsion in Rhodium-Catalyzed Asymmetric Hydrogenation. ChemCatChem 2016, 8, 3463–3465. [Google Scholar] [CrossRef]
- Zhang, J.; Jia, J.; Zeng, X.; Wang, Y.; Zhang, Z.; Gridnev, I.D.; Zhang, W. Chemo- and Enantioselective Hydrogenation of α-Formyl Enamides: An Efficient Access to Chiral α-Amido Aldehydes. Angew. Chem. Int. Ed. 2019, 58, 11505–11512. [Google Scholar] [CrossRef]
- Fan, D.; Zhang, J.; Hu, Y.; Zhang, Z.; Gridnev, I.D.; Zhang, W. Asymmetric Hydrogenation of α-Boryl Enamides Enabled by Nonbonding Interactions. ACS Catal. 2020, 10, 3232–3240. [Google Scholar] [CrossRef]
- Chen, J.; Zhang, Z.; Li, B.; Li, F.; Wang, Y.; Zhao, M.; Gridnev, I.D.; Imamoto, T.; Zhang, W. Pd (OAc)2-catalyzed asymmetric hydrogenation of sterically hindered N-tosylimines. Nat. Commun. 2018, 9, 5000. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, Z.; Zhang, J.; Liu, Y.; Gridnev, I.D.; Zhang, W. Cobalt-Catalyzed Asymmetric Hydrogenation of C= N Bonds Enabled by Assisted Coordination and Nonbonding Interactions. Angew. Chem. Int. Ed. 2019, 58, 15767–15771. [Google Scholar] [CrossRef]
- Li, B.; Chen, Z.; Zhang, Z.; Gridnev, I.D.; Zhang, W. Nickel-Catalyzed Asymmetric Hydrogenation of N-Sulfonyl Imines. Angew. Chem. Int. Ed. 2019, 131, 7407–7412. [Google Scholar] [CrossRef]
- Chen, J.; Liu, D.; Butt, N.; Li, C.; Fan, D.; Liu, Y.; Zhang, W. Palladium-Catalyzed Asymmetric Hydrogenation of α-Acyloxy-1-arylethanones. Angew. Chem. Int. Ed. 2013, 52, 11632–11636. [Google Scholar] [CrossRef]
- Chen, J.; Gridnev, I.D. Size is important: Artificial catalyst mimics behavior of natural enzymes. iScience 2020, 23, 100960. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, M.; La Manna, P.; Talotta, C.; Soriente, A.; Gaeta, C.; Neri, P. Supramolecular Organocatalysis in Water Mediated by Macrocyclic Compounds. Front. Chem. 2018, 6, 84. [Google Scholar] [CrossRef] [Green Version]
- Wagner, J.P.; Schreiner, P.R. London dispersion in molecular chemistry—Rreconsidering steric effects. Angew. Chem. Int. Ed. 2015, 54, 12274–12296. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).