Abstract
Natural zeolite clinoptilolite CLIN with a framework ratio of Si/Al ≥ 4 containing mainly potassium and calcium ions in its internal channel system was used as a starting material. The acidic HCLIN catalysts were prepared under soft conditions avoiding the use of environmental less-benign mineral acids. The starting material was ion exchanged using a 0.2 M aqueous ammonium nitrate solution at a temperature 80 °C for 2 h. The obtained NH4CLIN was converted into the acid HCLIN catalyst by calcination at 300–600 °C. The obtained samples were characterized by XRD, FTIR, SEM/TEM, AAS, and EDX element mapping. The state of aluminium and silicon was studied by 27Al- and 29SiMAS NMR spectroscopy. The textural properties of the catalysts were investigated by nitrogen adsorption and desorption measurements. The Brønsted acidity of the HCLIN catalysts was studied by temperature-programmed decomposition of the exchanged ammonium ions releasing ammonia as well as 1H MAS NMR, {1H–27Al} Trapdor, and {1H–27Al} Redor experiments. The strongly agglomerated samples were crystalline and thermally stable up to >500 °C. Although a part of the clinoptilolite framework is maintained up to 600 °C, a loss of crystallinity is already observed starting from 450 °C. The specific surface areas of the starting CLIN and ammonium exchanged NH4CLIN are low with ca. 26 m2/g. The pores are nearly blocked by the exchangeable cations located in the zeolite pores. The thermal decomposition of the ammonium ions by calcination at 400 °C causes an opening of the pore entrances and a markable increase in the specific micropore area and micropore volume to ca. 163 m2/g and 0.07 cm3/g, respectively. It decreases with further rising calcination temperature indicating some structural loss. The catalysts show a broad distribution of Brønsted acid sites (BS) ranging from weak to strong sites as indicated the thermal decomposition of exchanged ammonium ions (TPDA). The ammonium ion decomposition leaving BS, i.e., H+ located at Al–O–Si framework bridges, starts at ≥250 °C. A part of the Brønsted sites is lost after calcination specifically at 500 °C. It is related to the formation of penta-coordinated aluminium at the expense of tetrahedral framework aluminium. The Brønsted sites are partially recreated after repeated ammonium ion exchange. The catalytic performance of the acidic HCLIN catalysts was tested in the etherification of glycerol as a green renewable resource with different C1-C4 alcohols. The catalysts are highly active in the etherification of glycerol, especially with alcohols containing the branched, tertiary alkyl groups. Highest activity is observed with the soft activated catalyst HCLIN300 (300 °C, temperature holding time: 1 min). A total of 78% conversion of glycerol to mono and di ether were achieved with tert-butanol at 140 °C after 4 h of reaction. The mono- and di-ether selectivity were 75% and 25%, respectively. The catalyst can be reused.
1. Introduction
The boosting use of transportation fuel and the other energies is faced with the limited availability of fossil fuel resources and climate change due to global warming, which is fired by the increasing emissions of carbon dioxide [1,2]. Both facts require the enhanced use of renewable feedstock, which would decrease the use of limited fossils and, most importantly, reduce the emission of carbon dioxide into the atmosphere by CO2 cycling via photosynthesis [3].
This paper aims to use glycerol, a by-product of the bio-diesel production, as renewable feedstock for the synthesis of glycerol tert-butyl ether as component of new sustainable synthetic fuel. About 10 wt% of glycerol is obtained as a by-product of bio-diesel production in the transesterification of vegetable oils or suitable wastes with short chain alcohols [4,5,6].
In general, the synthesis of glycerol tert-butyl ether by etherification reaction requires the use of acid catalysts [7,8,9,10,11]. Among that, mineral acids are active catalysts for the etherification of glycerol; however, their application is not environmentally benign. Mineral acids are very corrosive and hardly separated and removed from the reaction products. The required washing and neutralization processes release salty wastewater into the environment. Therefore, heterogeneous acid catalysts are in the focus of interest.
Zeolites containing strong acid sites which are able to tune, and when showing selective properties were shown to be catalytic active in the etherification of glycerol. Some zeolites, which were reported to be active in the etherification of glycerol with tert-butanol, are zeolite HY, stabilized dealuminated HY, mordenite, zeolite Beta, or ZSM-5 with different catalytic activities, i.e., conversions. The selectivity to the wished di-ether should be further improved based on the zeolite properties. The natural zeolite clinoptilolite was, to the best of our knowledge, to date not catalytic tested in the etherification of glycerol, although the acidic material is also catalytic active. The advantage of the natural zeolite clinoptilolite is that it is cheap, widely available, and usable as a heterogeneous catalyst. The drawback of the natural material is the variance of chemical composition. Compared to synthetic zeolites, chemicals and energy required for the synthesis are saved by using natural zeolite [12].
Natural zeolite clinoptilolite has already found different applications in agriculture, e.g., in fertilizer and feed additive [13], water purification [14] from harmful metals, as drug support in pharmacy [15], as a drying agent for fuels [16], and as zeolites in general [17,18]. Modified clinoptilolites can be used as adsorbents and show catalytic properties [19], e.g., they are active in the selective catalytic reduction of NOx [20], the photocatalytic degradation of pollutants by using Fenton catalyst [21]. Acid and alkaline clinoptilolite catalysts were active in different reactions, such as the lactate formation from biomass [22], solvent-free isomerization of α-pinene [23], and the transesterification of fatty acids from vegetable (waste) oil [24] or aldol condensation [25].
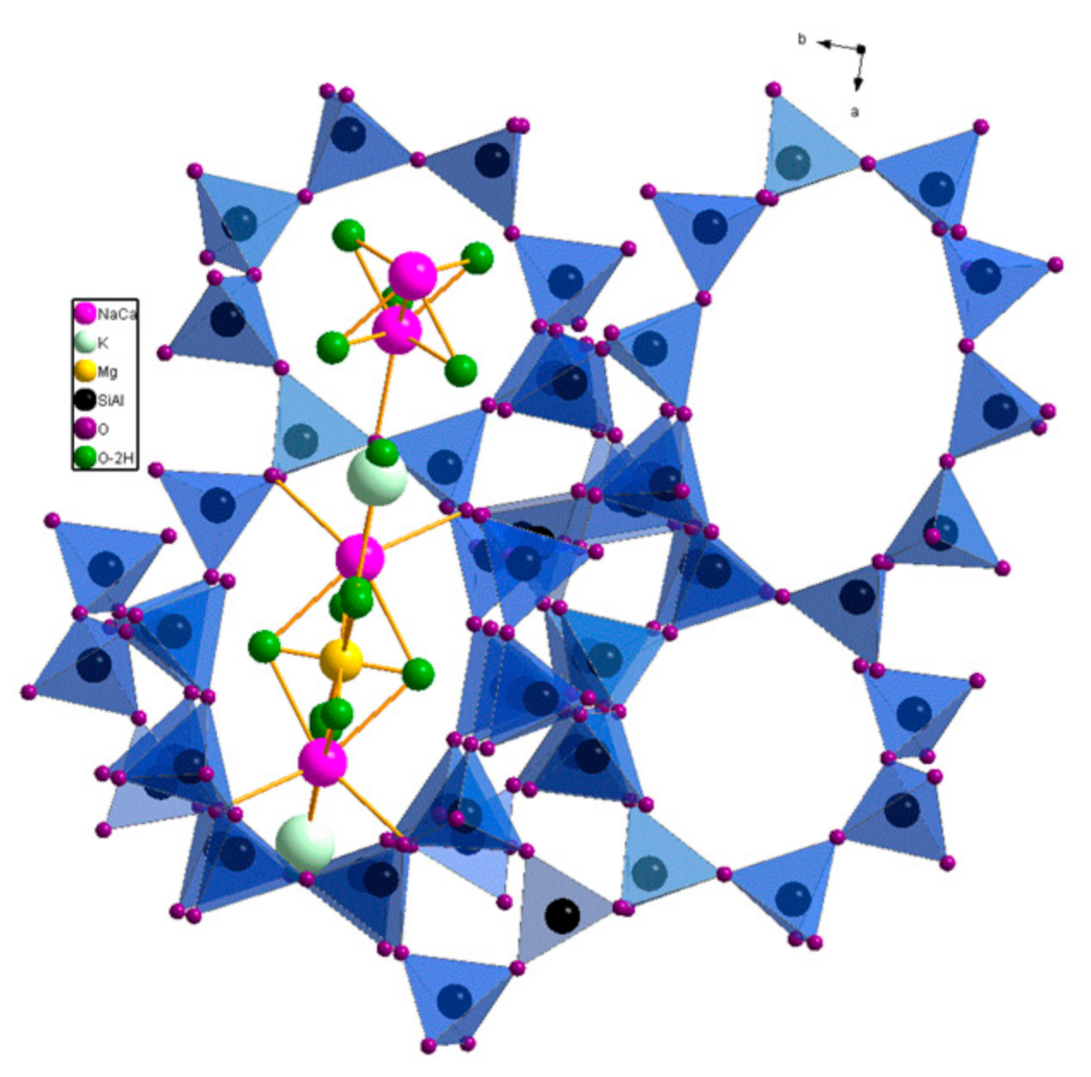
The crystalline aluminosilicate structure of clinoptilolite contains a two-dimensional pore system of partially interconnected, two types of eight and one type of ten, membered oxygen ring pores. Clinoptilolite has window sizes of ca. 0.26 × 0.47 nm, 0.33 × 0.46 nm, and 0.4 × 0.76 nm, respectively [26]. The cations of the clinoptilolite, located in the channels and windows, are difficult to exchange by ammonium ions (Figure 1). Therefore, in most cases the starting material is treated with mineral acids in order to replace the cations by Brønsted acidic protons H+. Thereby, the acid form of the natural zeolite clinoptilolite HCLIN is obtained. This approach suffers from environmental pollution and a part of the starting material lost by dissolution of the clinoptilolite. Additionally, it is difficult to remove the used mineral acid from the catalyst. Therefore, in this paper the acid form of the clinoptilolite was prepared to be more sustainable via ammonium ion exchange of the zeolite cations to obtain the NH4CLIN form. Thereafter, the ammonium ions are decomposed by calcination in air, leaving off the HCLIN catalyst [27].
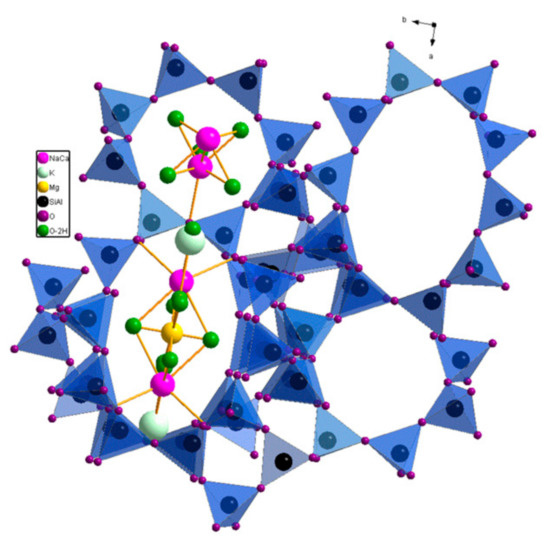
Figure 1.
Pore structure of clinoptilolite showing two parallel running oxygen-8-ring and oxygen- 10-ring pores with included cations and water molecules.
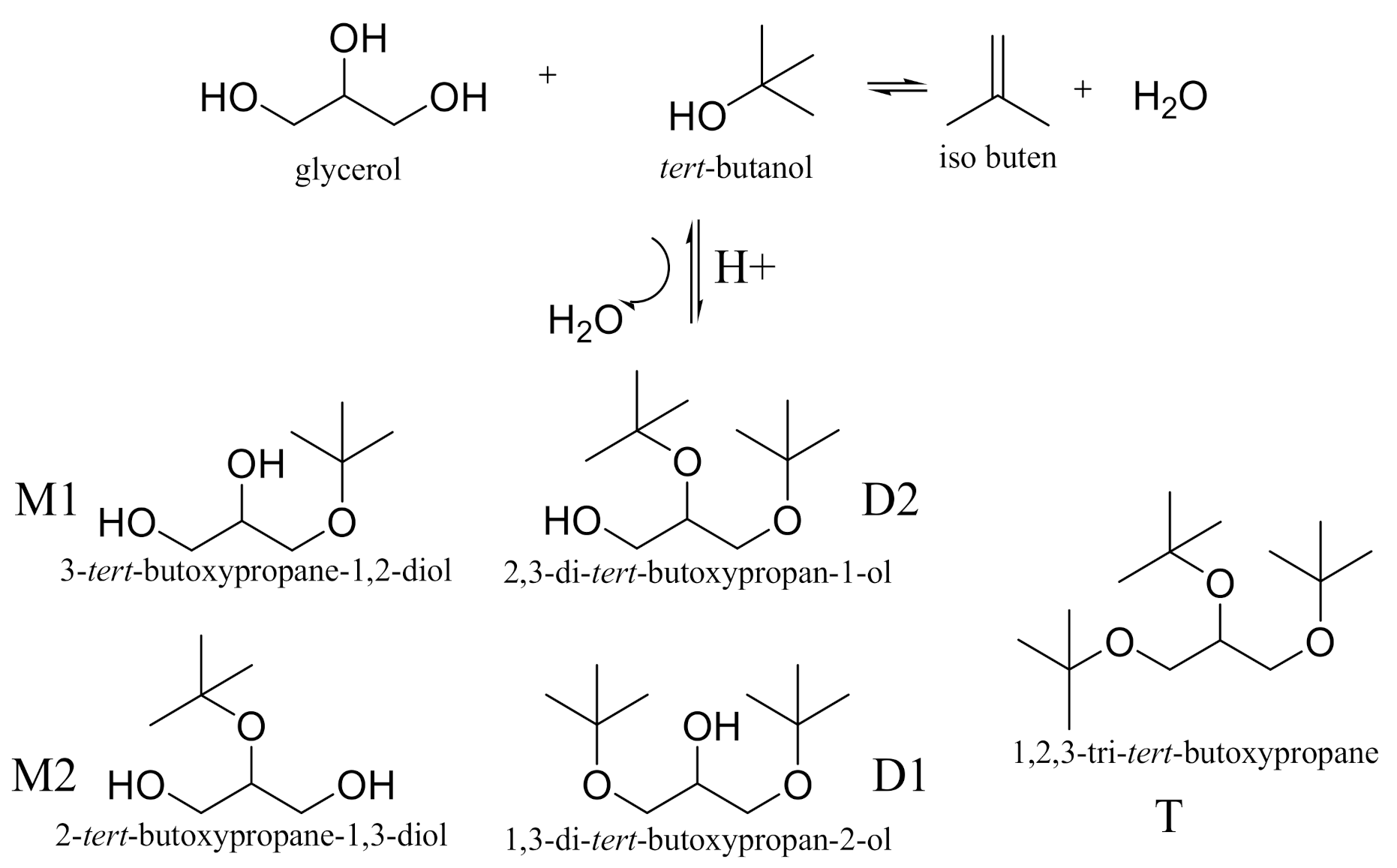
The paper aims the preparation of different activated acid clinoptilolite catalysts of HCLIN. The catalysts were characterized regarding the structure, porosity, stability, i.e., of the zeolite framework and Brønsted acid sites, and the acidity. The state of aluminium in the framework was studied by solid state NMR spectroscopy. The catalytic performance in the etherification was tested (Scheme 1).
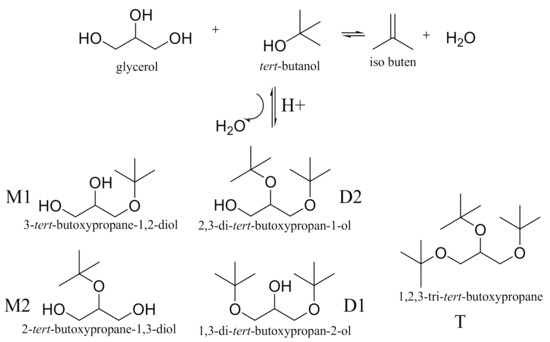
Scheme 1.
Etherification of Glycerol with tert-butanol (M- mono ether, D- di ether and T- tri ether).
2. Results and Discussion
2.1. Materials
The starting material for the preparation of heterogeneous acid catalysts was the natural zeolite clinoptilolite (clinofit® Si Premium, VITARING®- Biomedsystems GmbH, Kienberg, Germany) provided by Vitaring Biomed systems GmbH, ammonium nitrate NH4NO3 (Laborchemie Apolda, M = 80.05 g/mol, >99.9% purity) was used for the NH4+ ion exchange. Glycerol was provided by Sigma Aldrich (water content: <0.5%). The used alcohols were pure (>99%). The acids were of pure grade (Merck).
The chemical composition of the starting material and of the ammonium exchanged clinoptilolite was analysed by AAS (Table 1). The exchange of the zeolitic cations by ammonium ions in solutions leads to a decrease preferentially of the concentration potassium ions and calcium ions in the framework of NH4CLIN [18].

Table 1.
Chemical composition of the starting clinoptilolite CLIN and of ammonium exchanged NH4CLIN.
2.2. Preparation
2.2.1. Preparation of NH4CLIN
In detail, during ion exchange, the Vitaring clinoptilolite was continuously stirred in an aqueous 0.2 M NH4NO3 solution two times at 80 °C for 2 h each. Next, 250 mL of deionized water was heated to 80 °C. Then, 10 g of clinoptilolite powder was added, followed by the addition of 4 g NH4NO3. Thereafter, the solution was continuously stirred. The upper exchange solution was replaced a new 0.2 M NH4NO3 solution for the second ion exchange. The obtained samples were washed with deionized water several times and dried overnight at elevated temperature. An ion exchange degree of ca. <60% was achieved.
2.2.2. Preparation of the Acid H-form HCLIN
The acid forms were obtained by calcination of the NH4CLIN in air at different temperatures (200 °C, 300 °C, 400 °C, 500 °C, and 600 °C) with short temperature holding times (1 min and 30 min), respectively. The heating rate was 10 °C/min. The obtained acidic natural clinoptilolite zeolite catalysts were named HCLIN200, HCLIN300, HCLIN400, HCLIN500, and HCIN600, respectively.
2.3. Methods
The XRD pattern were recorded on a powder X-ray diffractometer (STADI-P, STOE, Darmstadt, Germany) using monochromatic Ni-filtered CuKα1 radiation (λ = 1.5406 Å). The diffraction angle range 2θ was 5–85°. The resolution was 0.02° (180 s per step). Atomic absorption spectrometry (contrAA800D, Fa. Analytik Jena, Jena, Germany) was used for the chemical analysis. The 20 mg samples were suspended in a mixture of concentrated hydrochloric acid HCl (36 wt%), 2 mL nitric acid HNO3 (65 wt%) and 2 mL hydrofluoric acid HF (40 wt%) provided by Fisher chemicals and hydrothermal treated in two steps at 155 °C and 200 °C. The ammonium content ammonium clinoptilolite and HCLIN samples was determined by TPDA.
A field emission scanning electron microscope (S4800 FE-SEM, Hitachi, Tokyo, Japan) at an accelerating voltage of 5 kV and a transmission electron microscope (TEM, EM 902A, Zeiss, Göttingen, Germany) were used to investigate the surface and morphology (size and shape) of the modified clinoptilolite. Sample imaging was obtained with a CCD camera (CCD-sensor THX 7888A, 14µm x 14µm pixel size, 1024 × 1024 pixel per mm2, Co. Proscan, Scheuring, Germany). Image processing was performed by iTEM software (Olympus soft imaging solutions GmbH, Münster, Germany). An FT-IR spectrometer (Nicolet 380, Bruker Alpha 2, Billerica, MA, USA) with a platinum ATR (Attenuated Total Reflection) device was used for the measurement of the infrared spectra in the spectral range of 400–4000 cm−1 covering the range of lattice (framework) vibrations of zeolites. For nitrogen adsorption and desorption isotherm measurements were carried out on a “Sorptomatic” instrument (Porotecc, Germany). The specific surface areas and pore volumes were determined by the BET and BJH method. The TGA curves were recorded on a TG/DSC device (Labsys 1600, Setaram, France) with a heating rate of 10 K/min in a helium flow of 20 mL/min. The thermal desorption of adsorbed ammonia and of ammonia released by the thermal decomposition of ammonium ions of the zeolite was performed with a TPDRO 1100 series device (Thermo Scientific, Schwerte, Germany). The 29Si MAS NMR measurements were recorded on an NMR spectrometer (DMX-400, Bruker), whereas the 27Al MAS NMR and 1H MAS NMR measurements were performed on the NMR spectrometer (AVANCE-600 spectrometer, Bruker). Before measurement, all samples were activated at 400 °C for 1 h, at a heating rate of 5 °C/min.
2.4. Catalysis
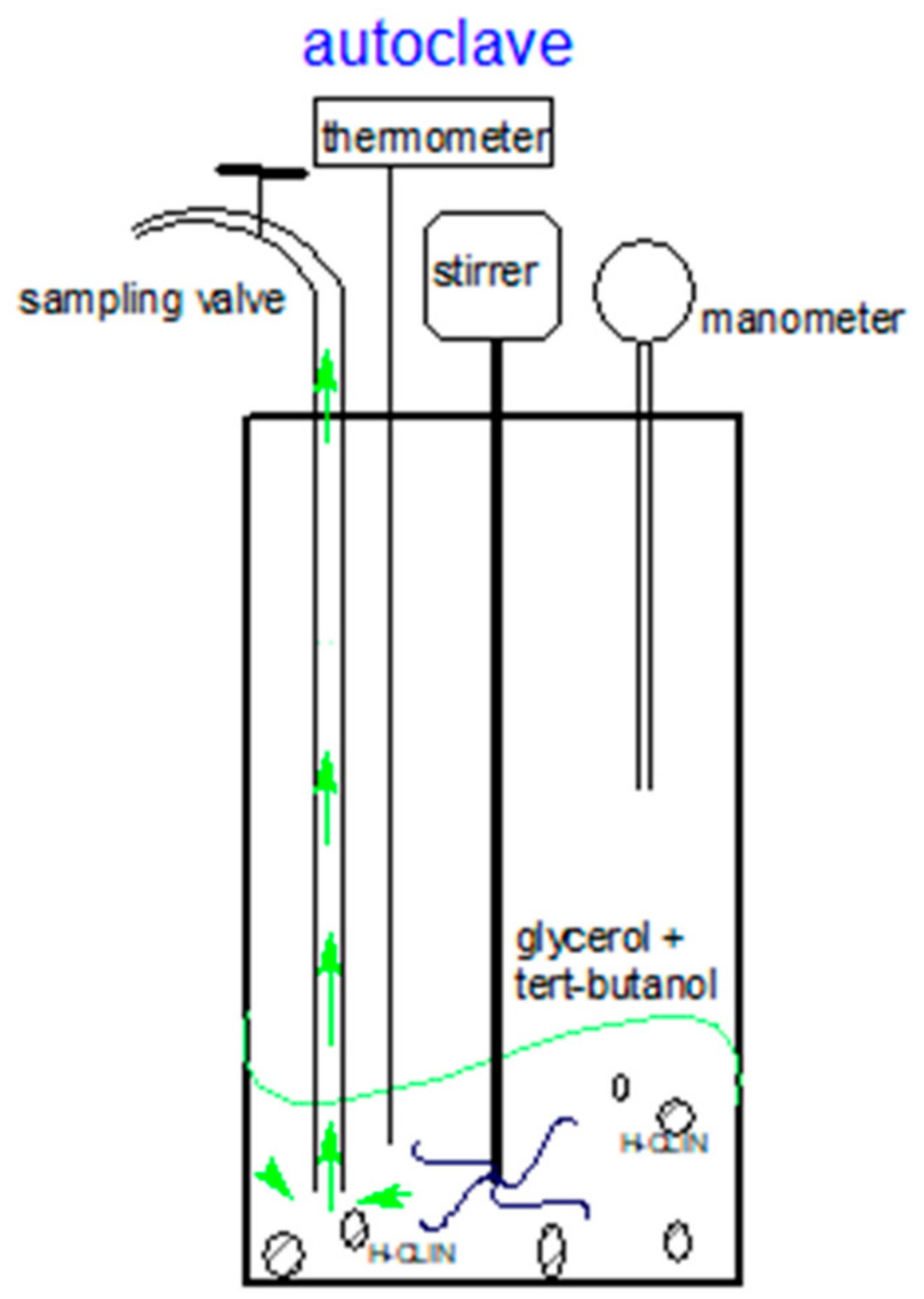
The catalytic testing of HCLIN catalysts in etherification of glycerol with tert-butanol in the liquid phase was carried in Teflon-lined stainless-steel autoclaves under autogenous pressure at elevated temperatures (Figure 2). Before catalytic testing, the samples were pre-treated in flowing helium at 110 °C to remove loosely bound ammonia and water.
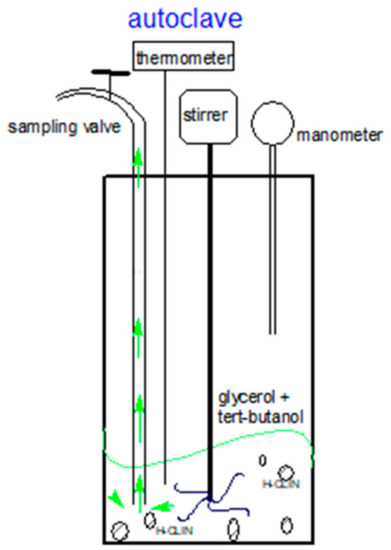
Figure 2.
Scheme of the autoclave reactor: High-pressure reactor with 150 mL volume capacity (5500 Parr autoclave) with a controller Parr 4848 used in catalyst testing.
A total of 10 g of glycerol and 32.6 g of t-butanol (Gly/TBA ratio = 1/4) was added into the Teflon beaker inside the autoclave then 0.5 g of catalyst (5 wt% regarding glycerol) was added before installation (Figure 2). The reaction mixture was heated to a temperature of 110 °C under stirring. The reaction temperature was reached after approximately 1 h and 10 min. Aliquots of 1.5 mL were taken from the reaction mixture using a sampling valve of the autoclave after 30 min, then 1, 2, 4, and 6 h of reaction, respectively. In order to avoid the presence of non-reacted solution inside the sample pipe, first, about 2 mL solution was released before keeping the solution for analysis. The experiments at 86 °C were carried out in a glass batch-reactor under normal pressure. It consisted of a 3-necked bulb equipped with a condenser, a thermometer, and an opening for aliquot samples.
For re-use testing and cycling experiments, the catalyst was separated from the reaction mixture and washed successively with water and ethanol 3 times. Then, the catalyst was dried at 110 °C overnight, before repeated use in a new reaction mixture.
2.5. Analysis
The chemical composition of reaction mixtures was analysed using a GC/FID device. A 10 m long 0.20 µm DB-Wax Gas Chromatography column with an open diameter of 0.1 mm was used to separate the reaction components. A GC-FID flame ionization detector was used. Further, 1 µL of analyte was used. The split rate was 1:50, the inlet temperature was set up at 240 °C. The column temperature program 40/5-6-180/10-8-240/5 was used. The initial temperature of 40 °C was held for 5 min. Then in the first ramp, the temperature was increased by 6 K per minute until it reached 180 °C and kept at this temperature for 10 min. In the second ramp, the temperature was increased by 8K per min to 240 °C, and kept for 5 min.
The conversion of the glycerol was determined from the change of the relative amount of glycerol in the reaction solution. For this, the GC peak areas of glycerol and of the reaction products, the mono, di, and tri-ether were summed up and set to 100%:
The selectivity of mono, di, and tri ethers ( was obtained from:
where are the peak areas of M1, M2, D1, D2, and T, components in the chromatograms.
2.6. Characterisation
The natural zeolite clinoptilolite precursor, ammonium exchanged clinoptilolite, and different calcined samples were characterized regarding their structure, crystallinity, morphology, stability, porosity, and acidic properties by XRD, SEM/TEM, FTIR lattice vibration spectra, as well as by nitrogen adsorption and desorption, TG-DSC and TPDA measurements. Additionally, 29Si MAS NMR, 27Al MAS NMR, 1H MAS NMR measurements as well as selected 1H- 29Si NMR Redor and 1H- 27Al NMR Trapdor experiments were performed.
2.6.1. XRD
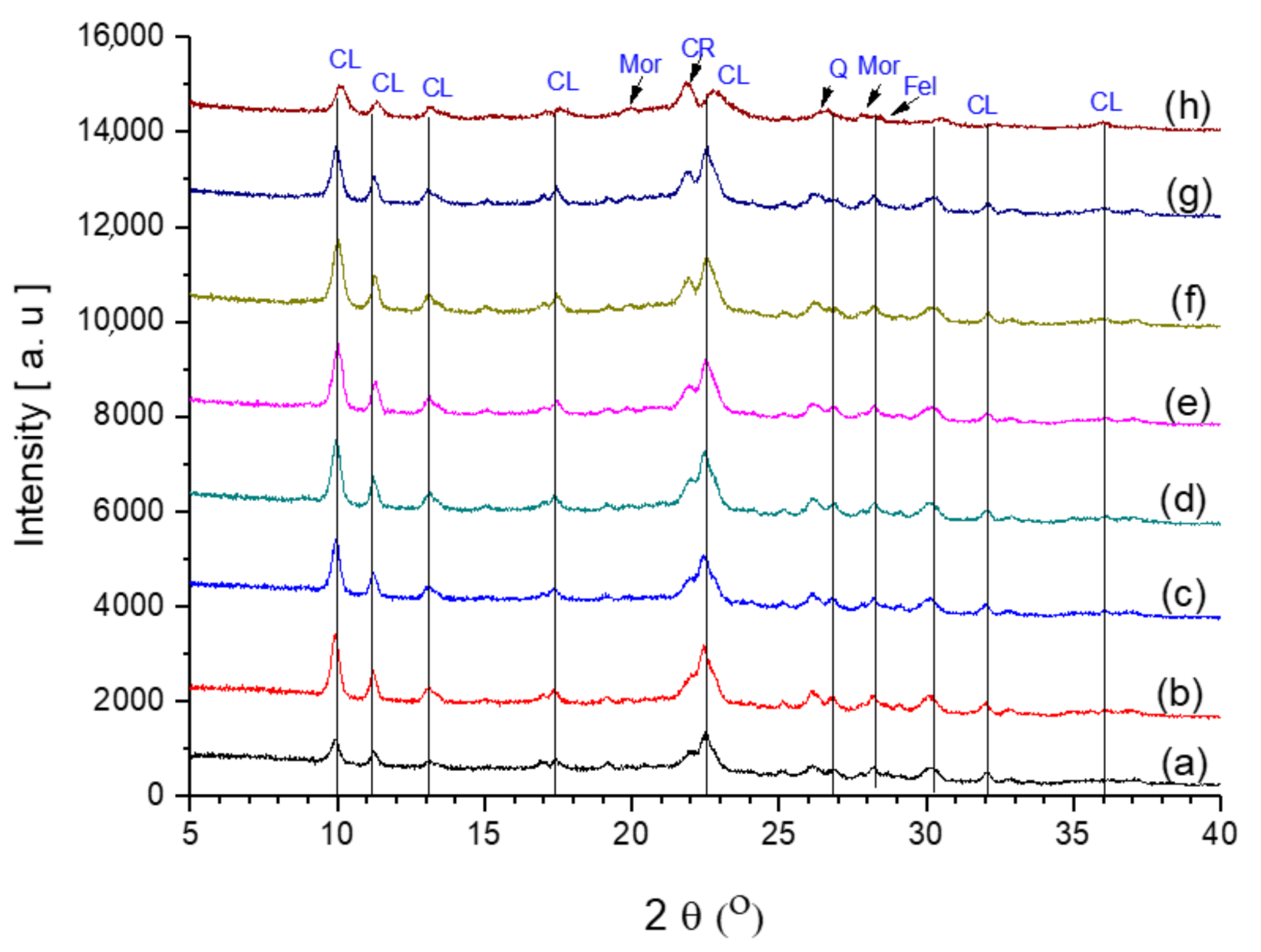
The obtained samples CLIN, NH4CLIN, and the thermal activated HCLNs, which were activated at different temperatures, namely 250, 300, 400, 450, 500, and 600 °C, were studied in order to investigate the crystallinity of modified clinoptilolite (Figure 3).
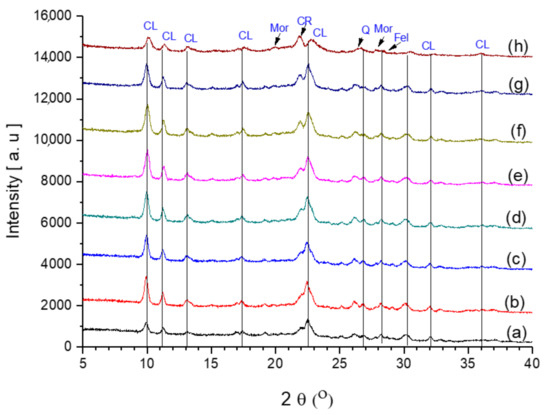
Figure 3.
Powder XRD patterns of (a) starting clinoptilolite CLIN, (b) ammonium exchanged NH4CLIN, and the thermal treated acidic forms: (c) HCLIN250, (d) HCLIN300, (e) HCLIN400, (f) HCLIN450, (g) HCLIN500, and (h) HCLIN600. “CL”—the typical reflections of CLIN, “CR”—Cristobalite, “Mor”—Mordenite, “Q”—Quartz, and “Fel”—Feldspar [28].
The X-ray diffraction patterns of the eight samples show that all samples contain clinoptilolite. Besides the clinoptilolite, the samples contain minor amounts of quartz, cristobalite, and feldspar as an impurity. The calcination temperature was increased up to 600 °C to study the thermal stability. The XRD pattern of the sample calcined at 600 °C still shows the reflections of the clinoptilolite structure, although the intensities of the reflections are lower compared to the sample calcined at 500 °C. Indeed, the background increases beyond heating to 400 °C, indicating some loss in the crystallinity. Additionally, the intensity of the XRD reflection of cristobalite at 2θ = 21.8° increases compared to the intensity of the neighboured clinoptilolite reflection. Normally, the clinoptilolite structure starts to collapse above 500 °C [29]. The calcination at 600 °C leads to partial destruction (and amorphization) of the clinoptilolite framework as indicated by the loss of intensity of the reflections and the marked increase in the background. In addition, the formation of the aluminosilicate labradorite was reported at 500 °C [30]. As shown in Figure 3, the crystallinity increases with NH4CLIN after washing and ion exchange with ammonium nitrate solution at elevated temperatures. This is likely due to the removal of amorphous impurities due to the treatment in the aqueous solution.
The crystallite size was determined by the Scherrer equation published in 1918 [31]:
where: D is the crystallite size (nm), K = 0.9 is a constant, λ = 0.15406 nm (wavelength of the x-ray radiation), β = FWHM is the full width at half maximum (FWHM) of the most intense diffraction peak (020 of CLIN), θ = peak position (radians).
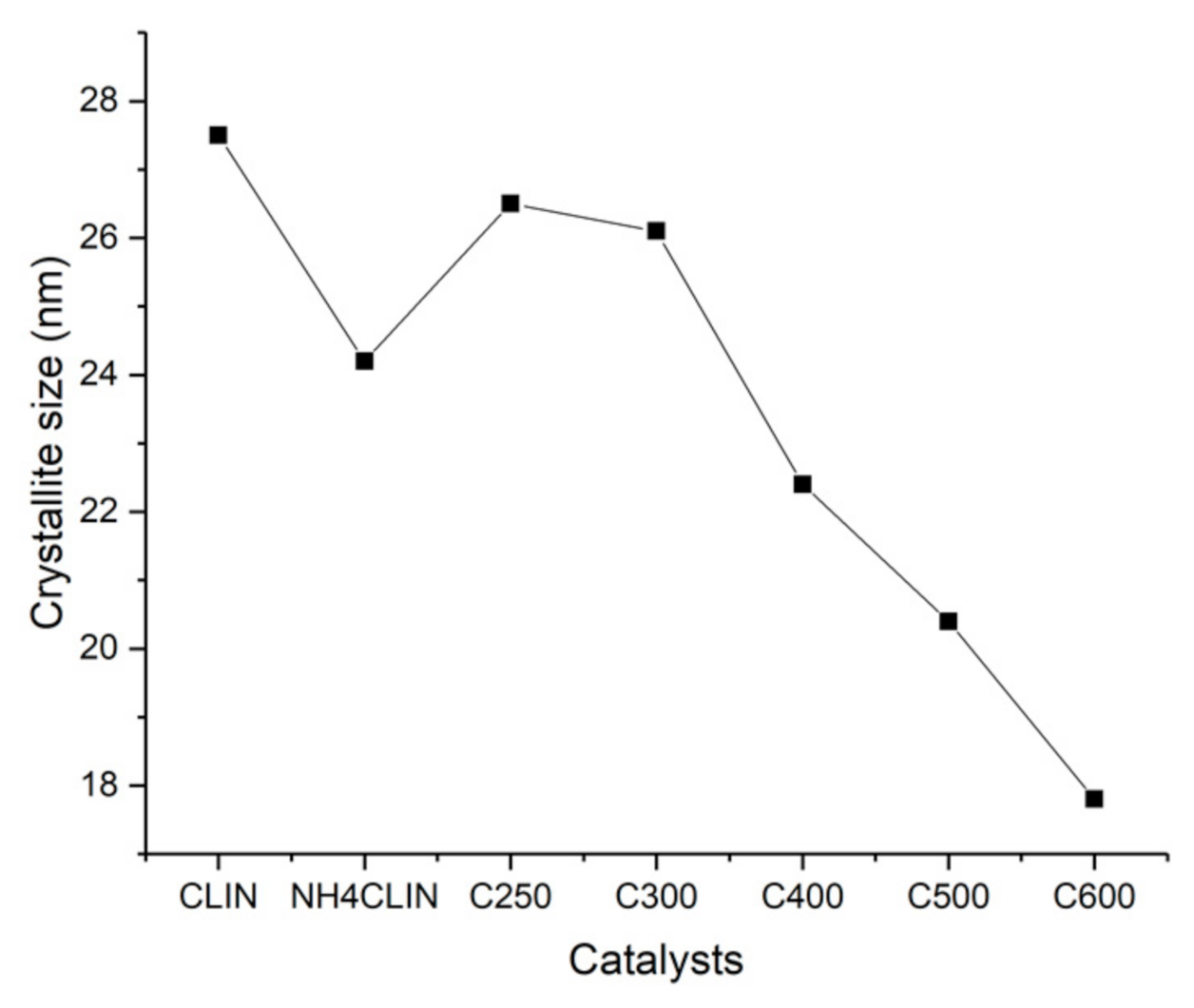
Figure 4 shows that the crystallite size of the samples which decreases with rising calcination temperature. The crystallite size decreases from ca. 26 to 18 nm with the samples HCLIN300 and HCLIN600, respectively. These values are close to the size of the primary aggregated nanoparticle found in TEM (ca. 20 × 50 nm).
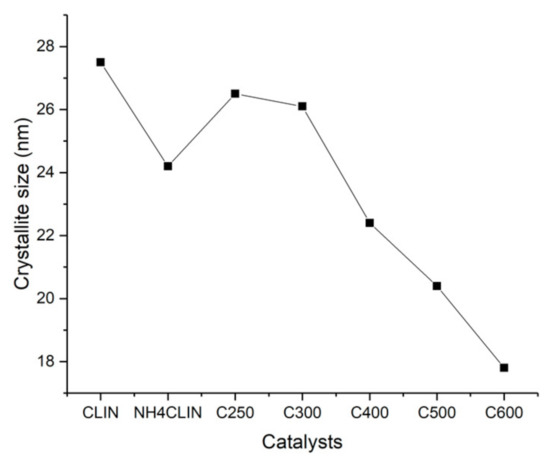
Figure 4.
Influence of the calcination temperature on the crystallite size of clinoptilolite: starting material CLIN, ammonium form NH4CLIN and calcined NH4CLIN (calcination temperatures C250, C300, C400, C500, C600).
In conclusion, the natural zeolite samples are crystalline and show high thermal stability close to 600 °C. They are composed mostly of strongly aggregated clinoptilolite nanocrystals as shown below.
2.6.2. Scanning Electron Microscopy (SEM)
The SEM images (Figure 5) at 1000 times magnification show that the morphologies of the starting CLIN and the NH4CLIN are similar. They consist of bulky particles of up to 10 (20) µm size and show a rough surface. The bulky particles consist of agglomerated nanoparticles. The appearance of the aggregated particle changes with thermal treatment. They disintegrate into smaller particles. The surface become more porous. In part, the particles look similar to nests with holes of µm size. The reason for the changes could be the rapid evaporation of loosely bound water and ammonium ions from inter and intra crystalline small pores. This causes internal stress and partial local collapses. The particles are more separated from each other. When the temperature increases to 500 or 600 °C, the surface changes again. More small particles are formed by the disintegration of larger ones.


Figure 5.
SEM images of (a) CLIN, (b) NH4CLIN, (c) HCLIN200, (d) HCLIN300, (e) HCLIN400, (f) HCLIN500, (g) HCLIN600 with 100 k of magnification.
The high magnification SEM images (100 k) confirm the aggregated structure of the material. Indeed, the µm-sized particles are composed of nanoparticles. They are stacked together in the samples CLIN, NH4CLIN, and HCLIN200. The HCLIN300 showed more holes and more flowery particle shapes. The thermal treatment markedly changes the morphology of sample particles. The changes are likely related to the fast decomposition of ammonium ions and the release of water from the internal pores at elevated temperatures.
2.6.3. TEM
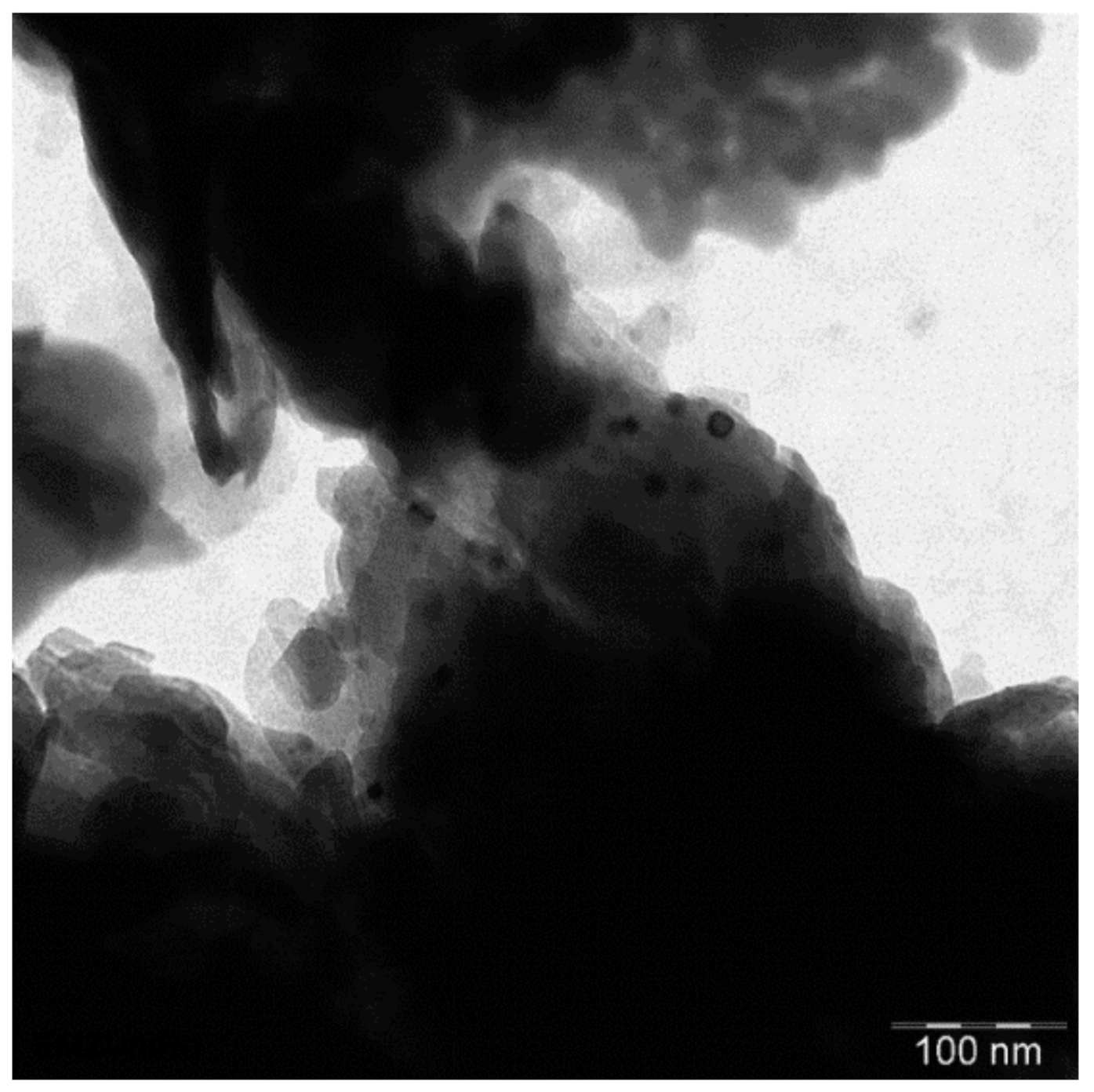
The large bulky particles of the natural clinoptilolite sample consist of a of smaller aggregated subunits as show by SEM (Figure 5). These aggregates decompose into smaller particles upon calcination, down to a size of ca. 1 µm. The TEM image of such a small particle shows that it is composed of strongly agglomerated nanoparticles (Figure 6). A part of the clinoptilolite nanoparticles looks round shaped with a size of ca. 40–60 nm (Figure 6). Another part is plate-like shaped with a thickness of ca. 10–20 nm and a length of ca. 50 nm and more (Figure 6). Hence, the clinoptilolite catalyst shows a hierarchical particle structure composed of primary crystalline clinoptilolite nanoparticles which are aggregated into µm-sized sub particles, forming finally the bulky material. The thereby formed macro pores facilitate greater accessibility to the catalyst surface.

Figure 6.
TEM image of HCLIN300.
2.6.4. EDX
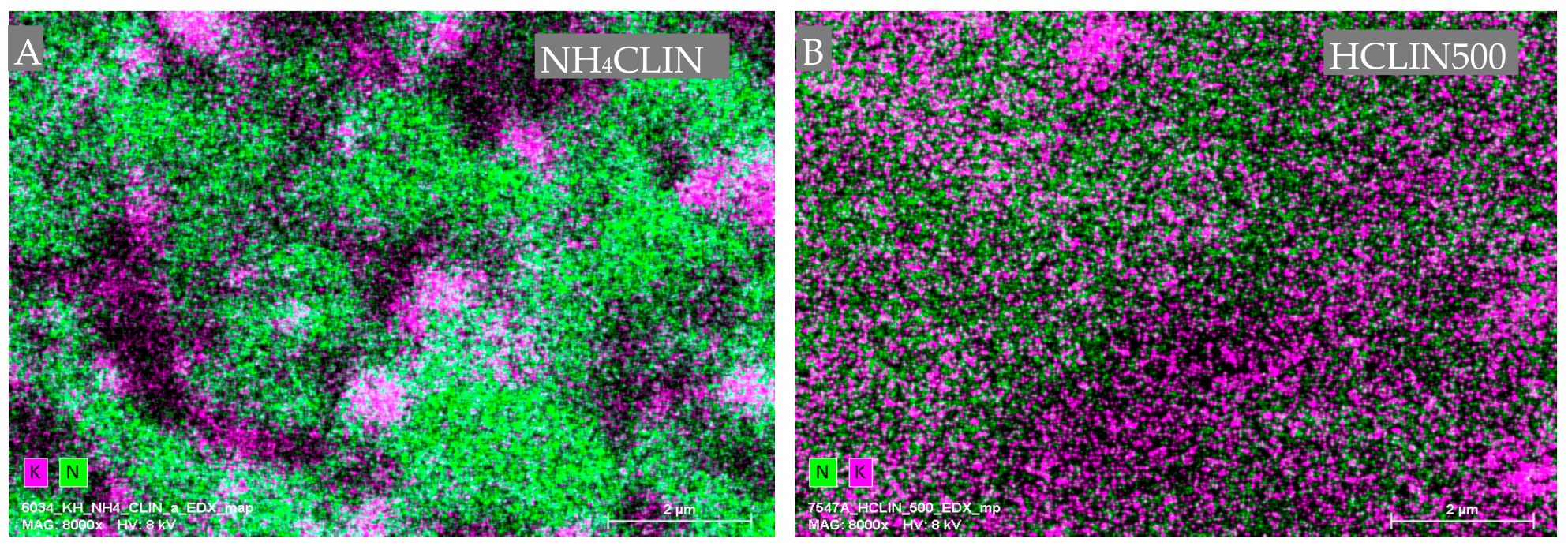
Figure 7 shows the nitrogen, i.e., ammonium ions (green dots) and potassium (pink dots) mapping images of NH4CLIN and HCLIN500. The intensity of the nitrogen dots decreases markedly after heating to 500 °C. This is in line with the observed decomposition of the ammonium ions after calcination at 500 °C as observed with FTIR, TGA and TPDA. Interestingly, the potassium ions are non-homogenously distributed and form “islands” in the image recorded with NH4CLIN. With HCLIN500, potassium dots are observed within all sample parts, however, with low density, as expected after ammonium ion exchange.
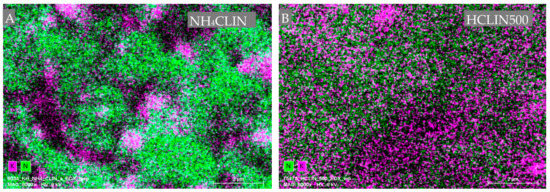
Figure 7.
Energy dispersive X-ray (EDX) element mapping analysis for nitrogen (green), and potassium (pink) element distribution in two samples: NH4CLIN (A), HCLIN500 (B).
Figure 8 shows the Ca ion mapping of the starting CLIN and after ammonium ion exchange and calcination at 500 °C. The Ca2+ ions (yellow dots) are homogenously distributed throughout both of the samples. However, the yellow dots are less dense, indicating a lower calcium content with HCLIN500 caused by the ammonium ion exchange. This finding is in line with the chemical analysis.
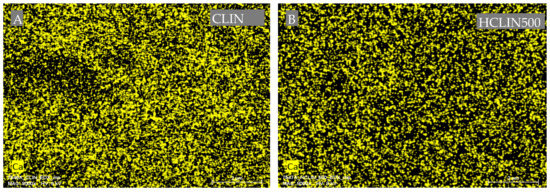
Figure 8.
Energy dispersive X-ray (EDX) mapping analysis for the element calcium of two samples: CLIN (A), HCLIN500 (B).
2.6.5. Nitrogen Adsorption and Desorption Measurements
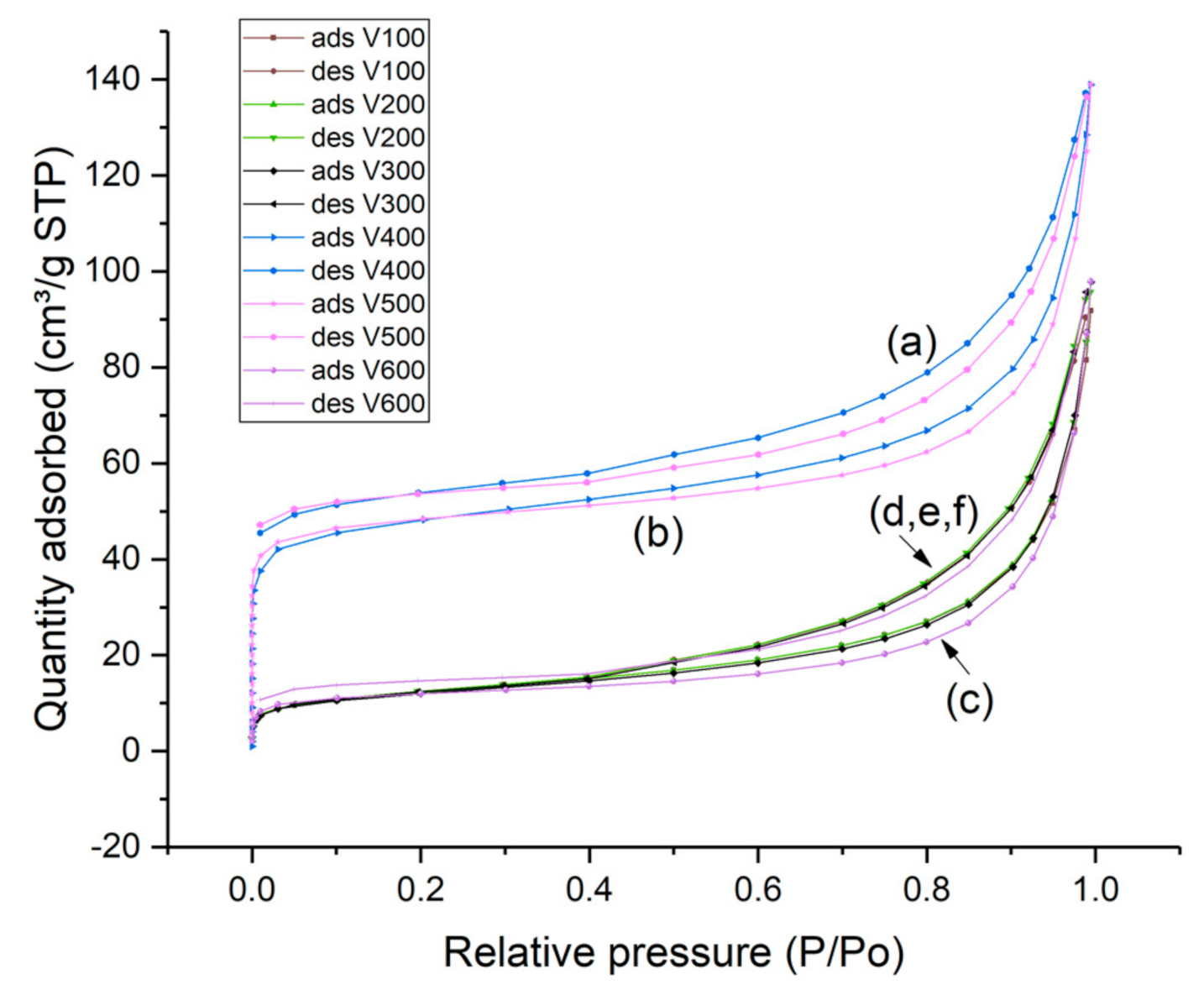
In Figure 9, the nitrogen adsorption and desorption isotherms of ammonium clinoptilolite and HCLIN catalysts obtained at different calcination temperature are shown. The isotherms are a combination of type I (microspores) and type II (macrospores) isotherms according to the IUPAC nomenclature [32]. The nitrogen uptake at very low relative pressure of p/p0 of 0 to 0.05 is due to the adsorption in the microspores of the zeolite framework. Uptakes at higher relative pressures are due to the filling of mesoporous multilayer adsorption. Adsorption at high relative pressures of p/p0 > 0.6 is due to adsorption in the interparticle macropores. The starting clinoptilolite and the ammonium exchanged clinoptilolite at a very low specific surface area. The pore system is closed by the exchangeable cations located in the pore entrances. Obviously, the highest microporosity and BET specific surface areas are found with HCLIN400 and HCLIN500, ca. 163 and 120 m2/g. In this case, the ammonium ions located in and blocking the micropores are decomposed by the thermal treatment. This way the internal micro pore system is opened. The pore system of samples calcined at a lower temperature is still blocked. Therefore, the samples NH4CLIN100, NH4CLIN200, and HCLIN300 show only a low increase in the adsorption isotherm at low relative pressure. The same holds with the sample calcined at 600 °C, although this temperature is sufficient for the decomposition of pore-blocking ammonium ions (Table 2, Figure 9). However, the framework destruction indicated by the XRD measurements also leads obviously to a loss and/or the blocking of the micropores in the clinoptilolite crystals. The microporosity is strongly decreased but the macroporosity (interparticle pores) is maintained. The latter is also confirmed by the SEM and TEM images.
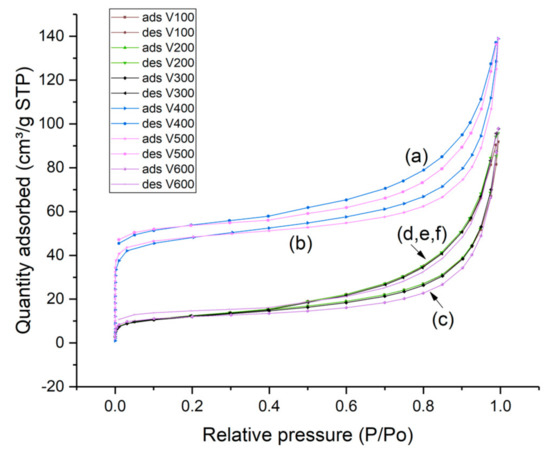
Figure 9.
Nitrogen adsorption and desorption isotherms of (a) HCLIN400, (b) HCLIN500, (c) HCLIN600, (d) NH4CLIN100, (e) NH4CLIN200, and (f) HCLIN300.
Table 2.
Specific surface area, micropore and macropore volumes of ammonium exchanged and thermally activated HCLIN.
2.6.6. FTIR
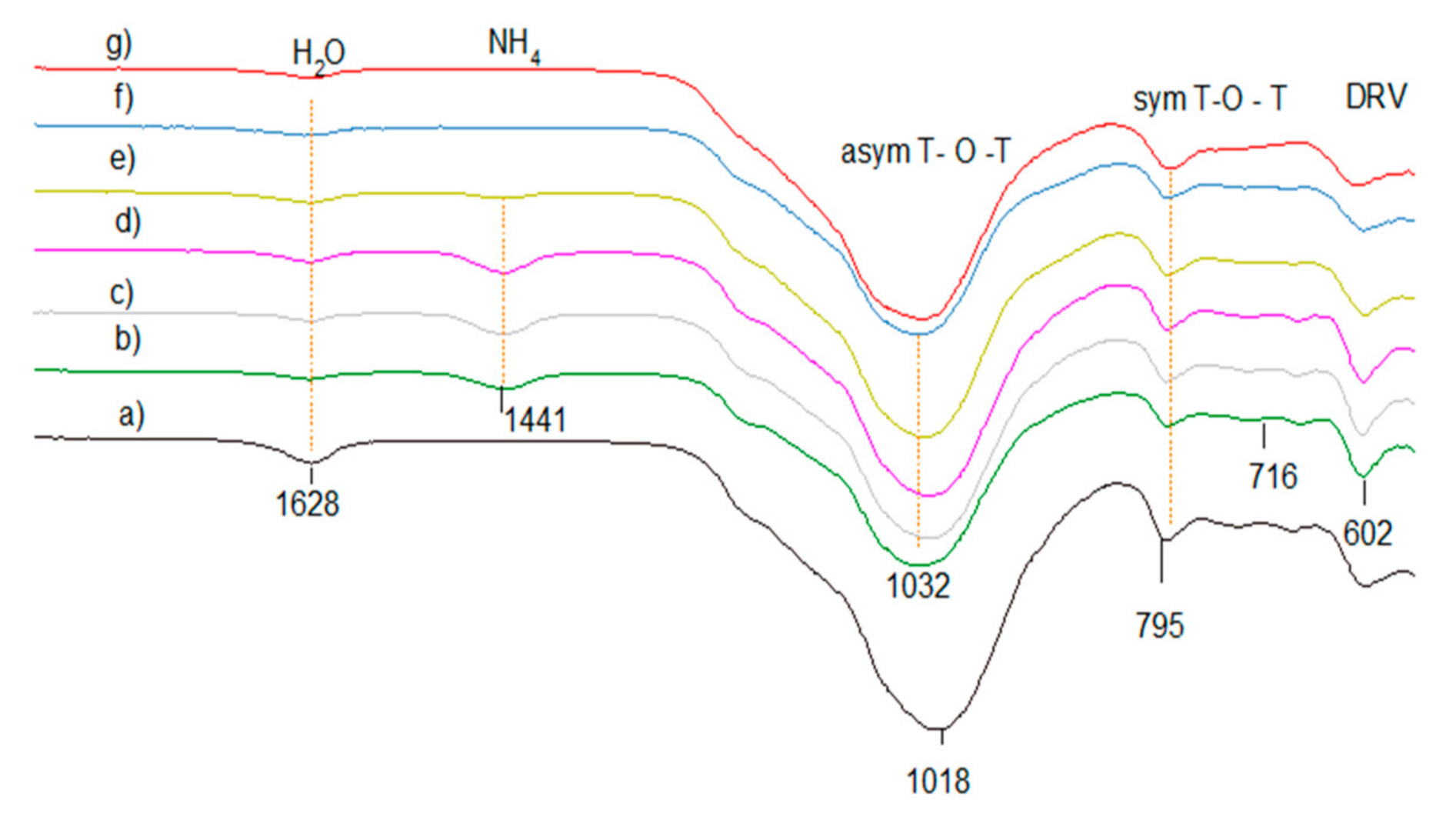
The starting clinoptilolite, the ammonium-exchanged form, and obtained acidic clinoptilolite catalysts HCLIN were investigated by FTIR spectroscopy in order to follow independently the decomposition of the ammonium ions by the decrease in the intensity of the ammonium mode at ca. 1440 cm−1 (Figure 10). Infrared spectroscopy IR also allows use to determine roughly the fraction of aluminium in the framework. The lattice vibration bands of the Si–O–Al framework appear in the mid-IR in the spectral range of 400–1200 cm−1. The vibration band at ca. 1250–950 cm−1 is assigned to anti-symmetric T–O–T stretching vibrations. Symmetrical stretching vibrations appear at 720–650 cm−1. The T–O bending vibration bands appear at 420–500 cm−1. Structurally sensitive vibration bands of secondary building units as the zeolite double-ring units (DRV) are observed at 650–500 cm−1. Deformation vibrations of adsorbed water molecules occur at ca. 1630 cm−1 [33].
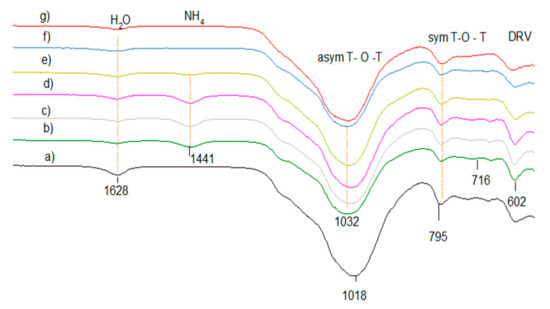
Figure 10.
FTIR lattice vibration spectra of clinoptilolite CLIN after different treatment: (a) starting material CLIN, (b) ammonium exchanged NH4CLIN, and (c) calcined HCLIN200, (d) HCLIN300, (e) HCLIN400, (f) HCLIN500, (g) HCLIN600.
The FTIR spectra of the starting clinoptilolite CLIN, ammonium exchanged NH4CLIN and thermally activated HCLINs are shown in Figure 10. The anti-symmetric T–O–T vibration band observed in the spectrum of the starting clinoptilolite is shifted to a higher wavenumber from 1018 to 1032 cm−1 (Figure 10) after exchange of the zeolite cations by ammonium ions and desorption of loosely bound water at 200 °C. A further shift from 1024 cm−1 (HCLIN300) to 1029 cm−1 HCLIN400 to 1034 with HCLIN500 is observed, which is related to the decomposition of the ammonium ions. This shift reflects the different interaction of the ammonium ions with the framework compared to Na, K, Ca, and Mg ions and the role of water. The framework shrinkage causes a decrease in the T–O bond lengths in the T–O–T bridges (T = Si, Al) tetrahedral atoms. Additionally, a new vibration band at 1441 cm−1 appears in the FTIR spectrum of NH4CLIN. It is assigned to the deformation vibration of the exchanged ammonium ions. The intensity of this band decreases with raising calcination temperature slowly. A strong decrease, i.e., decomposition of ammonium ions, is observed after raising the temperature to 400 °C. The ammonium ion absorption band nearly disappears after heating to 500 °C.
The FTIR results confirm that a major part of the ammonium ions is decomposed by heating the samples to 400–500 °C. Thereby, ammonia gas is released. The ammonium ions located in the pore windows are decomposed and replaced by acid protons. The pores are opened as observed by the nitrogen adsorption and desorption experiments.
2.6.7. TG-DSC
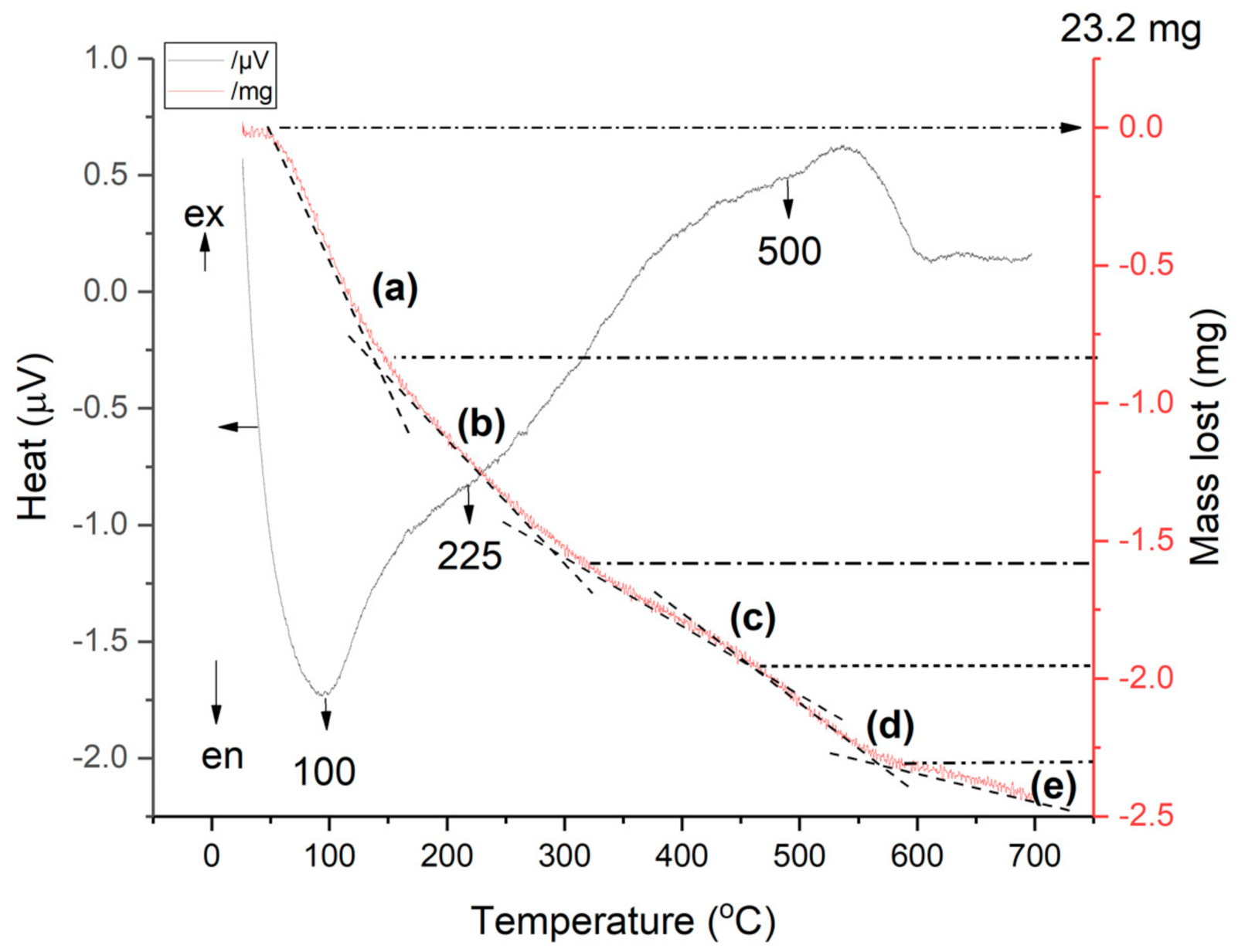
The combined (TG-DSC) curves of NH4CLIN are shown in Figure 11. A markedly endothermic weight loss is observed from 50 to 150 °C assigned to water removal by heating the sample to ca. 200 °C. It is associated with the loss of weakly bound water molecules as indicated by the low temperature of the peak maximum of 100 °C in the DSC curve. Above 150 °C (step from 150–280 °C) stronger bound water, e.g., bound to cations, is desorbed from the zeolite pores, giving rise to an endothermic shoulder in the DSC curve which is located at 225 °C. Step 3, starting from 280 °C to 450 °C, is assigned to the decomposition of ammonium ions and also some dehydroxylation or dehydration. The formation of BrØnsted acid sites by decomposition of the ammonium ions starts with HCLIN300. Step 4 is characterized by a slight decrease in the weight loss curve between ca. 450–570 °C. It is assigned to the decomposition of strongly bound ammonium ions with corresponding desorption of ammonia from stronger Brønsted sites. It is also an exothermic process. Further enhancement of the temperature causes dehydroxylation. At high calcination temperature, partial destruction of the clinoptilolite lattice occurs. At step 5, temperatures above 600 °C are assigned to the de-hydroxylation of the framework, which is in part connected with structural destruction, amorphization of the zeolite as indicated by XRD.
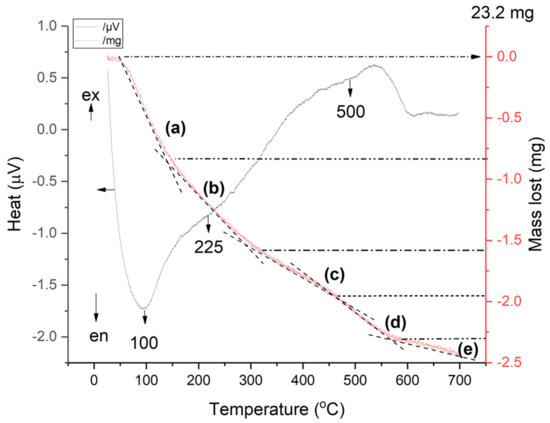
Figure 11.
Combined TG-DSC curves of ammonium exchanged NH4CLIN.
The weight loss of ammonia: less than 3.2 wt% (step c) and (step d). Because step (c) also contains the removal of some strongly bound water, the amount of ammonium is ca. 2.4 wt%. The maximum possible ammonium content achievable after complete ammonium ion-exchange of the clinoptilolite is ca. 4 wt% according to the theoretical formula of ammonium clinoptilolite (NH4)6 Al6Si30O72 · 24H2O. The reached ion exchange degree is ca. 60% which roughly agrees with the results of the ammonia TPD measurements and the chemical analysis. This roughly agrees with the results of the TPD and chemical analysis.
2.6.8. TPD of Ammonia
The acidity of different activated HCLIN catalysts was studied by temperature-programmed desorption of ammonia (Figure 11 and Figure 12). The maximum available acidity, concentration of BS, was determined by the thermal decomposition of the ammonium exchanged clinoptilolite by heating it to 700 °C (Figure 12). The decomposition of the ammonium ions starts at ca. 230 °C.
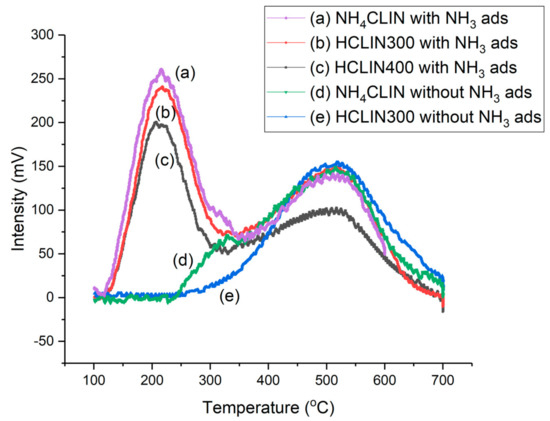
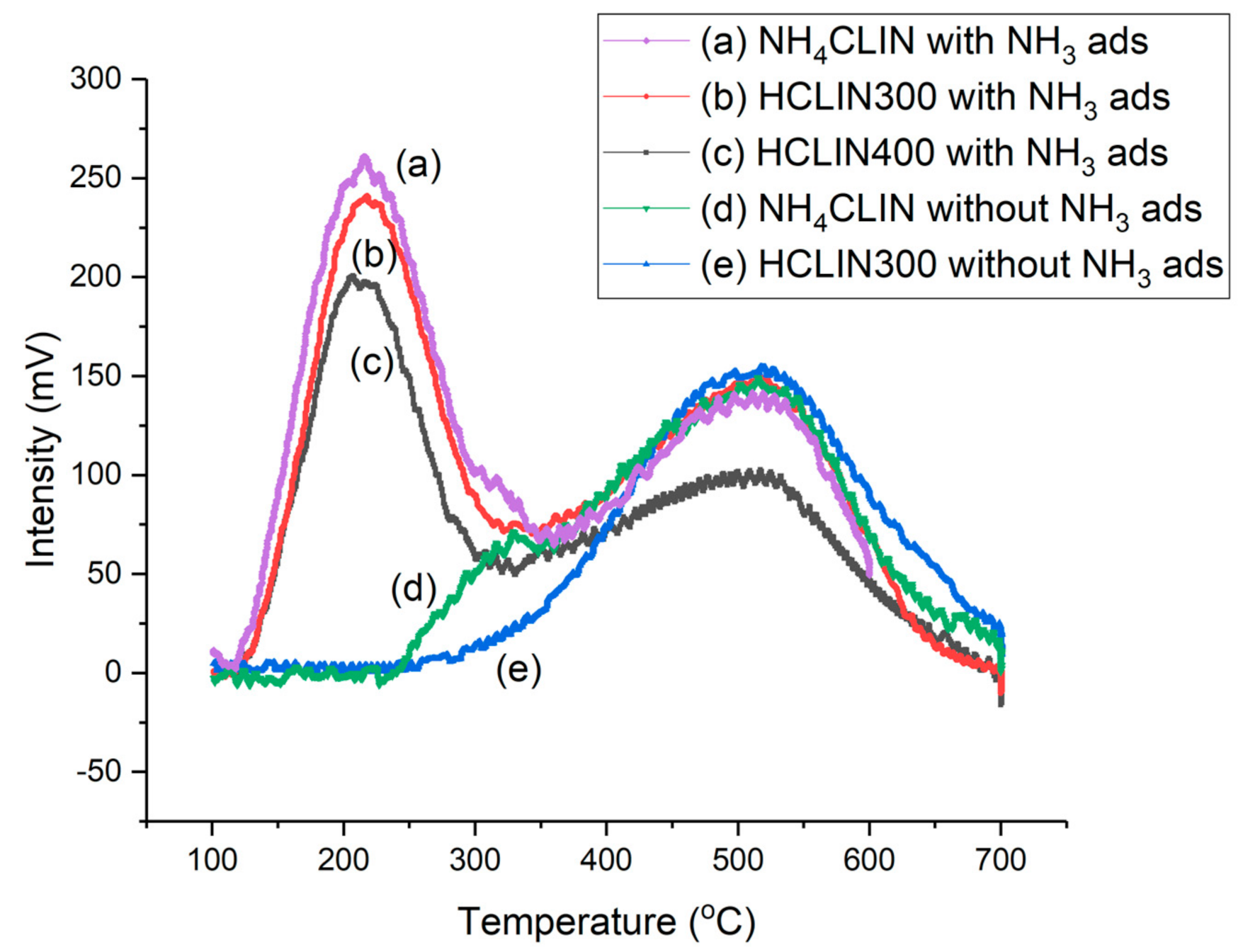
Figure 12.
Temperature-programmed desorption curves of NH4CLIN with NH3 flow treatment for (a) NH4HCLIN, (b) HCLIN300, (c) HCLIN400 (pre-treatment of samples at 180 °C, NH3 gas adsorption at 100 °C). Without NH3 adsorption: (d) NH4CLIN, (e) HCLIN300.
In the first step until 320 °C, ammonia is desorbed from weak BS. Thereafter, ammonia desorption continuous and reaches a maximum at ca. 520 °C followed by a decrease and tailing until 700 °C. The desorption of ammonia at higher temperatures is assigned to medium (400 °C) and strong (520 °C) acid sites. Correspondingly, the observed peak maximum indicates the presence of (very) strong BS in HCLIN catalysts. With HCLIN300, mainly weaker acid sites are liberated. These BS are stable. Re-adsorption of ammonia covers these sites again (Figure 12b) as well as in the NH4CLIN sample (Figure 12a). The total amount of desorbed ammonia with the ammonia-loaded sample is increased compared to the ammonium-exchanged sample, because ammonia adsorption occurs not only on the BS but also at weaker interacting adsorption sites in the pores. The temperature maximum of the low-temperature desorption is ca. 200 °C. Both desorption parts at low and high temperatures overlap. The low amount of ammonia desorbed from NH4HCLIN until 300 °C is in line with a low specific surface area of this catalyst. The micropores are still blocked by the ammonium ions. In contrast, ammonia desorption found with heating sample HCLIN400 to 400 °C is higher. The maximum acidity could be 1.0 mmol/g at HCLIN500 (Table 3). Microspores are opened, leading to high nitrogen uptake of these samples at low relative pressure in the micropores, i.e., large specific surface area.
Table 3.
Amounts of ammonia released from NH4CLIN, and from remaining ammonium ions of thermal activated catalysts HCLIN300, HCLIN400, and HCLIN500, and calculated acidity measured by TPD of ammonia. * Determined by TPDA.
2.6.9. Solid State NMR Studies
The Brønsted acidity of zeolites is directly connected with the incorporation of 3-valent Al instead of 4-valent silicon into tetrahedral coordinated framework positions of the aluminosilicate lattice. The created negative framework charge is balanced by cations or acidic protons H+. The protons, Brønsted active species, are located at the bridging oxygen atoms of the alumino–siloxane bridges, Si–O(H)–Al, connecting the SiO4 and AlO4 tetrahedra. The 29Si MAS NMR, 27Al MAS NMR and 1H MAS NMR spectra provide information about the environment of these atoms in the zeolites.
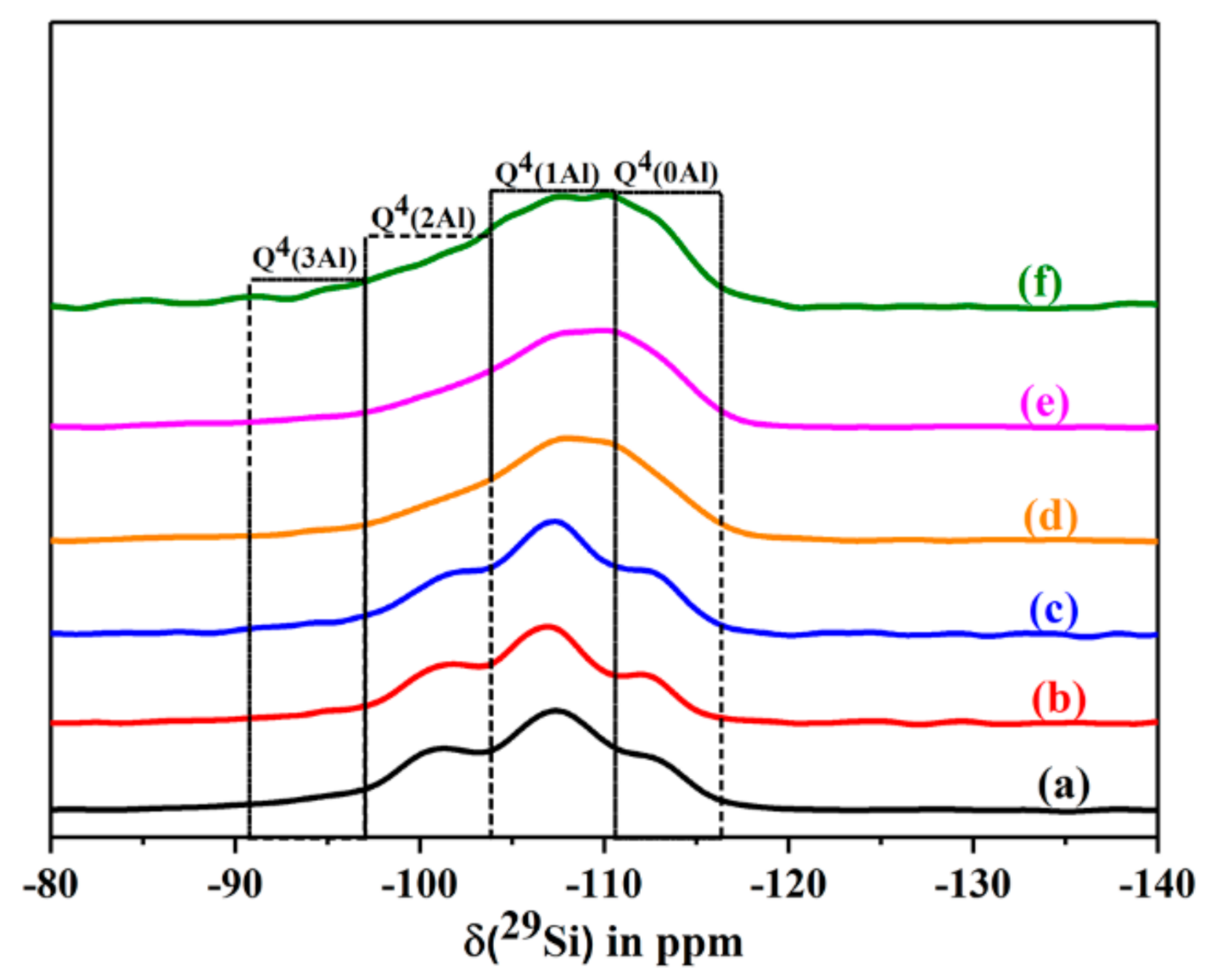
The 29Si MAS NMR spectra of the starting clinoptilolite, its ammonium exchanged form, and of different calcined HCLIN samples are shown in Figure 13. The spectra show four signals located at ca. 95, 102, 106, and 112 ppm, respectively. They belong to the framework silicon atoms Si(3Al), Si(2Al), Si(1Al), and Si(0Al) connected with 3, 2, 1, and 0 aluminium atoms in the neighbourhood, respectively. The relative intensity of the Si(1Al) signal increases at the expense of the weak Si(3Al) and medium intense Si(2Al) peaks after calcination of the ammonium form. The latter signal belongs to bridged Si–O–Al–O–Si–O–Al–O–Si units [34,35,36,37]. The Al-rich species are less thermally stable than the Si-rich ones. The Si NMR signals are markedly broadened after heating the sample beyond 400 °C, indicating structural disorder.
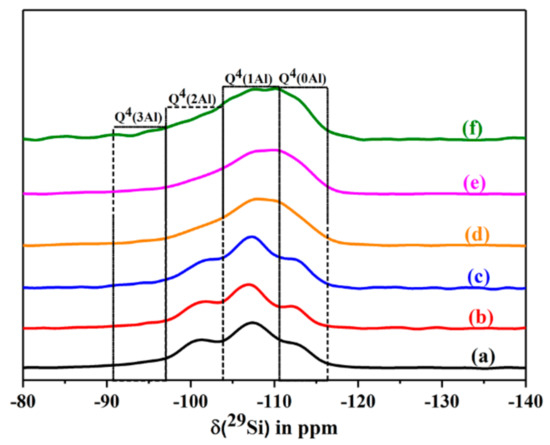
Figure 13.
The 29Si MAS NMR spectra of dehydrated CLIN, and calcined NH4CLIN (0.5M NH4NO3 at 80 °C for 1 h, 400-600 °C) samples in dehydrated state. (a) CLIN, (b) NH4CLIN, (c) HCLIN 400 °C, (d) HCLIN 450 °C, (e) HCLIN 500 °C, (f) HCLIN 600 °C.
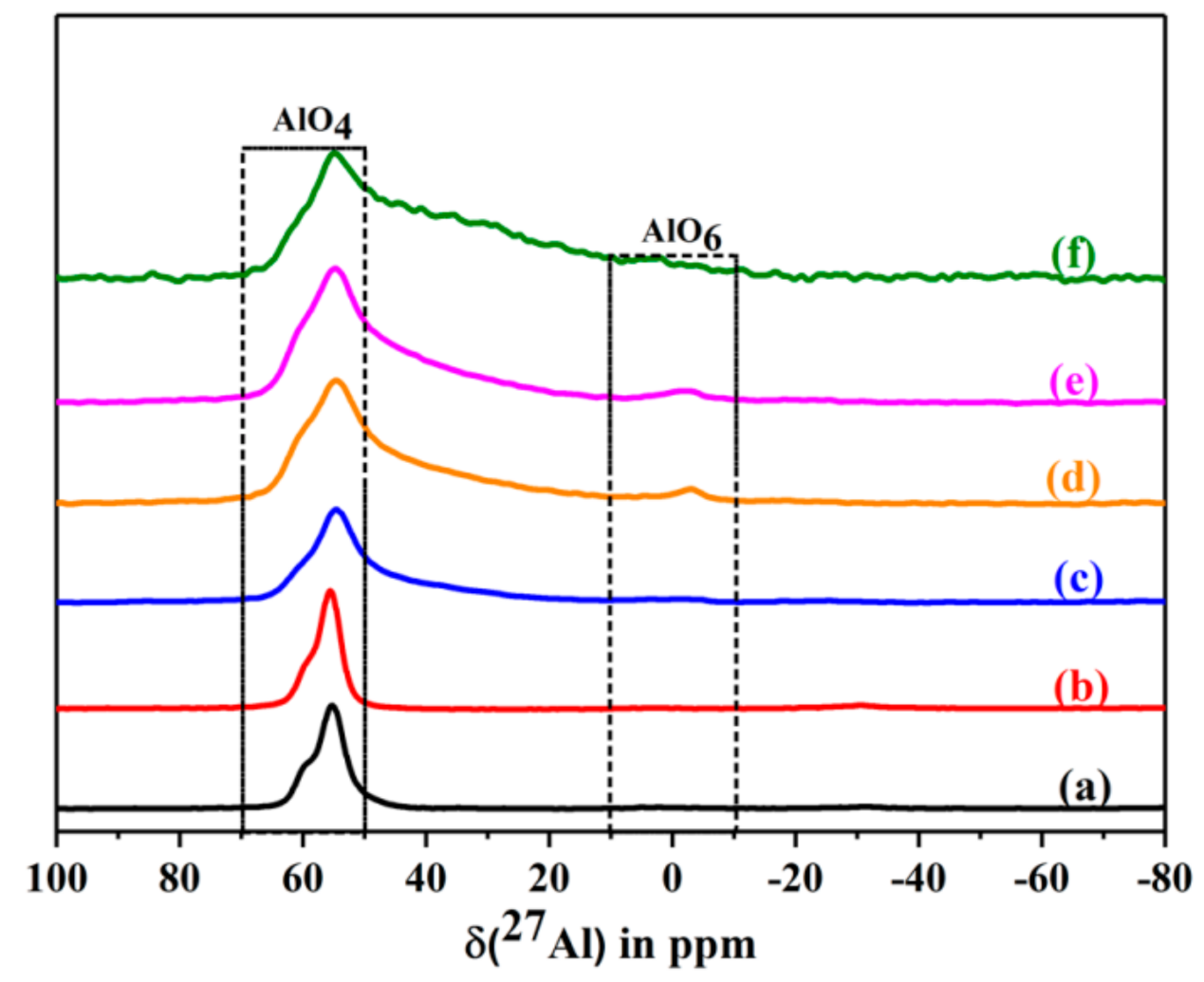
The corresponding 27Al MAS NMR spectra are shown in Figure 14. The spectra show a strong 27Al MAS NMR signal at ca. 56 ppm, which is assigned to a tetrahedrally coordinated framework aluminium. The signal is broadened after calcination at 400 °C and 500 °C. Further, a new signal of very low intensity appears at ca. 2 ppm. It is assigned to the formation of some extra-framework aluminium. An additional shoulder appears at ca. 30–40 ppm after calcination at 400 °C [37,38,39,40]. It belongs to penta-coordinated aluminium. Its intensity rises strongly after calcination at 500 °C. As a result, the relative portion of tetrahedral coordinated framework Al decreases by ca. 30%. Conclusively, the penta-coordinated aluminium is still connected with the framework. This assumption is supported by the fact that the crystal structure is maintained. No shrinkage of the lattice is indicated by the XRD pattern nor a corresponding high frequency shift of lattice vibration bands in the IR spectra.
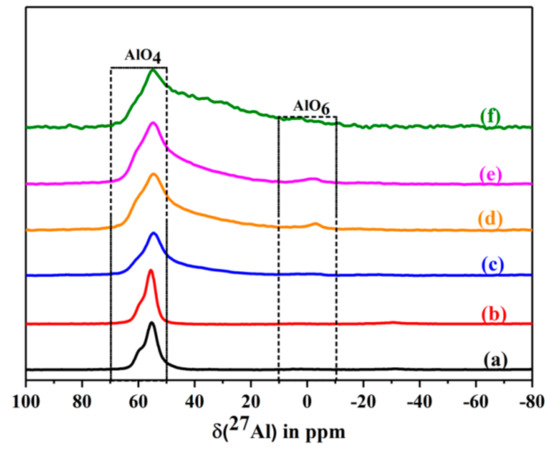
Figure 14.
The 27Al MAS NMR spectra of dehydrated CLIN, and calcined NH4CLIN (0.5M NH4NO3 at 80 °C for 1 h, 400–600 °C) samples. (a) CLIN, (b) NH4CLIN, (c) H-CLIN-400 °C, (d) H-CLIN-450°C, (e) H-CLIN-500 °C, (f) H-CLIN-600 °C.
The relative decrease in the tetrahedral framework aluminium by ca. 30% is in line with the loss of Brønsted sites found with ammonia TPD measurements. They showed a loss of 10% after heating to 400 °C of ca. 35% after calcination at 500 °C. The 27Al MAS NMR data are in qualitative and roughly quantitative agreement with the TPDA data.
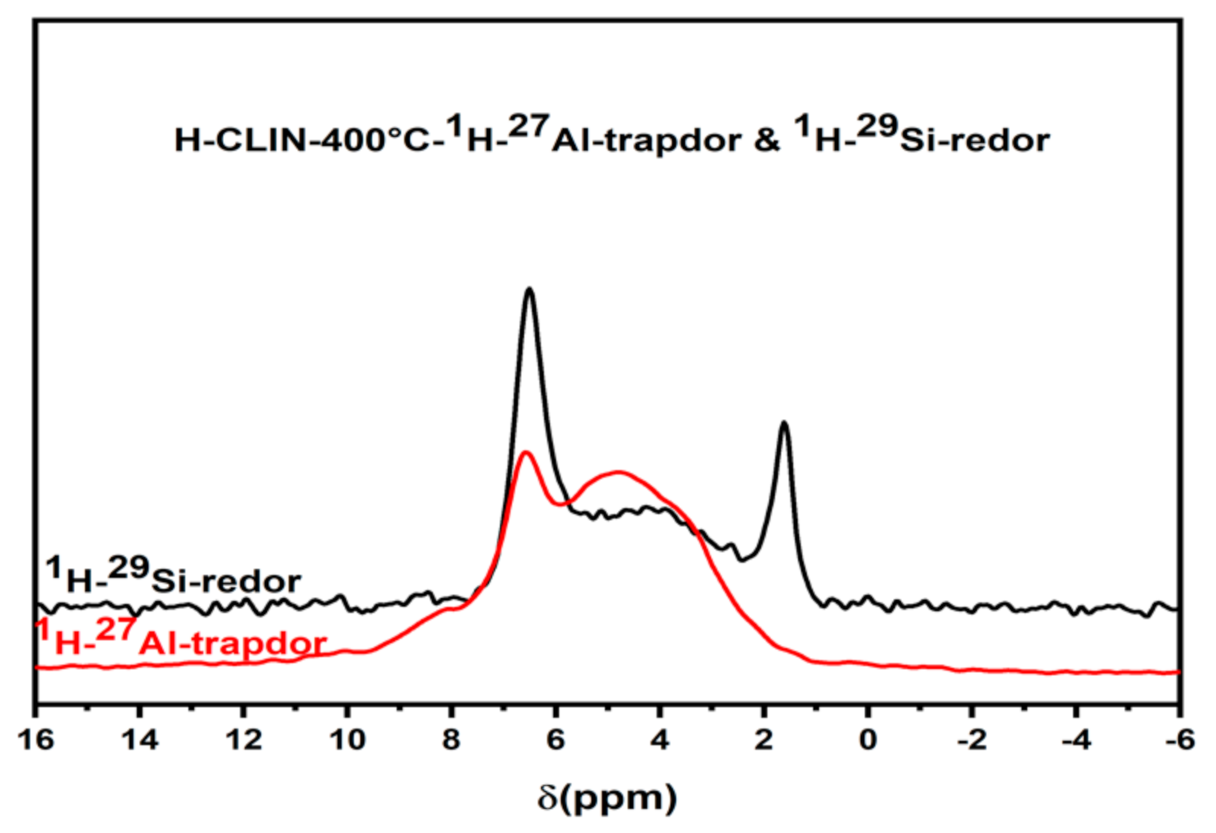
The 1H MAS NMR spectra show a broad peak in the chemical shift range between ca. 3–6 ppm which is usually assigned to zeolitic Bronsted acid sites created by Si–O(H)–Al bridges. Additionally, a signal of non-acidic isolated silanol groups SiOH appears at ca. 1.8 ppm. The signal of the protons of ammonium ions is observed at ca. 6.8 ppm. The 29Si MAS NMR Redor measurements and 27Al MAS NMR Trapdor measurements confirm the assignment (Figure 15). The signal of the silanol groups is excited via the silicon atoms in the Redor experiment but not via the aluminium atoms in the Trapdor experiment. Further, the broad signal of BS, Si–O(H)–Al groups, observed between ca. 3–6 ppm, is excited via both Al and Si atoms [40,41]. The broad distribution of BS is related with the structural disorder found with the clinoptilolite after calcination at elevated temperature. This is due to large variation of Si, Al–O–Sil bridging angles, and T–O bond distances in the clinoptilolite framework are observed. Additionally, the number of next nearest aluminium surrounding the silicon varies.
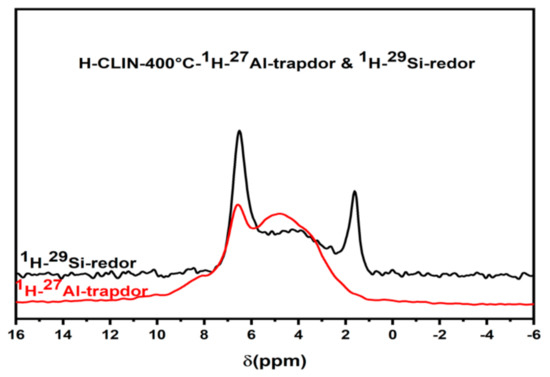
Figure 15.
The 1H–7Al Trapdor and 1H–29Si Redor experiments of HCLIN400.
2.7. Catalysis
2.7.1. Influence of the Catalyst Activation
Catalyst Activation Temperature
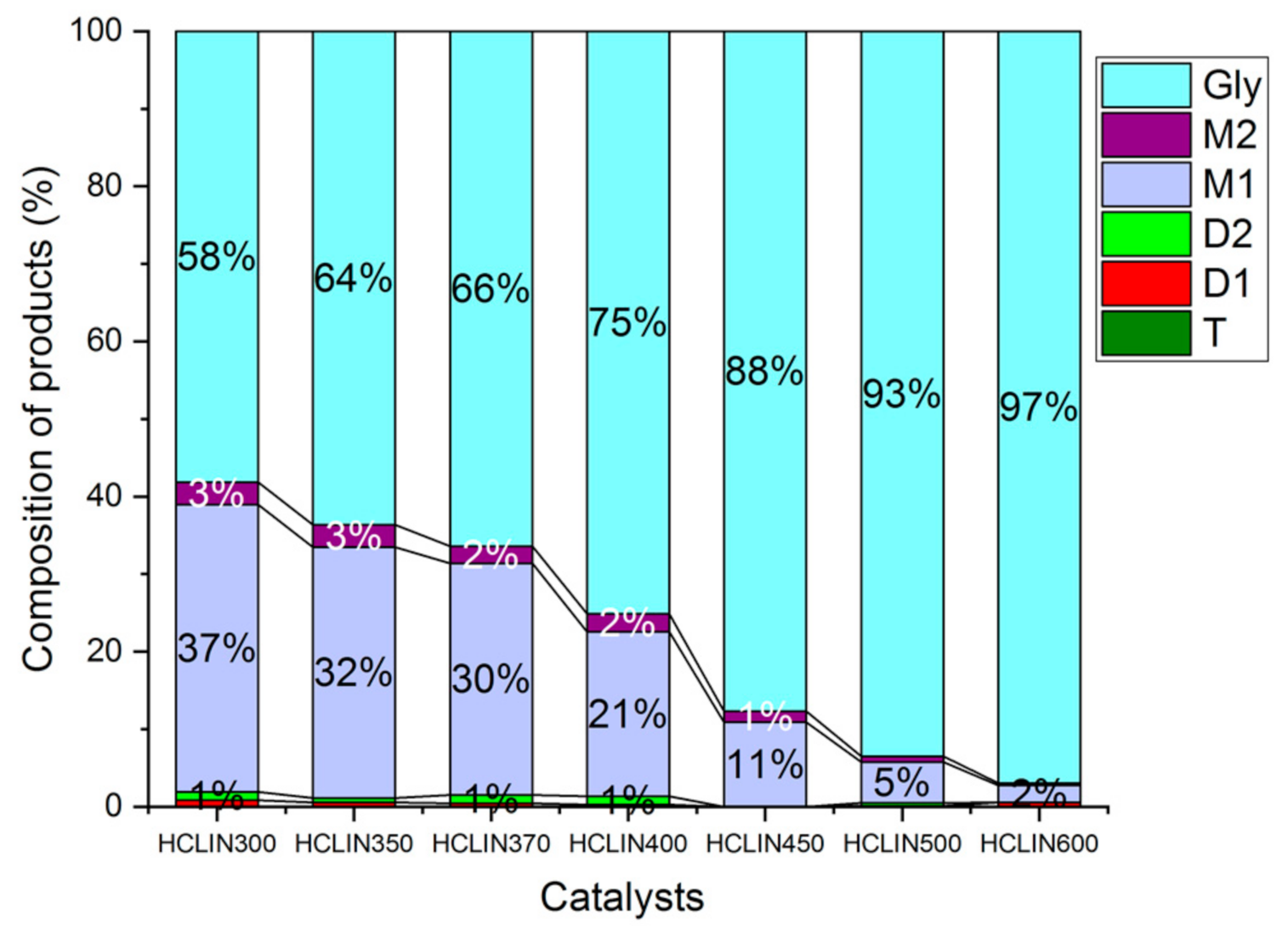
The influence of the calcination temperature of HCLIN catalysts on the catalytic performance was investigated. Figure 16 shows the composition of the reaction mixtures obtained with different activated HCLIN catalysts after 4 h of reaction at 110 °C. The highest conversion of glycerol (42%) is observed with HCLIN300, the catalyst was activated at 300 °C in short time. For the catalyst with the lowest acidity, further heating to 370 °C decreases the glycerol conversion to 34%. Interestingly, the lowest conversion of glycerol is observed with the catalysts HCLIN400 and HCLIN500. Mostly, the mono-ethers are formed. The selectivity to di-ether, the wished component, is also low. These results are surprising. The acidity of HCLIN300 is markedly lower (0.25 mmol/g) than that of HCLIN400 (0.47 mmol/g) and of HCLIN500 (0.76 mmol/g). At the same time, the specific surface area is increased from 36 m2/g to ca. 195 m2/g and 151 m2/g, respectively. Therefore, an increase in the conversion should be expected with the rising activation temperature of the heterogeneous catalyst. However, the opposite is observed. It is concluded that the strong acid sites formed at higher activation favours the formation of glycerol decomposition or polymerization products which block the active sites. This would explain the sharp decline in the conversion, especially after calcination beyond 400 °C.
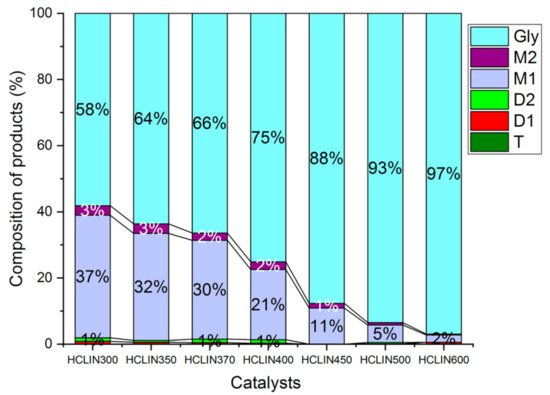
Figure 16.
Influence of the catalyst activation temperature on the glycerol conversion to M1, M2, D1, and D2 ethers after 4 h of reaction over different catalysts, HCLIN300 to HCLIN600 (short-time activation of 1 min). Reaction condition: 110 °C, Gly/TBA = 1/4, catalyst/Gly mass = 5% in reaction time: 4 h.
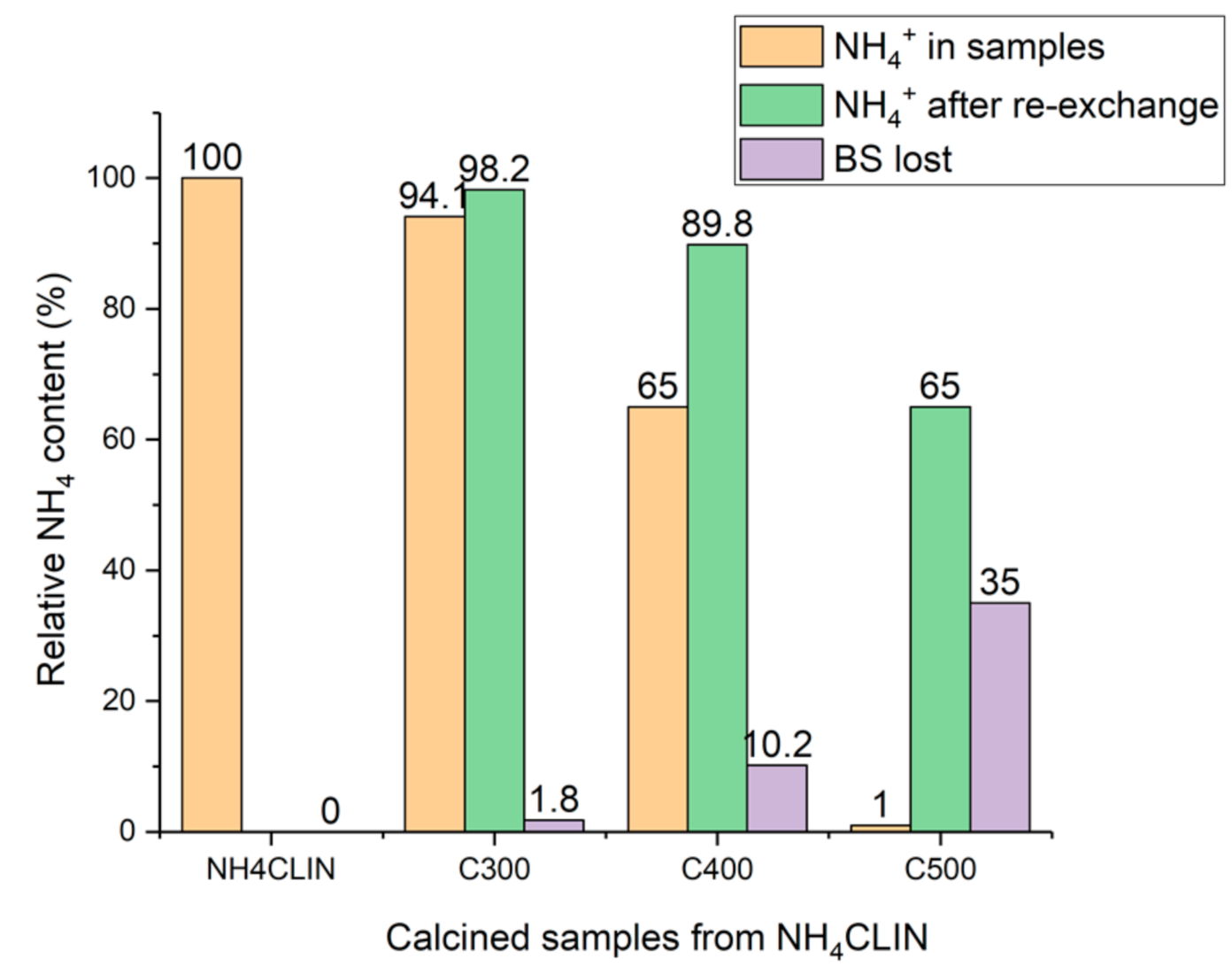
Another explanation for the loss of activity could be a limited stability of the BS. Generally, it is known that calcination of zeolites at high temperature can cause a destruction of BS by dihydroxylation. The thermal stability and loss of BS was studied using ammonium re-exchange experiments of BS protons (H+) by ammonium ions. The catalyst was treated with a slightly alkaline ammonium acetate solution in order to re-establish the ammonium form. The change of the ammonium ion content in the HCLIN catalysts was estimated from the relative change of the intensity of the ammonium vibration band in the FTIR spectra of the samples compared to the starting ammonium exchanged clinoptilolite.
With the catalyst HCLIN300, the re-exchange is nearly complete (Figure 17). However, with the catalysts HCLIN400 and HCLIN500 only a partial re-exchange to the NH4CLIN form could be achieved. Only 90% and 65% of ammonium sites could be re-established with HCLIN400 and HCLIN500, respectively. The loss of ion exchange sites (i.e., BS) with HCLIN300 was only ca. 2%. The incomplete re-exchange observed with HCLIN400 and HCLIN500 is assigned to a loss of ion exchange sites, i.e., tetrahedral coordinated at the framework aluminium by dehydroxylation of bridging acidic groups Al–O(H)–Si during the calcination process. The dehydroxylation increases with rising temperature and leads to a decrease in the acidity (Figure 17). However, this decrease is overcome by the overall increase in the acid site concentration. With HCLIN 500, the loss by dehydroxylation is ca. 35%. The total formed acidity is ca. 0.76 mmol/g. About ca. 0.5 mmol/g of BS remain after dehydroxylation. This is twice the concentration of the acid site with HCLIN300 (ca. 0.25 mmol/g).
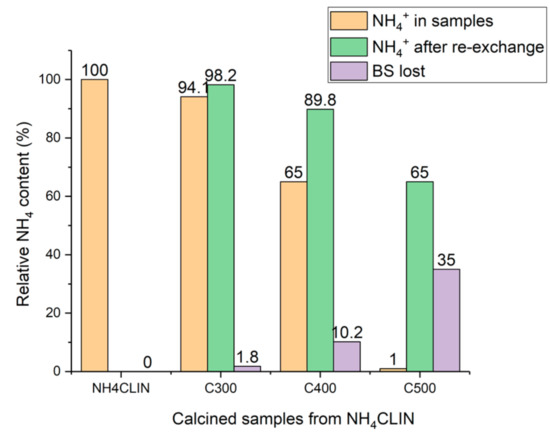
Figure 17.
Decrease in the ammonium ion content of HCLIN after thermal activation at 300 °C to 500 °C, recreation of sites by ammonium re-exchange, and the corresponding loss of BS by thermal dihydroxylation.
Finally, HCLIN300 shows the highest activity, although the pore system is still nearly blocked. Only ca. 15% of the ammonium ions are decomposed in the re-exchange experiment. Therefore, it is concluded that the etherification of glycerol with tert-butanol proceeds at or near the surface of the clinoptilolite catalyst and requires acid sites that are not very strong. After heating to 600 °C, the structure of the clinoptilolite is damaged to a great extent with a corresponding loss of the catalytic activity.
Catalyst Activation Time
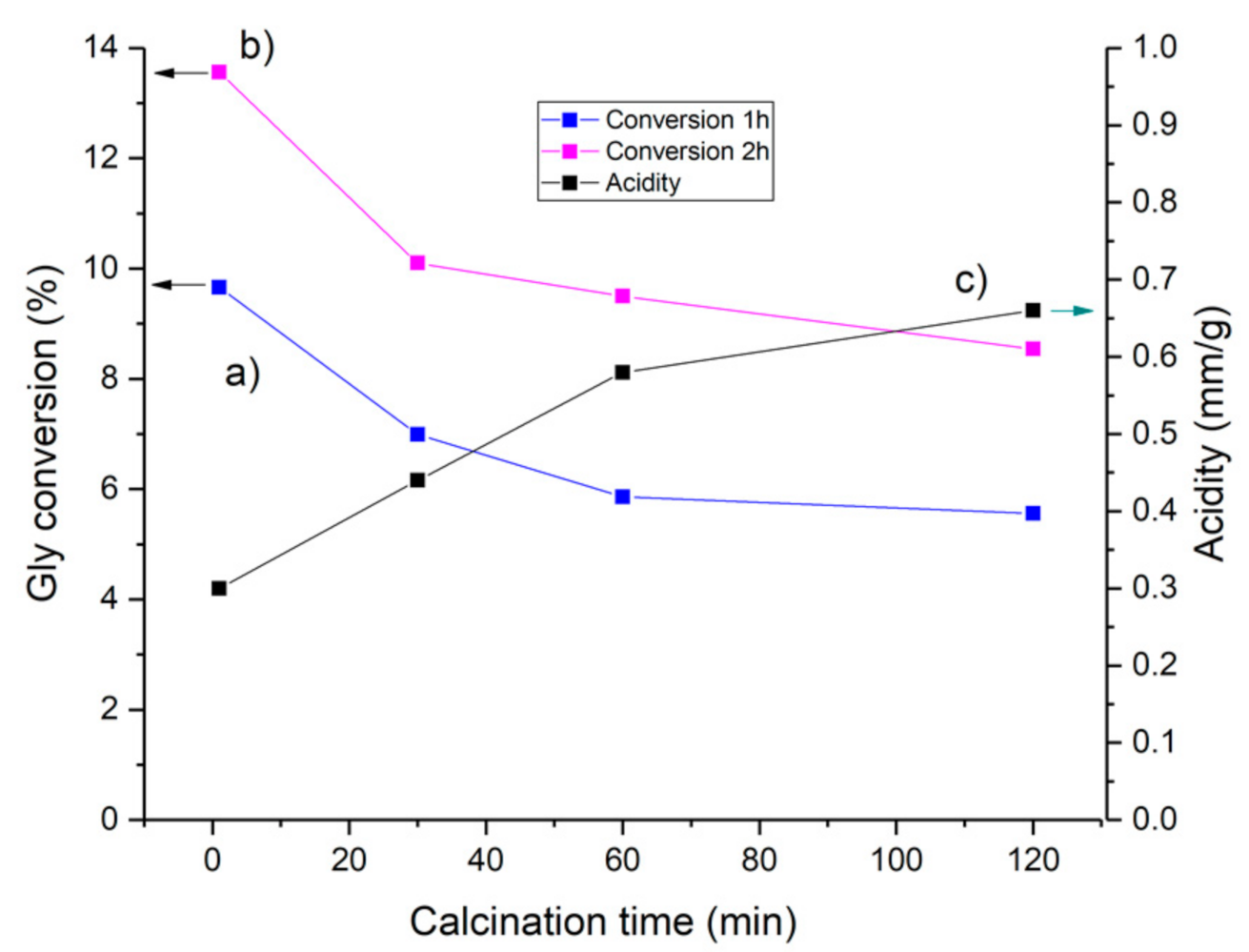
The influence of the activation time on the formation of acid sites and the conversion of glycerol with HCLIN300 is shown in Figure 18. The results show a marked influence of the calcination time on the acidity of the catalyst. Highest activity is achieved just after reaching the activation temperature (ca. 1 min). Prolonged calcination to 120 min causes a decrease in the conversion by ca. 30%. At the same time, the acid site concentration is nearly doubled. Again, the increase in the acidity of the catalyst is connected with a decrease in the catalytic activity in terms of conversion as observed with ring temperature.
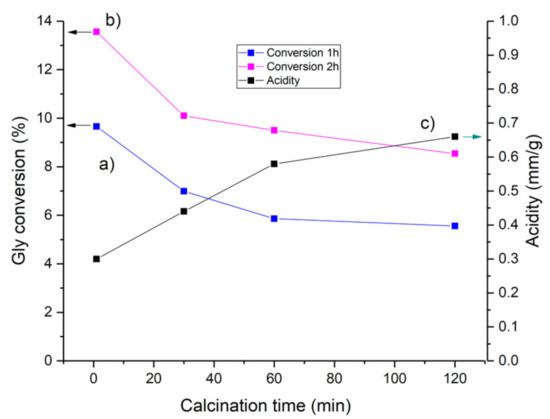
Figure 18.
Influence of the calcination time of the HCLIN300 catalyst on the conversion of glycerol with TBA after (a) 60 min and (b) 120 min of etherification reaction, (c) influence on acidity of catalyst.
The formation of BS in the HCLIN catalyst is very sensitive to the activation conditions, the activation temperature, and time of activation. The formation of BS by decomposition of the ammonium ions is continuous after reaching the calcination temperature with prolonged calcination time. As a result, the acidity is increased from ca. 0.25 to 0.67 mmol/g.
2.7.2. Influence of the Reaction Conditions
The influence of the reaction parameters, such as reaction time, the glycerol/tert-butanol ration, reaction temperature, and catalyst loading on the catalytic performance of HCLIN300, were investigated in detail.
Influence of the Reaction Time
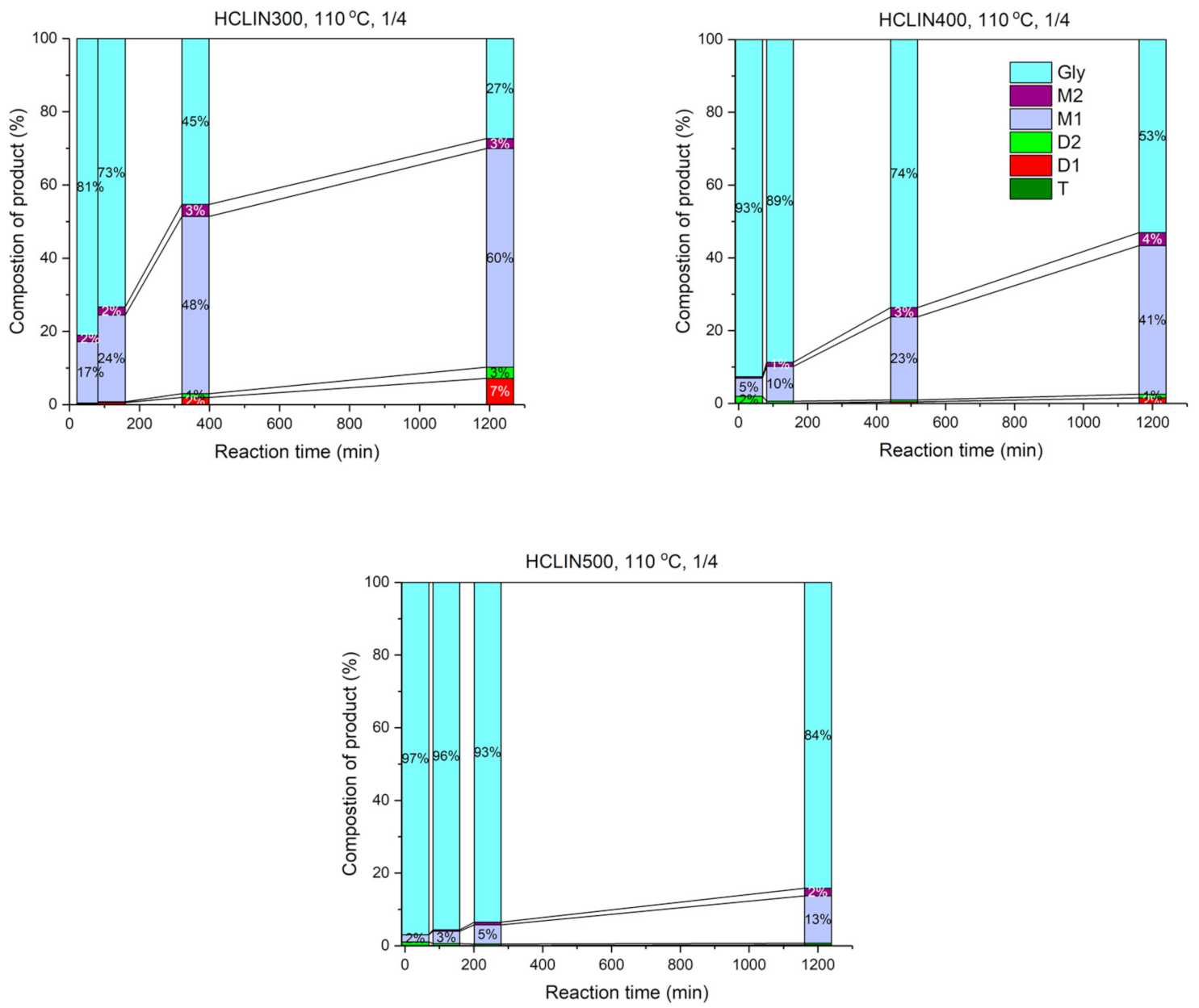
The influence of the reaction time on the catalytic performance is shown in Figure 19 in terms of product yields and remaining non-converted glycerol. The course of reaction was followed over a period of 24 h with the most active catalyst HCLIN300. After a rapid increase in conversion in the first 6 h of 55%, the reaction proceeded slower, reaching a conversion of glycerol of 73. Ca. 63% of mono-ether and ca. 10% of di-ether were formed. This confirms the stability of the catalyst during the course of reaction. The catalysts HCLIN400 and HCLIN500, which exhibit higher acidity and porosity, do not overcome the conversion found with HCLIN300. Conversions of 47% and 4% were reached after 24 h of reaction, respectively.
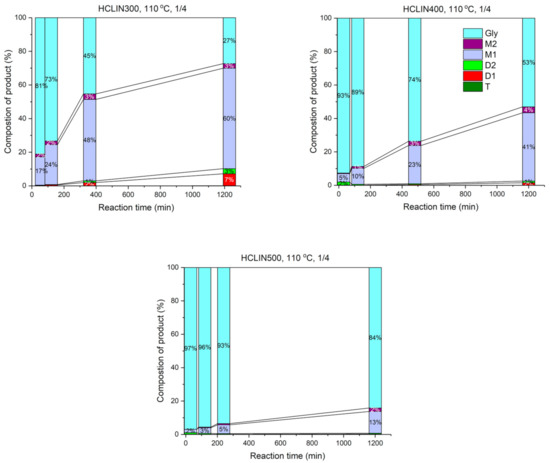
Figure 19.
Glycerol conversion vs. reaction time over HCLIN300, HCLIN400, and HCLIN500 catalysts (calcination time: 30 min).
The Influence of the Reaction Temperature
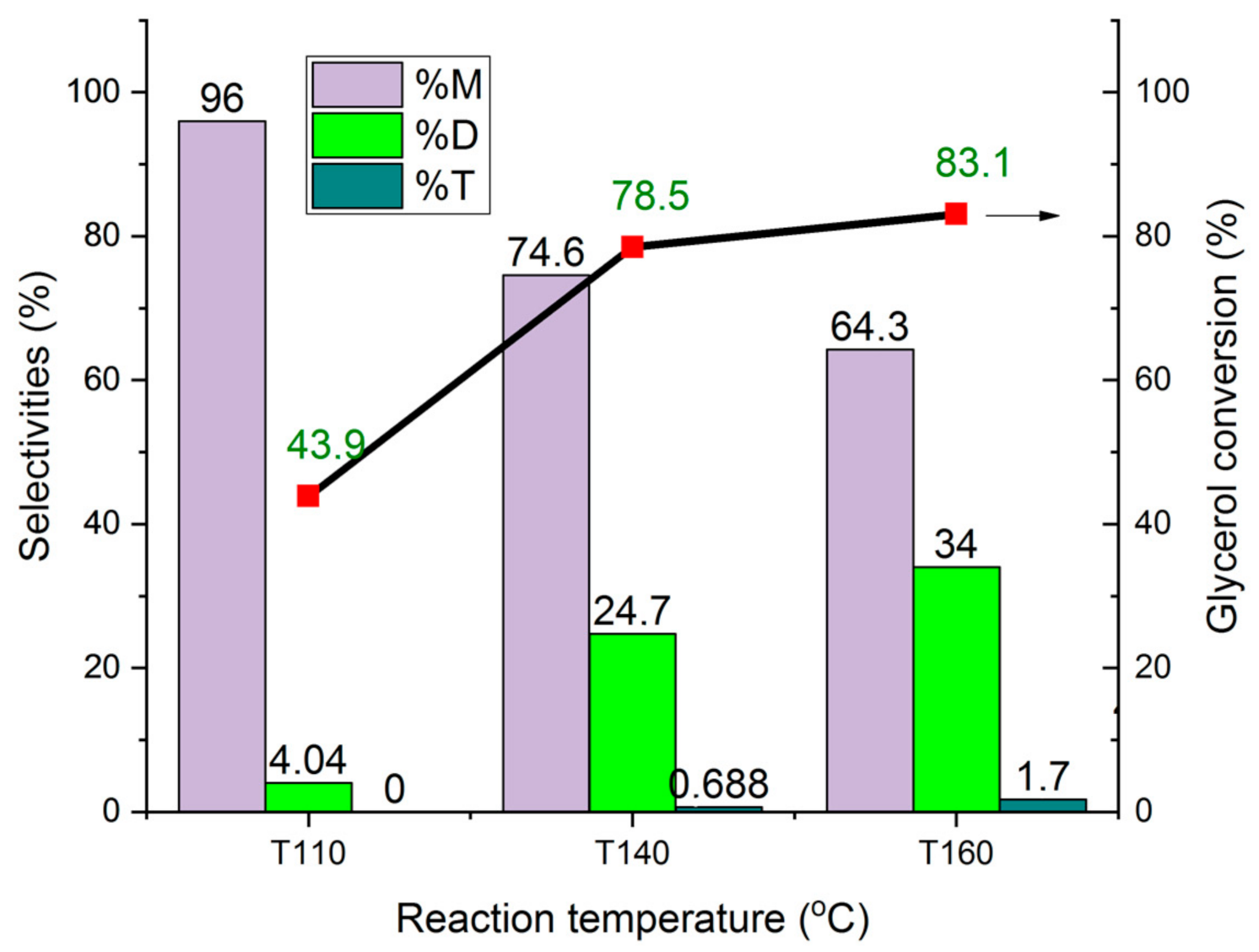
The influence of the reaction temperature on the conversion of glycerol and the formation of mono- and di-ether over HCLIN300 is shown in Figure 20. The reaction temperature was increased from 110 °C to 140 C and 160 °C, respectively. With an increase in the reaction temperature to 140 °C, a strong increase in the conversion from ca. 44% to ca. 78% is observed after 4 h of reaction. Further rise of the temperature to 160 °C leads only to a moderate increase in the conversion to ca. 85%. However, the temperature increase resulted in a marked increase in the selectivity of di-ether, from ca. 4% at 110 °C to 34% at the expense of the mono-ether. The formation of tri-ether reaches only 1.7% at 160 °C. A reason for the increased formation of di ether could be the lower adsorption of water at the catalyst surface at a high temperature, combined with the high formation of the mono-ether. Adsorbed mono-ether respells adsorbed water by the large hydrophobic tert-butyl group.
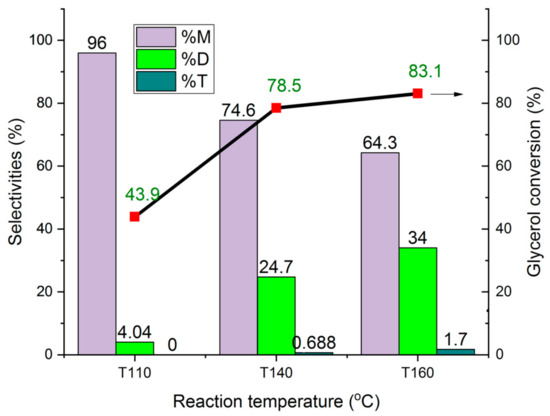
Figure 20.
Influence of the reaction temperature on the conversion of glycerol to M1, M2, D1, D2 ethers and on the selectivity over different catalysts.
Influence of the Gly/TBA Ratio
The reaction is usually carried out at a Gly/TBA ratio of 4, i.e., TBA is added in excess. The decrease in the glycerol to tert-butanol ratio, i.e., the enhancement of the excess of TBA from the standard condition 1/4 to 1/5 and 1/8, has only minor influence on the conversion of glycerol. This is likely due to the dilution of the reaction mixture by the added TBA and the corresponding decrease in the glycerol concentration. The remaining glycerol content changes from ca. 45%, 49% and 46% after 6 h, and from 27% to 28% and 22% after 24 h of reaction, respectively. Correspondingly, the yield of the mono- and di-ether varied from ca. 73% to 72% and 78% after 24 h, respectively. The selectivity to di- and tri-ether is ca. 13% after 24 h. In this case TBA acts as a solvent.
In summary, the influence of the Gly/TBA molar ratio is comparatively low. The conversions of glycerol reach 72% to 78% after 24 h of reaction. The yield of mono ether reaches 63% to 68%. The yield of di- and tri-ether reach ca. 10%. The M1 to M2 mass ratio is ca. 60/3 to 65/3, and the D1/D2 ratio is about 2, after 24 h of reaction.
Influence of the Catalyst Loading
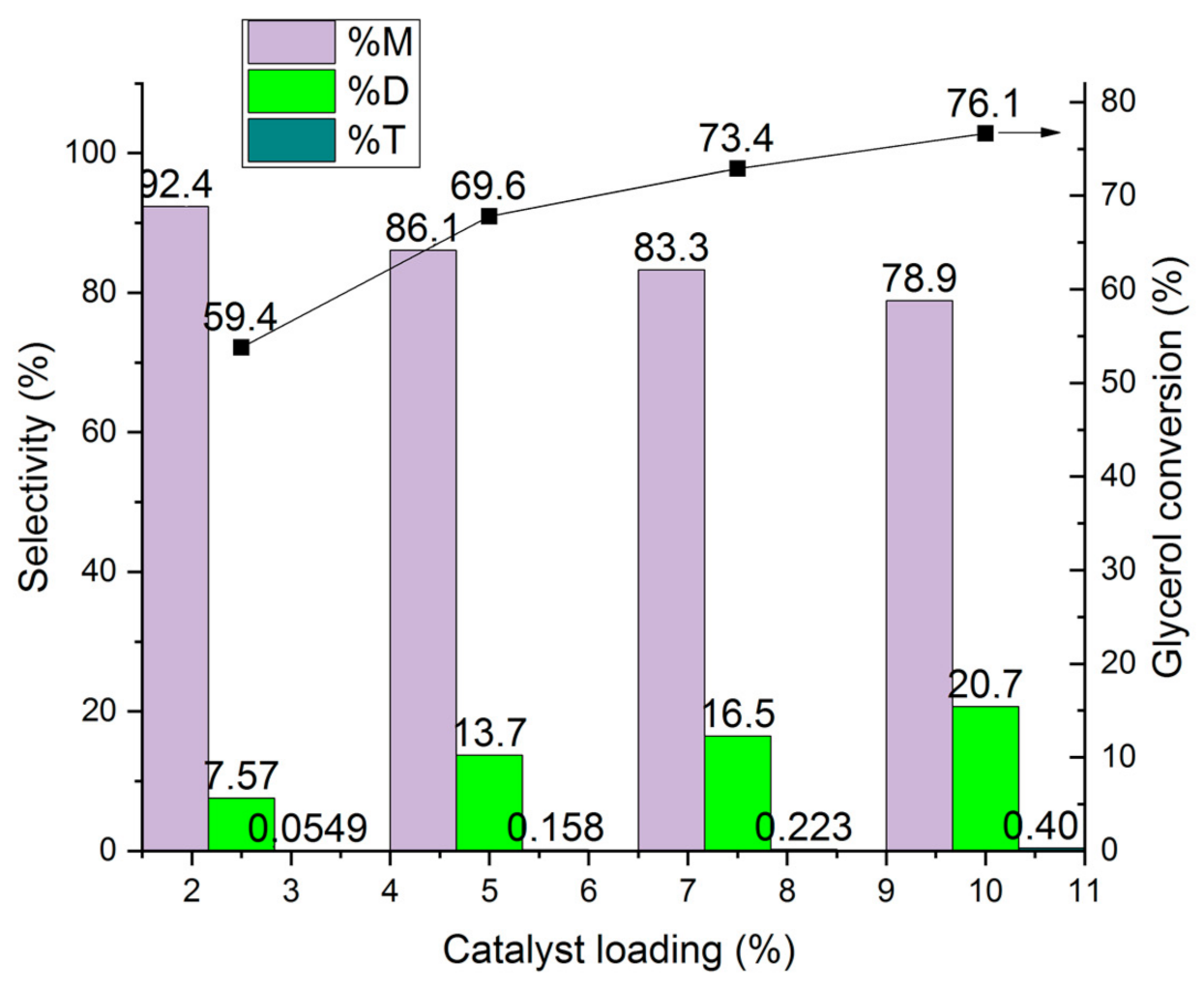
The influence of the catalyst loading on the conversion of glycerol and the selectivity to mono-, di- and tri ether is shown in Figure 21. The catalyst loading was varied from 2.5 to 10 ma%. The increase in the catalyst loading from 2.5 to 10 ma% lead to an increase in the conversion by ca. 25%, from 59.4 to 76.1%, whereas the selectivity to di-ether increased by a factor of three, from 7.57 to 20.7%, at the expense of the mono-ether. An explanation is that the increased amount of molecular sieve catalyst can decrease the solved water content in the reaction mixture. It works as drying agent [18]. Further, more dry catalyst is present which favour the formation of the di ether.
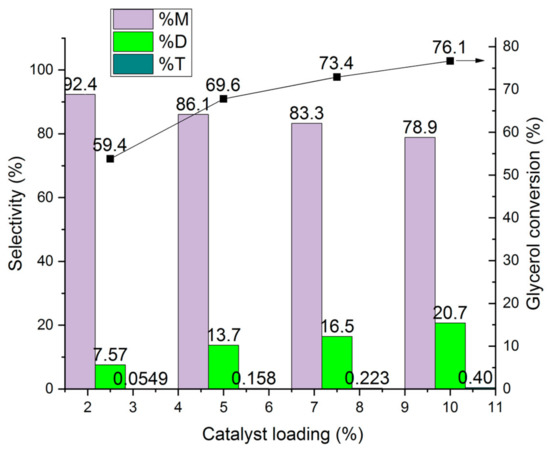
Figure 21.
Influence of the catalyst loading on glycerol conversion to mono-ether M, di- ethers D, and tri-ether T (rare to see <1%) over HCLIN300 (catalyst calcination: 30 min).
Comparison with Different Alcohols
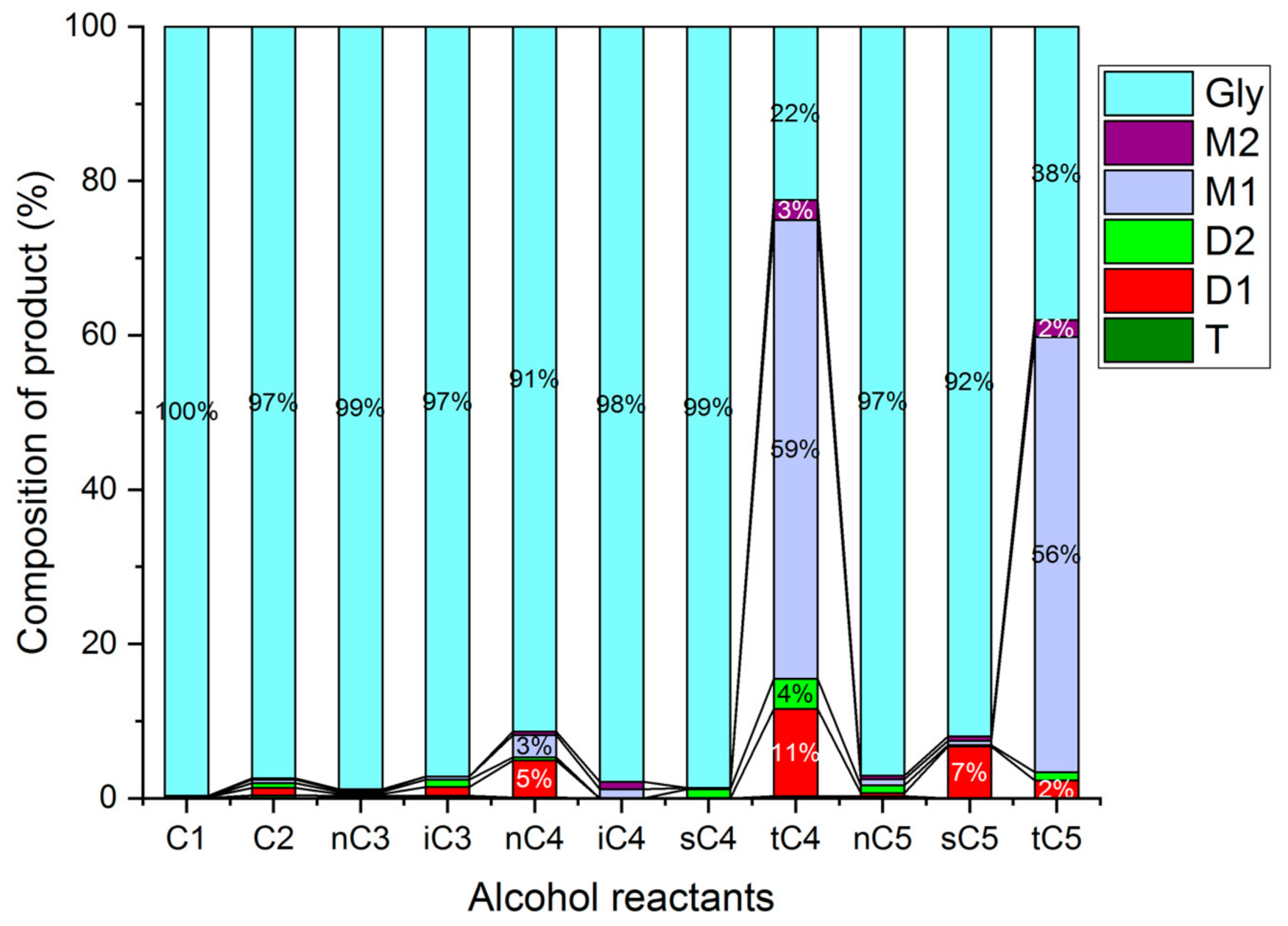
The influence of the alkyl chain length and of branched alkyl groups on the etherification of glycerol was tested using different C1 to C5 alcohols (Figure 22). The tests were carried out with the HCLIN300 catalyst. At 140 °C, the observed conversion with linear and branched alcohols was low. The highest conversions were found with alcohols containing tertiary alkyl groups. Relatively high selectivity to di-ether was found with the branched t-butanol and amyl alcohol. This finding is line with the stabilization of the formed intermediate alkyl carbocations. It is highest with tertiary alkyl groups. Additionally, a shielding of the reaction site on the catalyst from water by the umbrella of hydrophobic tertiary alkyl groups may facilitate the etherification. Furthermore, n-butanol is comparatively active and selective in the etherification of glycerol, similar to the long hydrophobic C4-chain shields the reaction side from competing water
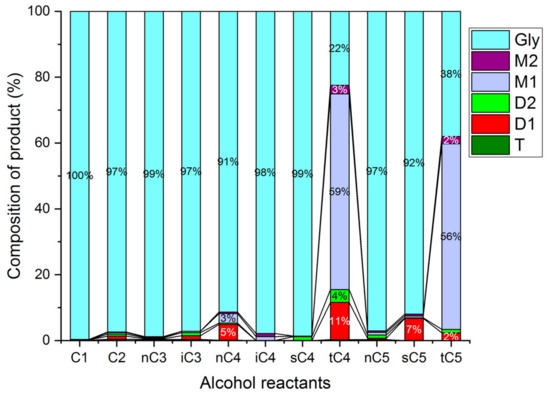
Figure 22.
Comparison of the etherification of glycerol with different alcohols over HCLIN300 (Reaction condition: 140 °C, 2 h).
2.7.3. Catalyst Re-Use
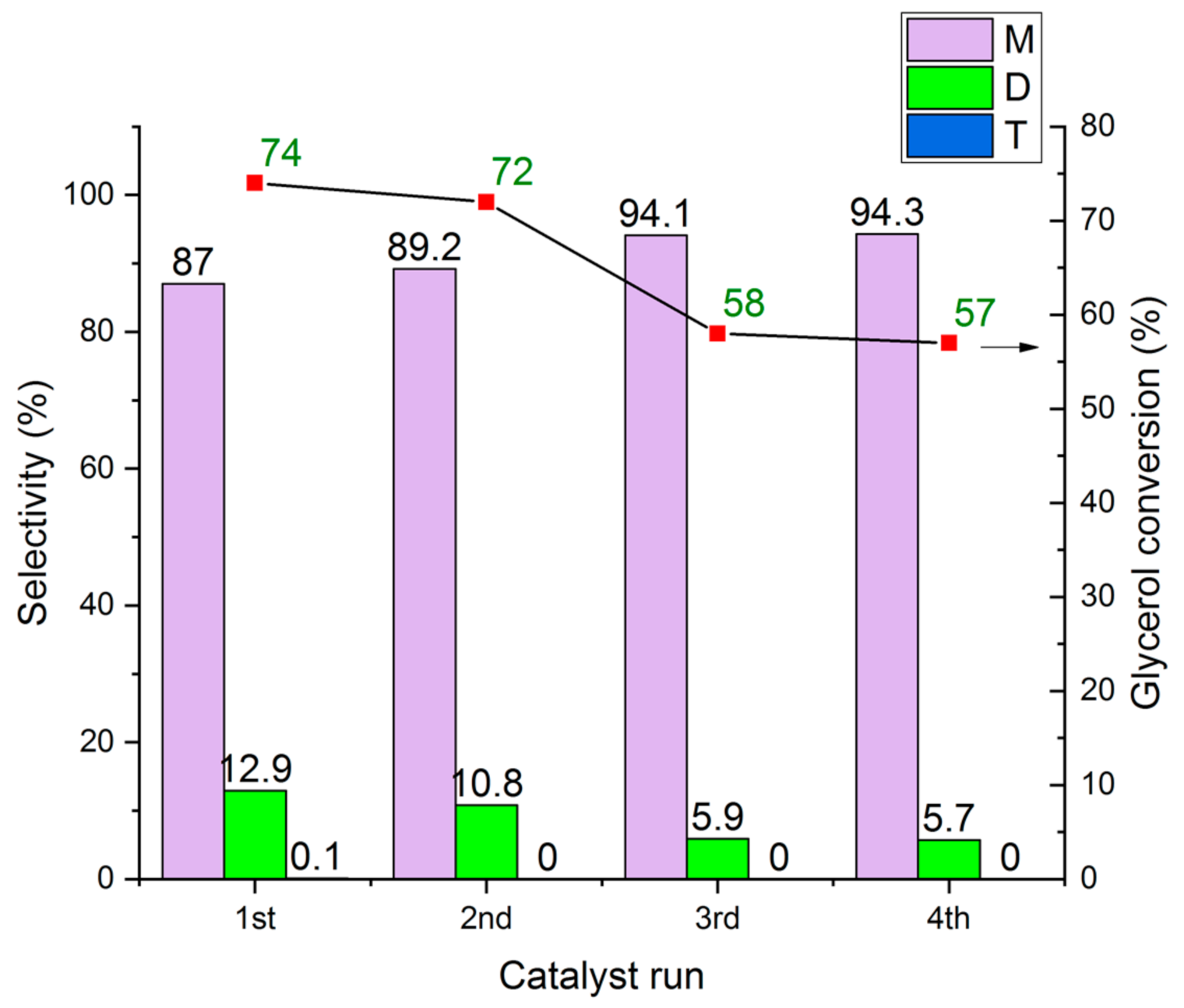
The re-use and cycling experiments of the HCLIN300 catalyst are shown in Figure 23. The results show that the catalyst is stable and shows only a moderate loss in the activity (in terms of conversion) after the cycling. The glycerol conversion decreases from ca. 74% to 57% after the 4th run. Besides, the selectivity of the mon-ether increases at the expense of the di ether. The latter decreases from 12.9% to 5.7%. This finding is likely due to the increasing amount of water adsorbed by the catalyst due to the washing operations and some poisoning. It was observed that the catalyst colour changed to “dark”.
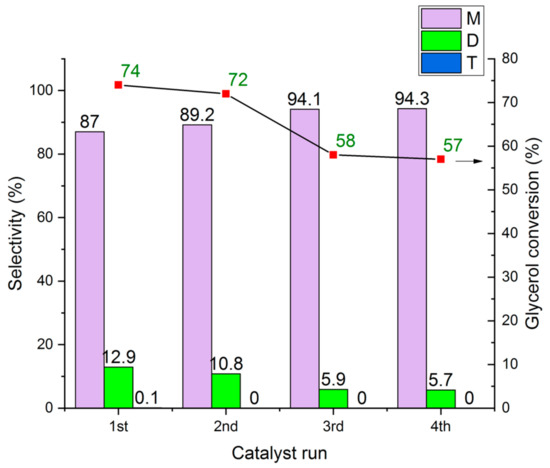
Figure 23.
Re-use and cycling of HCLIN300 in the etherification of glycerol and t-butanol (M, D, T – mono-, di, and tri-ether). Reaction condition: 110 °C in autoclave for 2 h, Gly/TBA = ¼, catalyst/Gly mass ratio = 0.3.
3. Conclusions
A green and improved natural zeolite acidic catalyst using clinoptilolite as starting material was prepared. The catalyst was prepared under mild conditions, low temperature, and low concentrated treatment solution, saving materials, waste, and energy. Additionally, the use of environmental hazardous and corrosive concentrated mineral acids was avoided. The washed material was simply ion exchanged with an aqueous, comparatively low concentrated, 0.2 M ammonium nitrate solution, dried and calcined heating to the low temperature of 300 °C for short time (holding time 1 min).
The material is crystalline and thermally stable. However, loss of crystallinity is observed starting at 450 °C, which leads to a partial loss of porosity and blocking of the pores. However, a part of the structure is maintained at a temperature of 600 °C. The micropore system of the mainly potassium and calcium ions containing natural zeolite and of the ammonium exchanged material NH4CLIN is nearly blocked by the cations and the ammonium ions located in the zeolite pores, as indicated by the nitrogen adsorption and desorption experiments. The thermal decomposition of the ammonium ions opens the pores, giving access to the pore system. The specific micropore area increases markedly to 163 m2/g with calcination at 400 °C without additional acid or base treatment. The clinoptilolite catalyst is highly acidic and contains weak, medium strong, and mainly strong acid sites desorbing ammonia from acid sites, i.e., released ammonia from decomposed ammonium ions, between 200–300 °C, 300–400 °C, and 400–550 °C, respectively. According to the TPDA, the NH4CLIN contains ca. 1.4 mmol/g of ammonium ions. Ca. 1 mmol/g of the ammonium ions are converted to acid sites by heating. The Brønsted sites show limited stability at a high temperature. A loss BS of up to 35% is found after heating to 500 °C. It is related to the change of the coordination of tetrahedral framework aluminium, counterbalancing the positive charge of the cations or acidic protons, to penta-coordinated aluminium. Nevertheless, the concentration of BS increases with calcination of up to 500 °C compared to calcination at 400 °C. The loss is partially reversible, and recreation of BS achieved after ammonium ion re-exchange. The concentration of BS is sensitive to the catalyst activation conditions, i.e., the temperature and holding time. The Brønsted acid HCLIN catalysts are catalytic highly active in the etherification of glycerol. High conversion of glycerol to ethers with short time (holding time: 1 min) and low temperature (300 °C) catalysts. Interestingly, highest catalytic activity is observed, with soft preparation, at 300 °C. Shortly activated HCLIN300 shows the lowest acidity. As the pore system of HCLIN300 is still blocked, it is concluded that the catalysis takes place at or near the external surface. Dried ammonium CLIN or K,Ca-clinoptilolite are not catalytic active in the etherification of glycerol. Acid sites are formed after heating of the NH4CLIN to ≥250 °C. Highest conversions to mono and specifically di ether are observed with t-butanol and amyl alcohol. A 78% conversion of glycerol to mono and di ether is achieved with t-butanol at 140 °C after 4 h of reaction. The mono and di ether selectivity are 75% and 25%, respectively. Other, shorter chain C1–C4 alcohols show markedly lower conversions. This finding is assigned to the enhanced stability of the intermediate formed carbocations. The bulky hydrophobic alkyl groups shield the reaction site from water facilitating the etherification reaction. Water molecules adsorbed at the catalyst can block the hydrophilic active site and glycerol hydroxy groups from approaching alcohol molecules. The catalyst can be reused after washing.
Author Contributions
Catalysis, M.R. and D.T.H.; Conceptualization, H.K., C.J. and A.S. (Axel Schulz); NMR, C.J.; analysis, N.T.M.T. and L.T.S.; TEM and SEM investigation, A.S. (Armin Springer) and M.F.; synthesis, M.R. and D.T.H.; writing—original draft preparation, D.T.H. and H.K.; writing—review and editing, H.K. and A.S.; visualization, supervision, H.K. and A.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The excellent contributions, assistance, discussion, and measurements are gratefully acknowledged: Christine Fischer (GC/Ms measurement and data evaluation), Astrid Lehmann (AAS analysis), Ulrike Schümann (fuel testing laboratory), Dipl.-Chem. (FH) Reiner Eckelt (assistance with BET measurements) and Alexander Villinger (XRD). The work was funded by the DAAD in the frame of the ROHAN Programme which is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pfeiffer, J. Fossil Resources and Climate Change—The Green Paradox and Resource Market Power; Ifo Beträge zur Wirtschaftsforschung; Ifo Institute: Munich, Germany, 2017. [Google Scholar]
- Graves, C.; Ebbesen, S.D.; Mogensen, M.; Lackner, K.S. Sustainable hydrocarbon fuels by recycling CO2 and H2O with renewable or nuclear energy. Renew. Sustain. Energy Rev 2011, 15, 1–23. [Google Scholar] [CrossRef]
- Melis, A. Photosynthesis-to-fuels: From sunlight to hydrogen, isoprene, and botryococcene production. Energy Environ. Sci. 2012, 5, 5531–5539. [Google Scholar] [CrossRef]
- Lotero, E.; Liu, Y.; Lopez, D.E.; Suwannakarn, K.; Bruce, D.A.; Goodwin, J.G. Synthesis of biodiesel via acid catalysis. Ind. Eng. Chem. Res. 2005, 44, 5353–5363. [Google Scholar] [CrossRef]
- Leung, D.Y.C.; Wu, X.; Leung, M.K.H. A review on biodiesel production using catalyzed transesterification. Appl. Energy 2010, 87, 1083–1095. [Google Scholar] [CrossRef]
- Hoekman, S.K.; Broch, A.; Robbins, C.; Ceniceros, E.; Natarajan, M. Review of biodiesel composition, properties, and specifications. Renew. Sustain. Energy Rev. 2012, 16, 143–169. [Google Scholar] [CrossRef]
- Frusteri, F.; Arena, F.; Bonura, G.; Cannilla, C.; Spadaro, L.; Di Blasi, O. Catalytic etherification of glycerol by tert-butyl alcohol to produce oxygenated additives for diesel fuel. Appl. Catal. A Gen. 2009, 367, 77–83. [Google Scholar] [CrossRef]
- Cannilla, C.; Bonura, G.; Frusteri, L.; Frusteri, F. Batch reactor coupled with water permselective membrane: Study of glycerol etherification reaction with butanol. Chem. Eng. J. 2015, 282, 187–193. [Google Scholar] [CrossRef]
- Magar, S.; Kamble, S.; Mohanraj, G.T.; Jana, S.K.; Rode, C. Solid-Acid-Catalyzed Etherification of Glycerol to Potential Fuel Additives. Energy Fuels 2017, 31, 12272–12277. [Google Scholar] [CrossRef]
- Estevez, R.; Aguado-Deblas, L.; Luna, D.; Bautista, F.M. An overview of the production of oxygenated fuel additives by glycerol etherification, either with isobutene or tert-butyl alcohol over heterogeneous catalysts. Energies 2019, 12, 2364. [Google Scholar] [CrossRef] [Green Version]
- Aguado-Deblas, L.; Estevez, R.; Russo, M.; La Parola, V.; Bautista, F.M.; Testa, M.L. Microwave-assisted glycerol etherification over sulfonic acid catalysts. Materials 2020, 13, 1584. [Google Scholar] [CrossRef] [Green Version]
- Mansouri, N.; Rikhtegar, N.; Panahi, A.H.; Atabi, F.; Shahraki, B.K. Porosity, characterization and structural properties of natural zeolite–Clinoptilolite—As a sorbent. Environ. Prot. Eng. 2013, 39, 139–152. [Google Scholar]
- Polat, E.; Karaca, M.; Demir, H.; Onus, A.N. Use of natural zeolite (clinoptilolite) in agriculture. J. Fruit Ornam. Plant Res. 2004, 12, 183–189. [Google Scholar]
- Çulfaz, M.; Yağız, M. Ion exchange properties of natural clinoptilolite: Lead–sodium and cadmium–sodium equilibria. Sep. Purif. Technol. 2004, 37, 93–105. [Google Scholar] [CrossRef]
- Rivera, A.; Farias, T. Clinoptilolite–surfactant composites as drug support: A new potential application. Microporous Mesoporous Mater. 2005, 80, 337–346. [Google Scholar] [CrossRef]
- Favvas, E.P.; Tsanaktsidis, C.G.; Sapalidis, A.A.; Tzilantonis, G.T.; Papageorgiou, S.K.; Mitropoulos, A.C. Clinoptilolite, a natural zeolite material: Structural characterization and performance evaluation on its dehydration properties of hydrocarbon-based fuels. Microporous Mesoporous Mater. 2016, 225, 385–391. [Google Scholar] [CrossRef]
- Rabo, J.A. Zeolite Chemistry and Catalysis (ACS Monograpgh, 171); American Chemical Society: Washington, DC, USA, 1976. [Google Scholar]
- Breck, D.W. Zeolite Molecular Sieves: Structure, Chemistry and Use; John Wiley & Sons Inc.: Hoboken, NJ, USA, 1974. [Google Scholar]
- Dziedzicka, A.; Sulikowski, B.; Ruggiero-Mikołajczyk, M. Catalytic and physicochemical properties of modified natural clinoptilolite. Catal. Today 2016, 259, 50–58. [Google Scholar] [CrossRef]
- Saramok, M.; Szymaszek, A.; Inger, M.; Jurak, K.A.; Samojeden, B.; Motak, M. Modified Zeolite Catalyst for a NOx Selective Catalytic Reduction Process in Nitric Acid Plants. Catalysts 2021, 11, 450–469. [Google Scholar] [CrossRef]
- Alvarez-Aguiñaga, E.A.; Elizalde-González, M.P.; Sabinas-Hernández, S.A. Unpredicted photocatalytic activity of clinoptilolite–mordenite natural zeolite. RSC Adv. 2020, 10, 39251–39260. [Google Scholar] [CrossRef]
- Sobuś, N.; Czekaj, I. Comparison of Synthetic and Natural Zeolite Catalysts‘ Behavior in the Production of Lactic Acid and Ethyl Lactate from Biomass-Derived Dihydroxyacetone. Catalysts 2021, 11, 1006–1023. [Google Scholar] [CrossRef]
- Miądlicki, P.; Wróblewska, A.; Kiełbasa, K.; Koren, Z.C.; Michalkiewicz, B. Sulfuric acid modified clinoptilolite as a solid green catalyst for solvent-free α-pinene isomerization process. Microporous Mesoporous Mater. 2021, 324, 111266–111279. [Google Scholar] [CrossRef]
- Mohadesi, M.; Aghel, B.; Maleki, M.; Ansari, A. The use of KOH/Clinoptilolite catalyst in pilot of microreactor for biodiesel production from waste cooking oil. Fuel 2020, 263, 116659–116668. [Google Scholar] [CrossRef]
- Balou, J.; Khalilzadeh, M.A.; Zareyee, D. KF/Nano-clinoptilolite Catalyzed Aldol-Type Reaction of Aldehydes with Ethyl Diazoacetate. Catal. Lett. 2017, 147, 2612–2618. [Google Scholar] [CrossRef]
- Chmielewská, E. Natural Zeolites as Sustainable and Environmental Inorganic Resources over the History to Present Natural Zeolites as Sustainable and Environmental Inorganic Resources over the History to Present. Gen. Chem. 2019, 5, 190001–190006. [Google Scholar] [CrossRef]
- Corma, A.; Zones, S.; Cejka, J. Zeolites and Catalysis; Wiley: Hoboken, NJ, USA, 2010. [Google Scholar]
- Adinehvand, J.; Shokuhi Rad, A.; Tehrani. A. S. Acid-treated zeolite (clinoptilolite) and its potential to zinc removal from water sample. Int. J. Environ. Sci. Technol 2016, 13, 2705–2712. [Google Scholar] [CrossRef]
- Vlad, E.; Bildea, C.S.; Bozga, G. Design and control of glycerol-tert-butyl alcohol etherification process. Sci. World J. 2012, 2012, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ozbay, N.; Oktar, N.; Dogu, G.; Dogu, T. Effects of sorption enhancement and isobutene formation on etherification of glycerol with tert-butyl alcohol in a flow reactor. Ind. Eng. Chem. Res. 2012, 51, 8788–8795. [Google Scholar] [CrossRef]
- Scherrer, P. Bestimmung der Größe und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Nachr. Ges. Wiss. Göttingen 1918, 26, 98–100. [Google Scholar]
- Sultan, M.; Miyazaki, T.; Koyama, S. Optimization of adsorption isotherm types for desiccant air-conditioning applications. Renew. Energy 2018, 121, 441–450. [Google Scholar] [CrossRef]
- Flanigen, E.M.; Khatami, H.; Szymanski, H.A. Infrared Structural Studies of Zeolite Frameworks. In Advances in Chemistry; Flanigen, E.M., Sand, L.B., Eds.; American Chemical Society: Washington, DC, USA, 1974; Volume 101, pp. 201–229. [Google Scholar]
- Van Aelst, J.; Haouas, M.; Gobechiya, E.; Houthoofd, K.; Philippaerts, A.; Sree, S.P.; Kirschhock, C.E.A.; Jacobs, P.; Martens, J.A.; Sels, B.F.; et al. Hierarchization of USY zeolite by NH4OH. A postsynthetic process investigated by NMR and XRD. J. Phys. Chem. C 2014, 118, 22573–22582. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Jiménez, A.; Palomo, A. Nanostructure/microstructure of fly ash geopolymers. In Geopolymers Structures, Processing, Properties and Industrial Applications; Provis, J.L., Van Deventer, S.J., Eds.; CRC Press: Boca Raton, FL, USA, 2009; pp. 89–114. [Google Scholar]
- Blumenfeld, A.L.; Coster, D.; Fripiat, J.J. Brønsted acid sites and surface structure in zeolites: A high-resolution 29Si NMR REDOR study. J. Phys. Chem. 1995, 99, 15181–15191. [Google Scholar] [CrossRef]
- Gackowski, M.; Kuterasiński, Ł.; Podobiński, J.; Sulikowski, B.; Datka, J. IR and NMR studies of hierarchical material obtained by the treatment of zeolite Y by ammonia solution. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 193, 440–446. [Google Scholar] [CrossRef]
- Vlasenko, N.V.; Kochkin, Y.N.; Telbiz, G.M.; Shvets, O.V.; Strizhak, P.E. Insight into the active site nature of zeolite H-BEA for liquid phase etherification of isobutylene with ethanol. RSC Adv. 2019, 9, 35957–35968. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Mao, D.; Zhai, X. Selective conversion of bio-ethanol to propene over nano-HZSM-5 zeolite: Remarkably enhanced catalytic performance by fluorine modification. Fuel Process. Technol. 2017, 167, 50–60. [Google Scholar] [CrossRef]
- Deng, F.; Yue, Y.; Ye, C. 1H/27Al TRAPDOR NMR studies on aluminum species in dealuminated zeolites. Solid State Nucl. Magn. Reson. 1998, 10, 151–160. [Google Scholar] [CrossRef]
- Greiser, S.; Gluth, G.J.G.; Sturm, P.; Jäger, C. 29Si{27Al}, 27Al{29Si} and 27Al{1H} double-resonance NMR spectroscopy study of cementitious sodium aluminosilicate gels (geopolymers) and gel-zeolite composites. RSC Adv. 2018, 8, 40164–40171. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).