
High-Performance Ligand-Protected Metal Nanocluster Catalysts for CO2 Conversion through the Exposure of Undercoordinated Sites


{kind=link}
{kind=link}
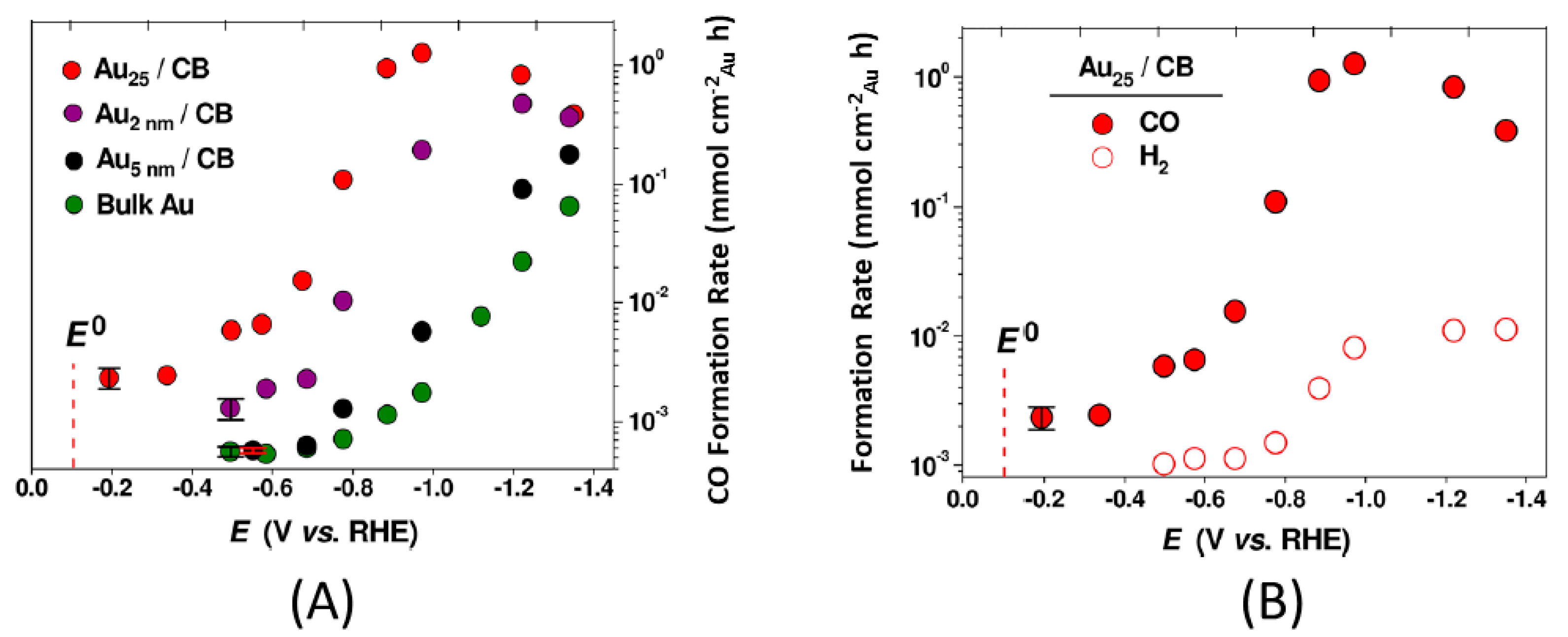
{kind=link}
{kind=link}
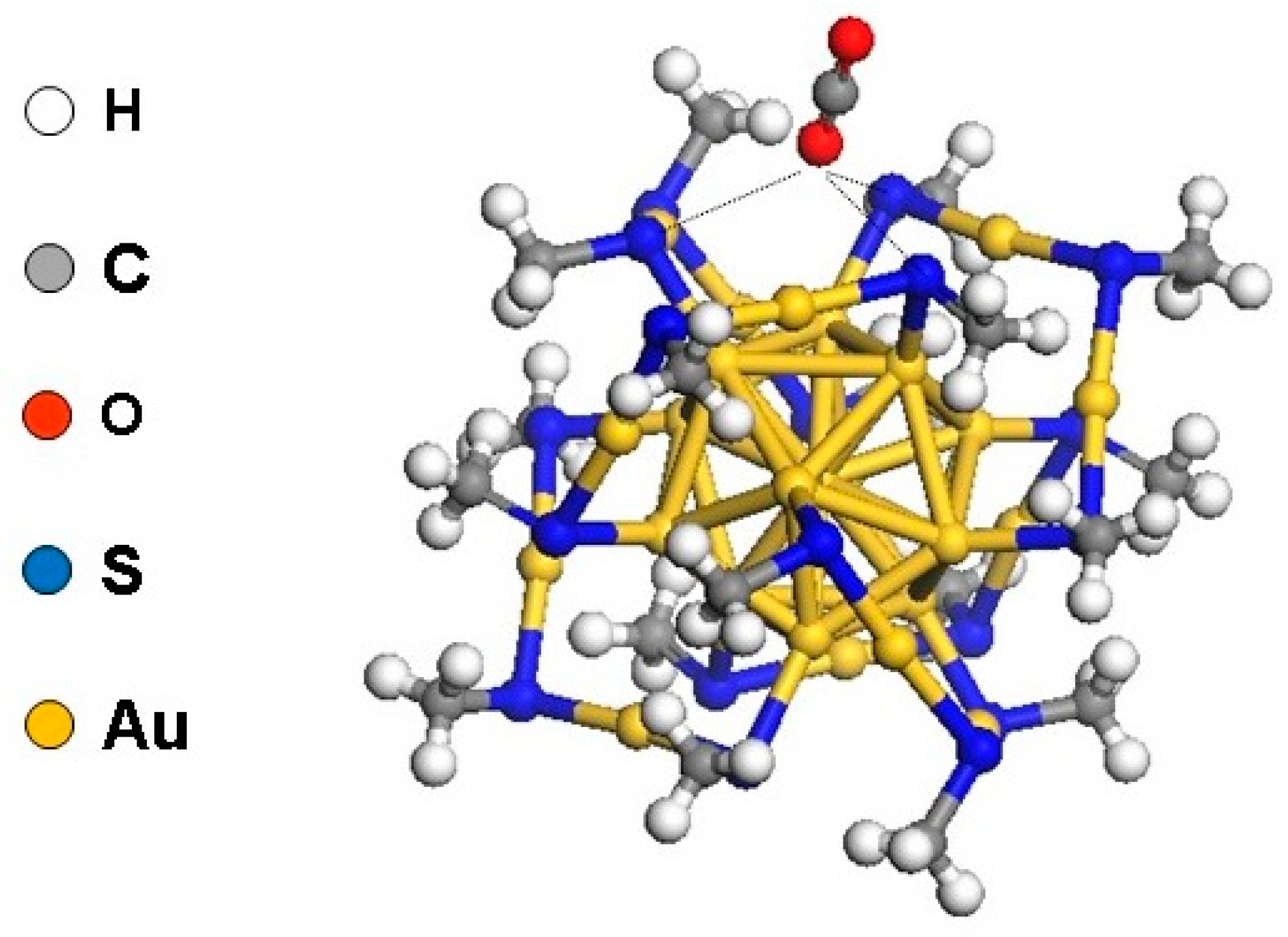
{kind=link}
{kind=link}
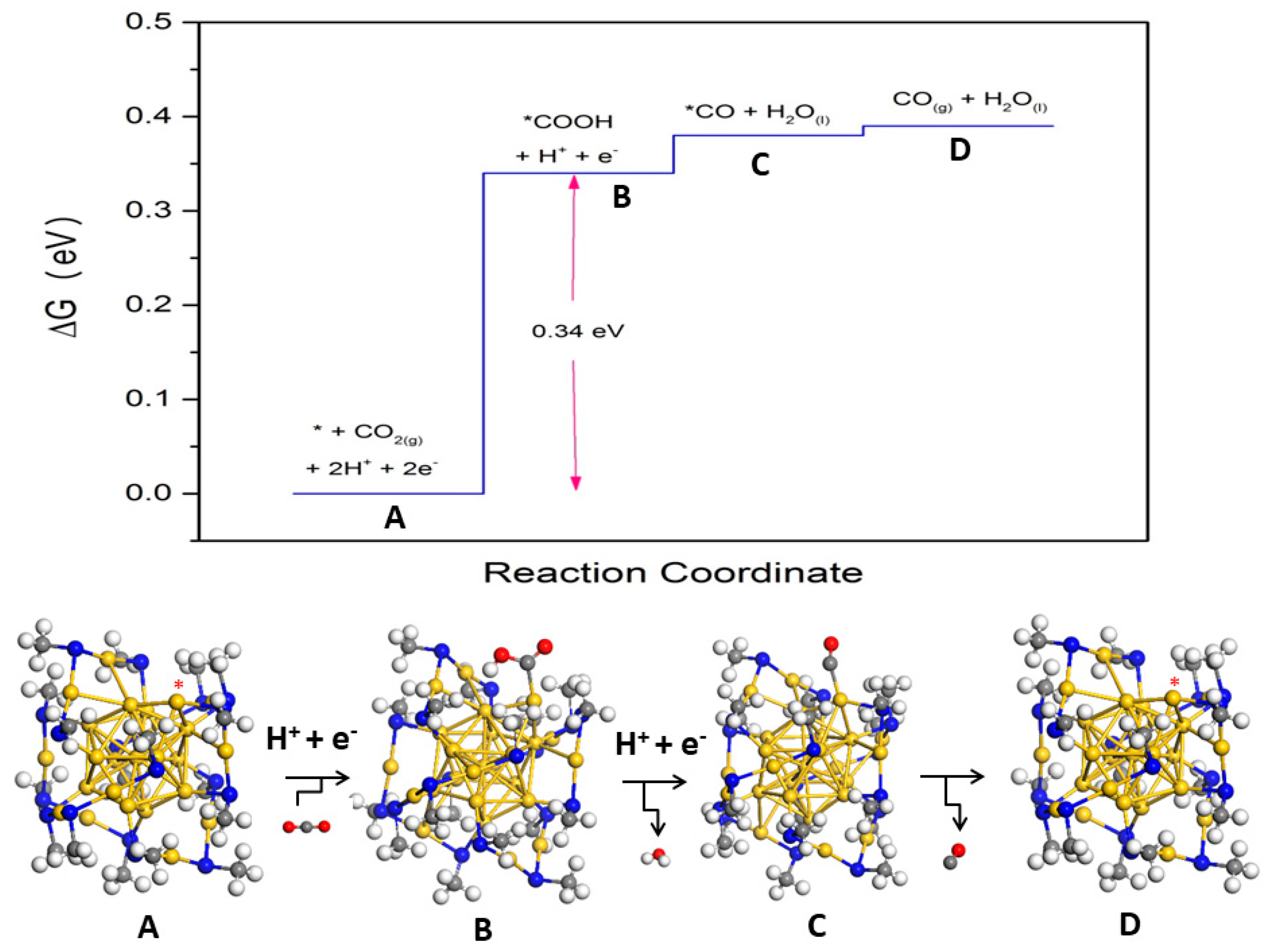
{kind=link}
{kind=link}
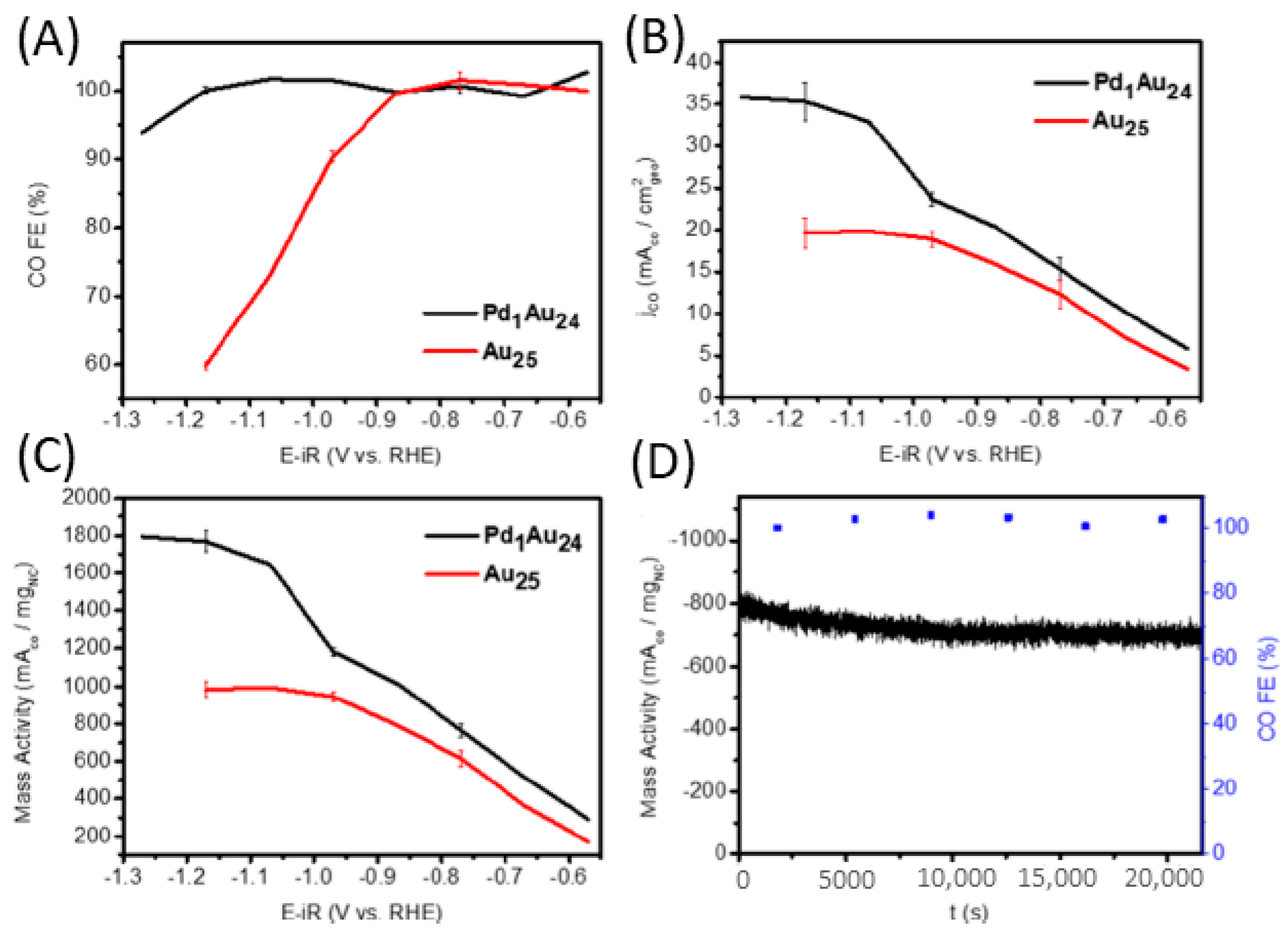
{kind=link}
Abstract
:1. Introduction
2. Theoretical Studies of Au-Based Nanoclusters
2.1. Aspects of Modeling
2.2. Theoretical Insights
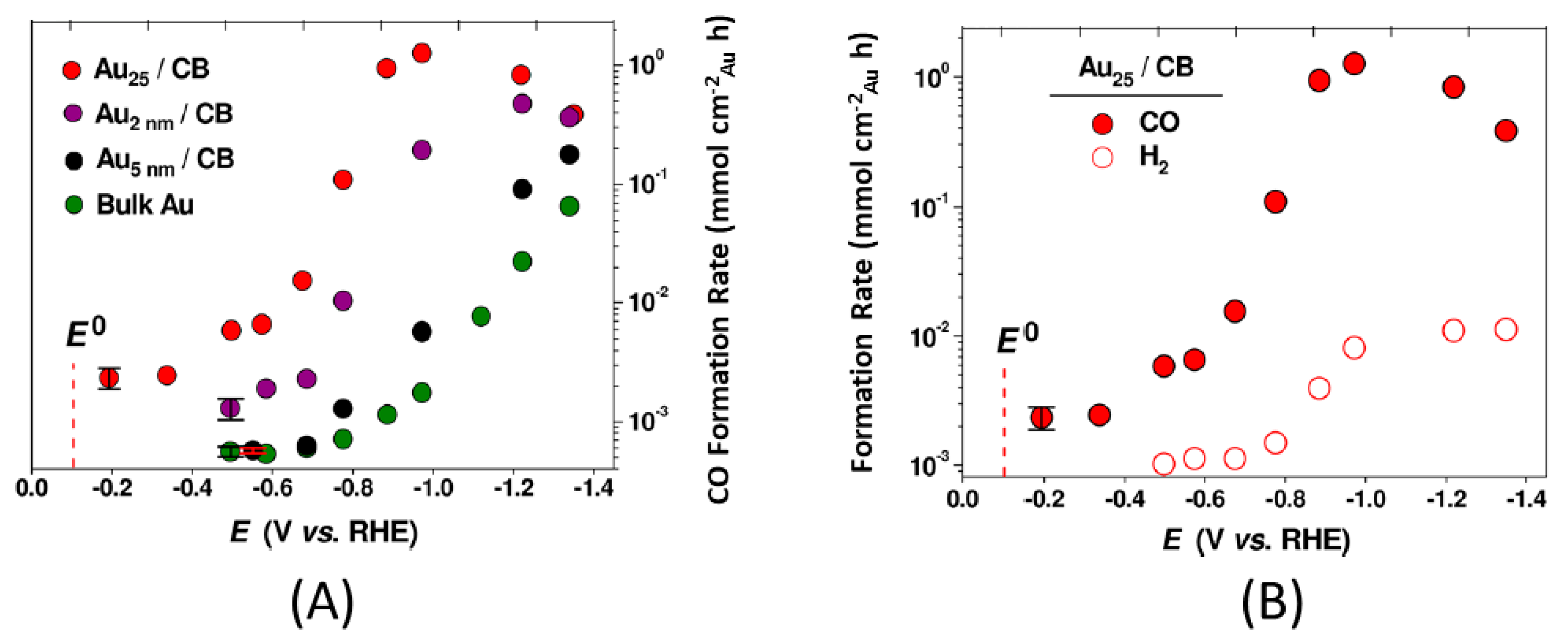
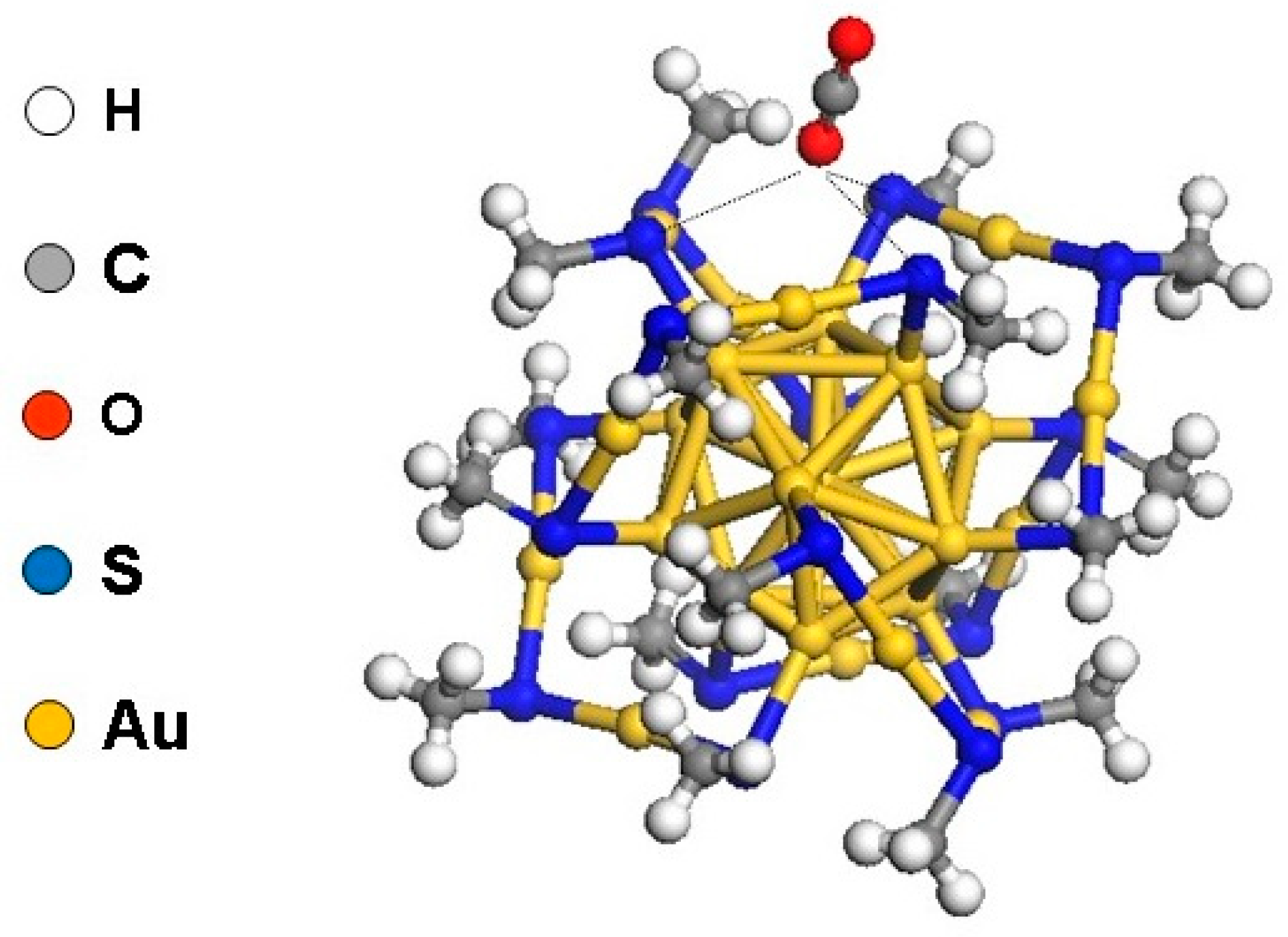
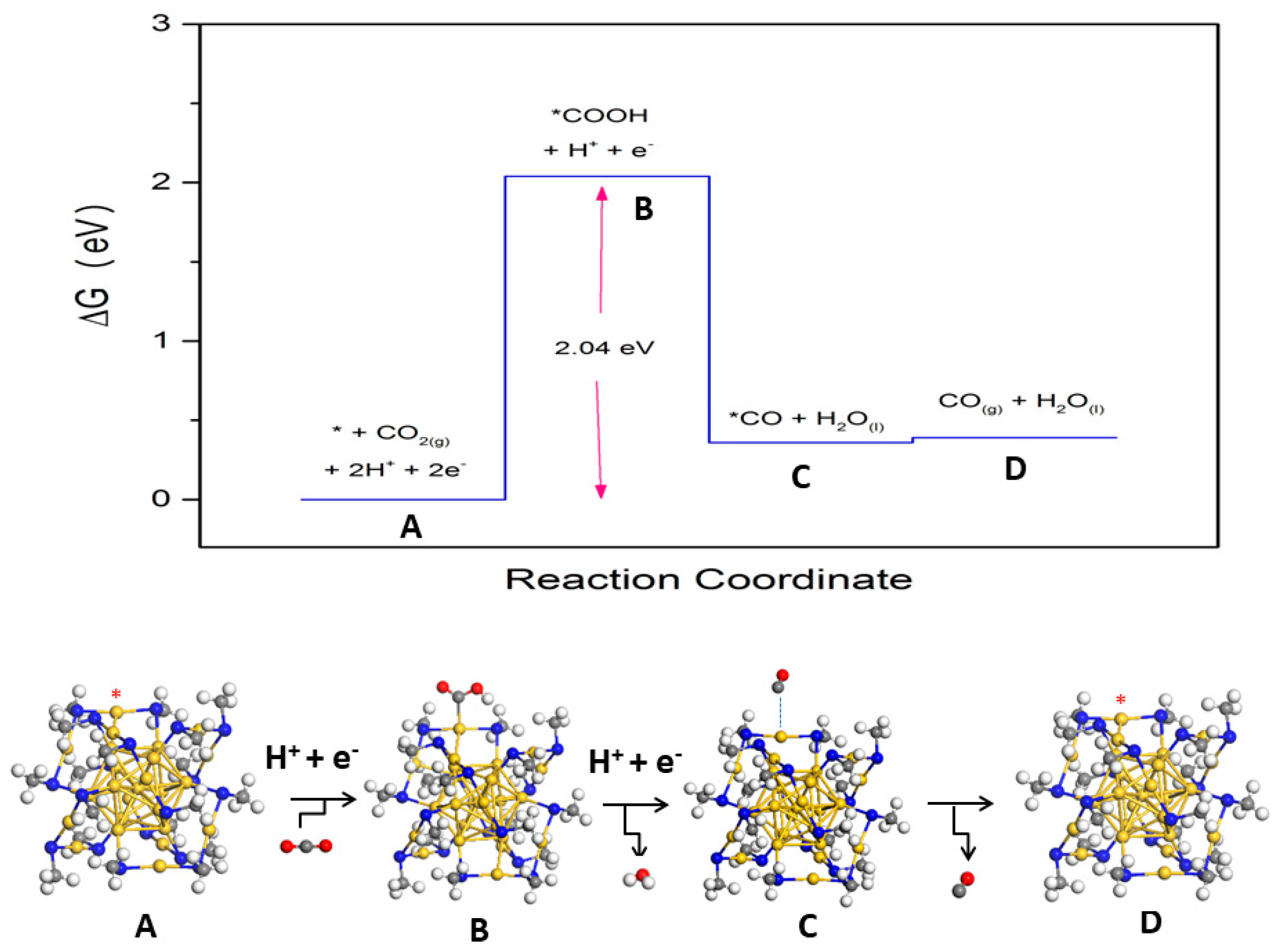
2.2.1. Carbon Dioxide Reduction on Au25(SR)18−
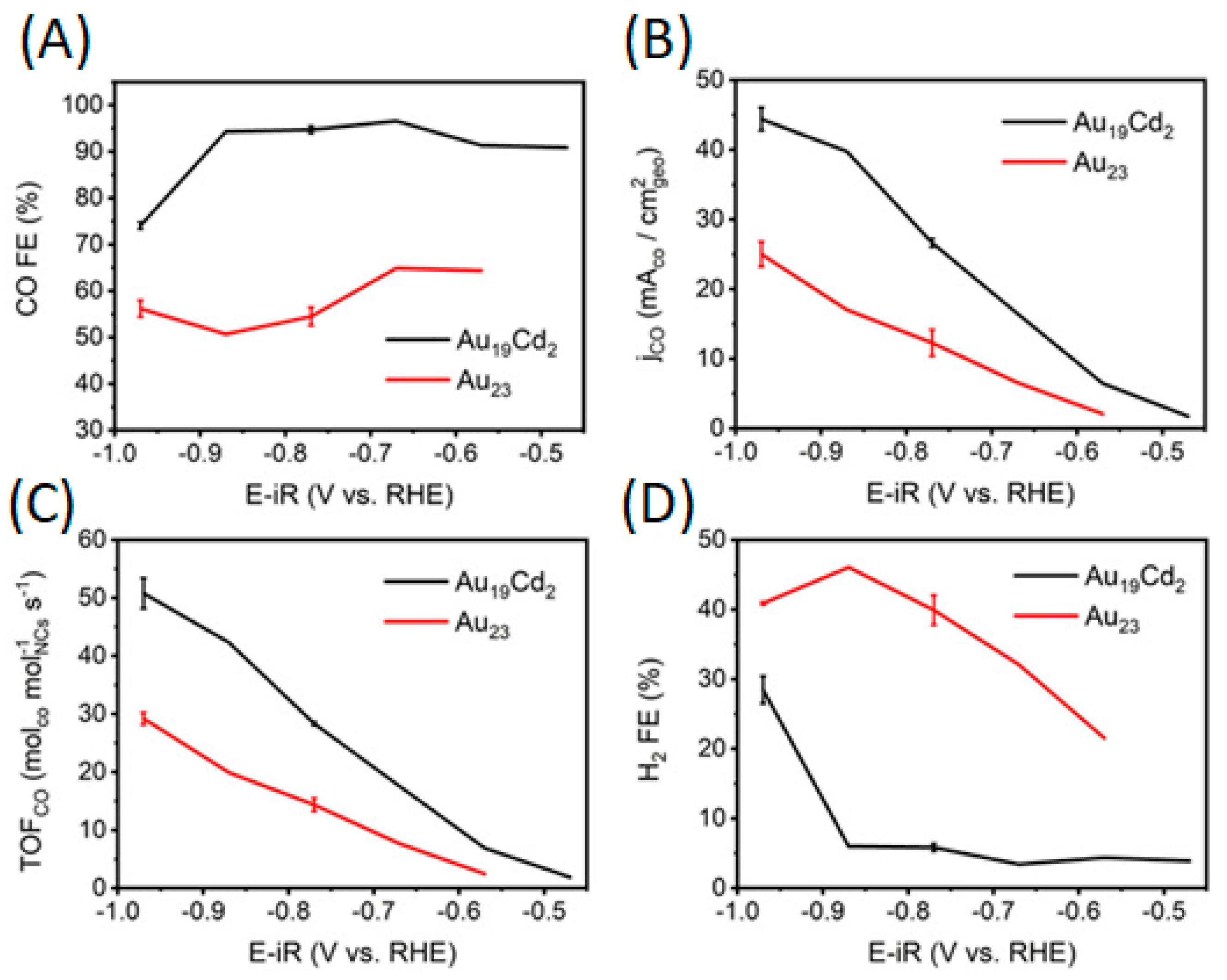
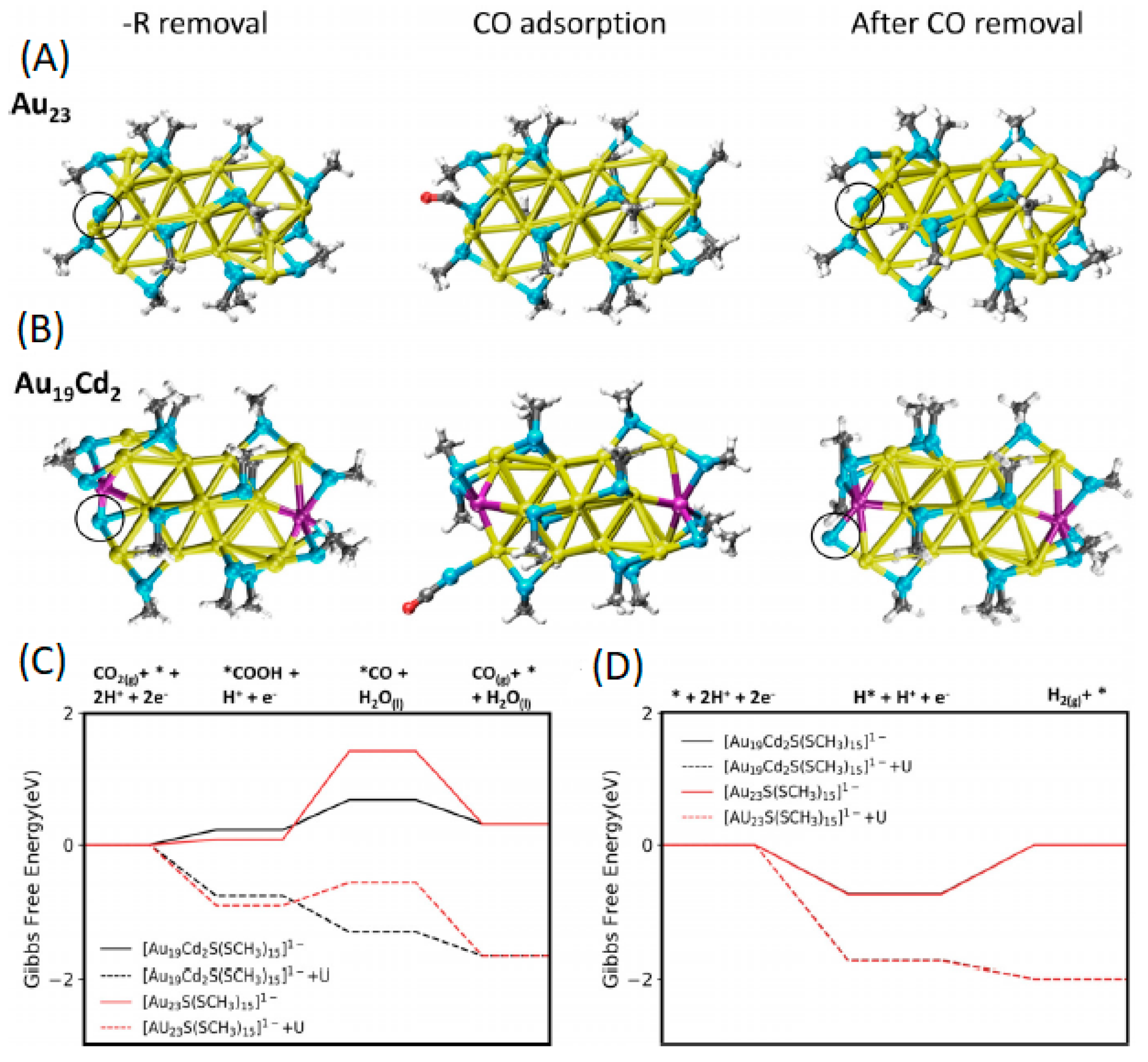
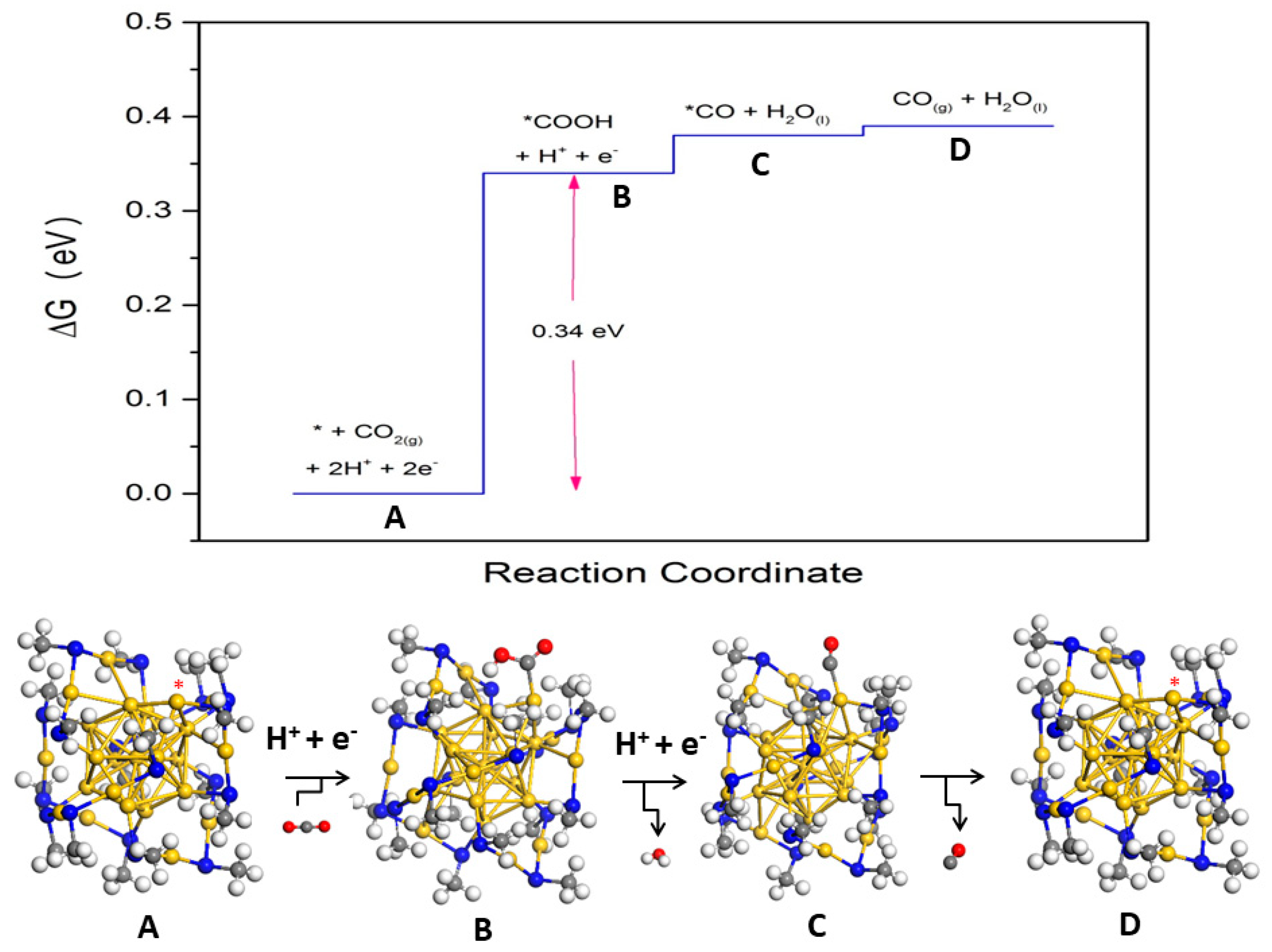
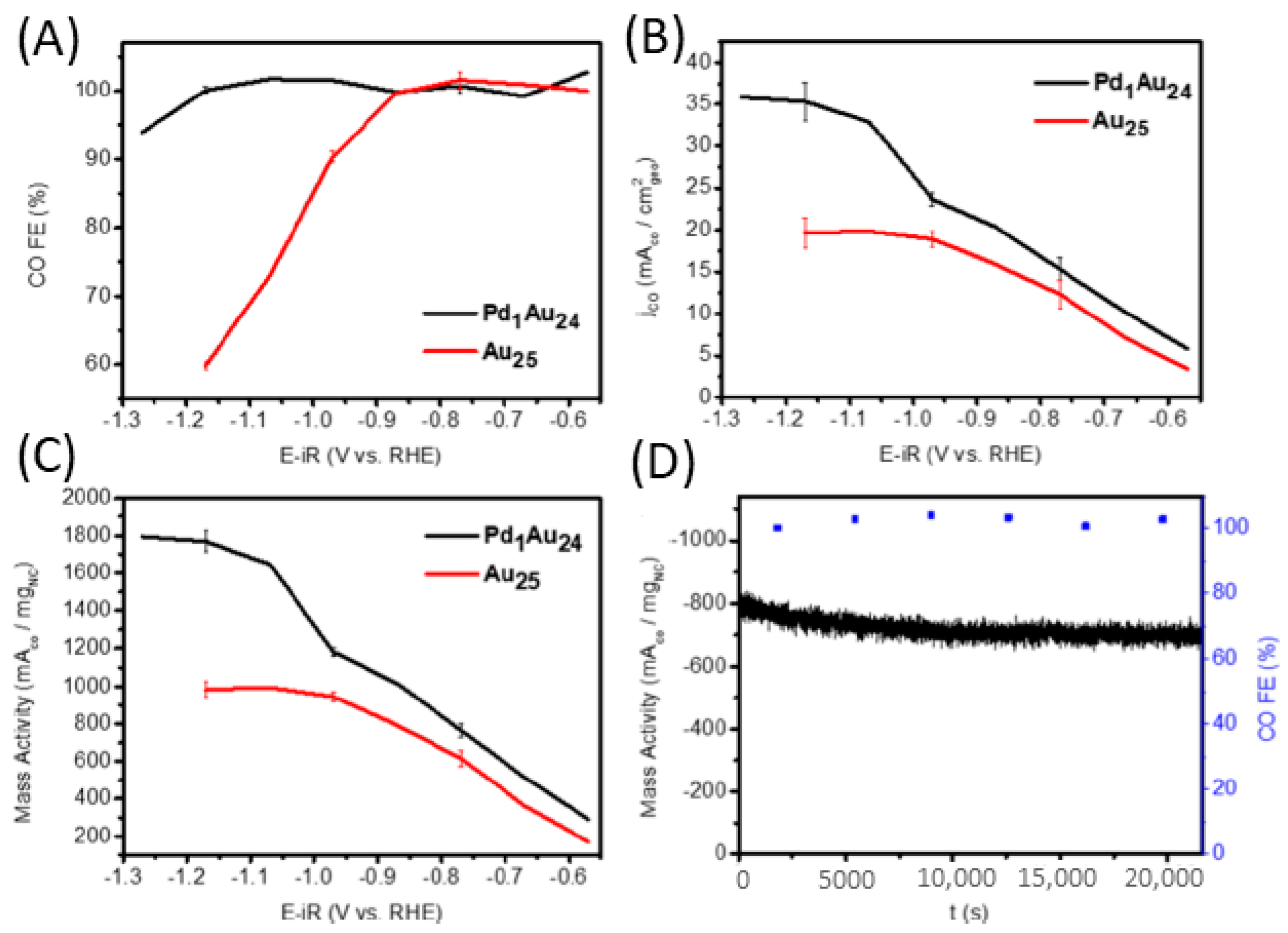
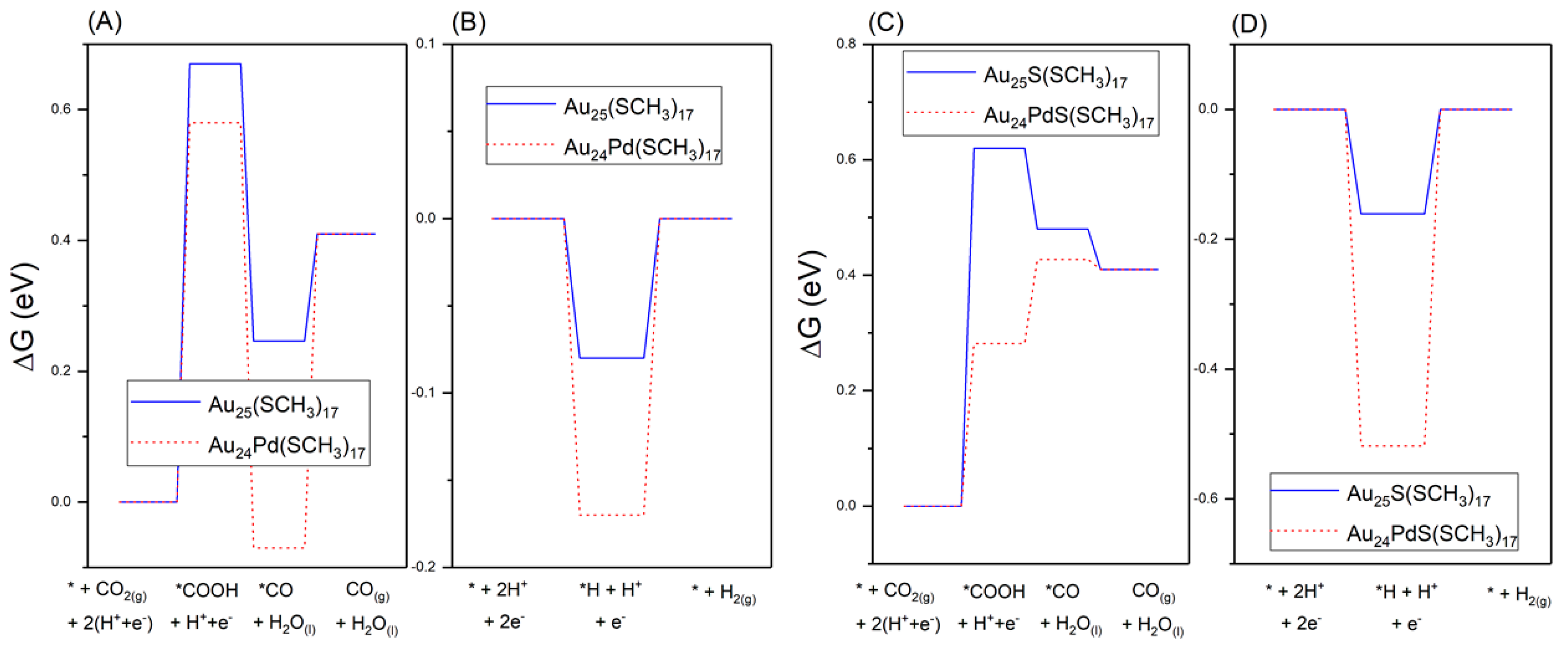
2.2.2. Promotion of Carbon Dioxide Reduction through Doping
3. Summary and Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Sakakura, T.; Choi, J.-C.; Yasuda, H. Transformation of carbon dioxide. Chem. Rev. 2007, 107, 2365. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y. Electrochemical CO2 Reduction on Metal Electrodes. In Modern Aspects of Electrochemistry; Vayenas, C.G., White, R.E., Gamboa-Aldeco, M.E., Eds.; Springer: New York, NY, USA, 2008. [Google Scholar]
- Mikkelsen, M.; Jørgensen, M.; Krebs, F.C. The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 2010, 3, 43–81. [Google Scholar] [CrossRef]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.E.; Kubiak, C.P.; Sathrum, A.J.; Smieja, J.M. Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem. Soc. Rev. 2009, 38, 89–99. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, Y.; Ma, L.; Zhu, G.; Wang, Y.; Xue, X.; Chen, R.; Yang, S.; Jin, Z. Progress and perspective of electrocatalytic CO2 reduction for renewable carbonaceous fuels and chemicals. Adv. Sci. 2018, 5, 1700275. [Google Scholar] [CrossRef]
- Dry, M.E. The fischer—Tropsch process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar] [CrossRef]
- Schulz, H. Short history and present trends of Fischer—Tropsch synthesis. Appl. Catal. A 1999, 186, 3–12. [Google Scholar] [CrossRef]
- Lan, G.; Yang, J.; Ye, R.P.; Boyjoo, Y.; Liang, J.; Liu, X.; Li, Y.; Liu, J.; Qian, K. Sustainable carbon materials toward emerging applications. Small Methods 2021, 5, 2001250. [Google Scholar] [CrossRef]
- Liu, J.; Cai, C.; Wang, Y.; Liu, Y.; Huang, L.; Tian, T.; Yao, Y.; Wei, J.; Chen, R.; Zhang, K. A biomimetic plasmonic nanoreactor for reliable metabolite detection. Adv. Sci. 2020, 7, 1903730. [Google Scholar] [CrossRef]
- Zu, Y.; Yao, H.; Wang, Y.; Yan, L.; Gu, Z.; Chen, C.; Gao, L.; Yin, W. The age of bioinspired molybdenum-involved nanozymes: Synthesis, catalytic mechanisms, and biomedical applications. View 2021, 2, 20200188. [Google Scholar] [CrossRef]
- Chu, S.; Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 2012, 488, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Graves, C.; Ebbesen, S.D.; Mogensen, M.; Lackner, K.S. Sustainable hydrocarbon fuels by recycling CO2 and H2O with renewable or nuclear energy. Renew. Sustain. Energy Rev. 2011, 15, 1–23. [Google Scholar] [CrossRef]
- Singh, M.R.; Clark, E.L.; Bell, A.T. Thermodynamic and achievable efficiencies for solar-driven electrochemical reduction of carbon dioxide to transportation fuels. Proc. Natl. Acad. Sci. USA 2015, 112, E6111–E6118. [Google Scholar] [CrossRef] [Green Version]
- Lopez, N.; Nørskov, J.K. Catalytic CO oxidation by a gold nanoparticle: A density functional study. J. Am. Chem. Soc. 2002, 124, 11262–11263. [Google Scholar] [CrossRef]
- Lopez, N.; Janssens, T.; Clausen, B.; Xu, Y.; Mavrikakis, M.; Bligaard, T.; Nørskov, J.K. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Liang, Y.; Li, Y.; Wang, H.; Zhou, J.; Wang, J.; Regier, T.; Dai, H. Co3O4 nanocrystals on graphene as a synergistic catalyst for oxygen reduction reaction. Nat. Mater. 2011, 10, 780–786. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Vogel, W.; Lamy, C.; Alonso-Vante, N. Structure and electrocatalytic activity of carbon-supported Pt− Ni alloy nanoparticles toward the oxygen reduction reaction. J. Phys. Chem. B 2004, 108, 11024–11034. [Google Scholar] [CrossRef]
- Chen, S.; Ferreira, P.J.; Sheng, W.; Yabuuchi, N.; Allard, L.F.; Shao-Horn, Y. Enhanced activity for oxygen reduction reaction on “Pt3Co” nanoparticles: Direct evidence of percolated and sandwich-segregation structures. J. Am. Chem. Soc. 2008, 130, 13818–13819. [Google Scholar] [CrossRef]
- Mazumder, V.; Chi, M.; More, K.L.; Sun, S. Core/shell Pd/FePt nanoparticles as an active and durable catalyst for the oxygen reduction reaction. J. Am. Chem. Soc. 2010, 132, 7848–7849. [Google Scholar] [CrossRef]
- Li, Y.; Wang, H.; Xie, L.; Liang, Y.; Hong, G.; Dai, H. MoS2 nanoparticles grown on graphene: An advanced catalyst for the hydrogen evolution reaction. J. Am. Chem. Soc. 2011, 133, 7296–7299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popczun, E.J.; McKone, J.R.; Read, C.G.; Biacchi, A.J.; Wiltrout, A.M.; Lewis, N.S.; Schaak, R.E. Nanostructured nickel phosphide as an electrocatalyst for the hydrogen evolution reaction. J. Am. Chem. Soc. 2013, 135, 9267–9270. [Google Scholar] [CrossRef] [PubMed]
- Salehi-Khojin, A.; Jhong, H.-R.M.; Rosen, B.A.; Zhu, W.; Ma, S.; Kenis, P.J.; Masel, R.I. Nanoparticle silver catalysts that show enhanced activity for carbon dioxide electrolysis. J. Phys. Chem. C 2013, 117, 1627–1632. [Google Scholar] [CrossRef]
- Lu, Q.; Rosen, J.; Zhou, Y.; Hutchings, G.S.; Kimmel, Y.C.; Chen, J.G.; Jiao, F. A selective and efficient electrocatalyst for carbon dioxide reduction. Nat. Commun. 2014, 5, 3242. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tao, H.; Zeng, L.; Liu, Q.; Xu, Z.; Liu, Q.; Luo, J.-L. Shape-dependent electrocatalytic reduction of CO2 to CO on triangular silver nanoplates. J. Am. Chem. Soc. 2017, 139, 2160–2163. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Michalski, R.; Metin, O.; Lv, H.; Guo, S.; Wright, C.J.; Sun, X.; Peterson, A.A.; Sun, S. Monodisperse Au Nanoparticles for Selective Electrocatalytic Reduction of CO2 to CO. J. Am. Chem. Soc. 2013, 135, 16833–16836. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Reske, R.; Zeng, Z.; Zhao, Z.-J.; Greeley, J.; Strasser, P.; Roldan Cuenya, B. Exceptional Size—Dependent Activity Enhancement in the Electroreduction of CO2 over Au Nanoparticles. J. Am. Chem. Soc. 2014, 136, 16473–16476. [Google Scholar] [CrossRef]
- Gates, B.C. Supported gold catalysts: New properties offered by nanometer and sub-nanometer structures. Chem. Commun. 2013, 49, 7876–7877. [Google Scholar] [CrossRef]
- Herzing, A.A.; Kiely, C.J.; Carley, A.F.; Landon, P.; Hutchings, G.J. Identification of active gold nanoclusters on iron oxide supports for CO oxidation. Science 2008, 321, 1331–1335. [Google Scholar] [CrossRef]
- Corma, A.; Concepción, P.; Boronat, M.; Sabater, M.J.; Navas, J.; Yacaman, M.J.; Larios, E.; Posadas, A.; López-Quintela, M.A.; Buceta, D. Exceptional oxidation activity with size-controlled supported gold clusters of low atomicity. Nat. Chem. 2013, 5, 775–781. [Google Scholar] [CrossRef] [Green Version]
- Oliver-Meseguer, J.; Cabrero-Antonino, J.R.; Domínguez, I.; Leyva-Pérez, A.; Corma, A. Small gold clusters formed in solution give reaction turnover numbers of 107 at room temperature. Science 2012, 338, 1452–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.M.; Khoury, J.T.; Schaaff, T.G.; Shafigullin, M.N.; Vezmar, I.; Whetten, R.L. Optical absorption spectra of nanocrystal gold molecules. J. Phys. Chem. B 1997, 101, 3706–3712. [Google Scholar] [CrossRef] [Green Version]
- Jin, R. Quantum sized, thiolate-protected gold nanoclusters. Nanoscale 2010, 2, 343–362. [Google Scholar] [PubMed]
- Wu, Z.; Jiang, D.-E.; Mann, A.K.; Mullins, D.R.; Qiao, Z.-A.; Allard, L.F.; Zeng, C.; Jin, R.; Overbury, S.H. Thiolate ligands as a double-edged sword for CO oxidation on CeO2 supported Au25 (SCH2CH2Ph) 18 nanoclusters. J. Am. Chem. Soc. 2014, 136, 6111–6122. [Google Scholar] [CrossRef]
- Gaur, S.; Wu, H.; Stanley, G.G.; More, K.; Kumar, C.S.; Spivey, J.J. CO oxidation studies over cluster-derived Au/TiO2 and AUROlite™ Au/TiO2 catalysts using DRIFTS. Catal. Today 2013, 208, 72–81. [Google Scholar] [CrossRef]
- Zhu, Y.; Qian, H.; Zhu, M.; Jin, R. Thiolate-Protected Aun Nanoclusters as Catalysts for Selective Oxidation and Hydrogenation Processes. Adv. Mater. 2010, 22, 1915–1920. [Google Scholar] [CrossRef]
- Nie, X.; Qian, H.; Ge, Q.; Xu, H.; Jin, R. CO oxidation catalyzed by oxide-supported Au25(SR)18 nanoclusters and identification of perimeter sites as active centers. ACS Nano 2012, 6, 6014–6022. [Google Scholar] [CrossRef]
- Kauffman, D.; Alfonso, D.; Matranga, C.; Qian, H.; Jin, R. Experimental and Computational Investigation of Au25 Clusters and CO2: A Unique Interaction and Enhanced Electrocatalytic Activity. J. Am. Chem. Soc. 2012, 134, 10237–10243. [Google Scholar] [CrossRef]
- Kauffman, D.R.; Alfonso, D.; Matranga, C.; Ohodnicki, P.; Deng, X.; Siva, R.C.; Zeng, C.; Jin, R. Probing active site chemistry with differently charged Au25q nanoclusters (q = −1, 0, +1). Chem. Sci. 2014, 5, 3151–3157. [Google Scholar] [CrossRef]
- Vickers, J.W.; Alfonso, D.; Kauffman, D.R. Electrochemical carbon dioxide reduction at nanostructured gold, copper, and alloy materials. Energy Technol. 2017, 5, 775–795. [Google Scholar] [CrossRef] [Green Version]
- Norskov, J.K.; Rossmeisl, J.; Logadottir, A.; Linqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonnson, H.J. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Norskov, J.K. How Copper Catalyzes the Electroreduction of Carbon Dioxide into Hydrocarbon Fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Hansen, H.A.; Varley, J.B.; Peterson, A.A.; Nørskov, J.K. Understanding trends in the electrocatalytic activity of metals and enzymes for CO2 reduction to CO. J. Phys. Chem. Lett. 2013, 4, 388–392. [Google Scholar] [CrossRef]
- Zhu, W.; Zhang, Y.-J.; Zhang, H.; Lv, H.; Li, Q.; Michalsky, R.; Peterson, A.A.; Sun, S. Active and selective conversion of CO2 to CO on ultrathin Au nanowires. J. Am. Chem. Soc. 2014, 136, 16132–16135. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Effeciency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comp. Mat. Sci. 1996, 6, 15–50. [Google Scholar]
- Perdew, J.P.; Burke, K.; Enzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High Precision Sampling for Brillouin Zone Integration for Metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [Green Version]
- Matthew, K.; Sundararaman, R.; Lechtworth-Weaver, K.; Arias, T.A.; Hennig, R.J. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. Chem. Phys. 2014, 140, 84106. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Acevedo, O.; Kacprzak, K.A.; Akola, J.; Hakkinen, H. Quantum size effects in ambient CO oxidation catalysed by ligand-protected gold clusters. Nat. Chem. 2010, 2, 329–334. [Google Scholar] [CrossRef]
- Alfonso, D.; Kauffman, D.; Matranga, C.J. Active sites of ligand-protected Au25 nanoparticle catalysts for CO2 electroreduction to CO. Chem. Phys. 2016, 144, 184705. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Abroshan, H.; Chen, Y.; Jin, R.; Kim, H.J. Experimental and mechanistic understanding of aldehyde hydrogenation using Au25 nanoclusters with Lewis acids: Unique sites for catalytic reactions. J. Am. Chem. Soc. 2015, 137, 14295–14304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Kaziz, S.; Li, H.; Hevia, M.G.; Wodka, D.; Mazet, C.; Burgi, T.; Barrabés, N. Modulation of Active Sites in Supported Au38(SC2H4Ph)24 Cluster Catalysts: Effect of Atmosphere and Support Material. J. Phys. Chem. C 2015, 119, 11193–11199. [Google Scholar] [CrossRef]
- Austin, N.; Zhao, S.; McKone, J.; Jin, R.; Mpourmpakis, G. Elucidating the active sites for CO2 electroreduction on ligand-protected Au25 nanoclusters. Catal. Sci. Technol. 2018, 8, 3795–3805. [Google Scholar] [CrossRef]
- Varley, J.; Hansen, H.; Ammitzbøll, N.L.; Grabow, L.; Peterson, A.; Rossmeisl, J.; Nørskov, J. Ni-Fe-S cubanes in CO2 reduction electrocatalysis: A DFT study. ACS Catal. 2013, 3, 2640–2643. [Google Scholar] [CrossRef]
- Chan, K.; Tsai, C.; Hansen, H.A.; Norskov, J. Molybdenum Sulfides and Selenides as Possible Electrocatalysts for CO2 Reduction. ChemCatChem 2014, 6, 1899. [Google Scholar] [CrossRef]
- Li, S.; Alfonso, D.; Nagarajan, A.V.; House, S.D.; Yang, J.C.; Kauffman, D.R.; Mpourmpakis, G.; Jin, R. Monopalladium substitution in gold nanoclusters enhances CO2 electroreduction activity and selectivity. ACS Catal. 2020, 10, 12011–12016. [Google Scholar] [CrossRef]
- Li, S.; Nagarajan, A.V.; Alfonso, D.R.; Sun, M.; Kauffman, D.R.; Mpourmpakis, G.; Jin, R. Boosting CO2 electrochemical reduction with atomically precise surface modification on gold nanoclusters. Angew. Chem. Int. Ed. 2021, 60, 6351–6356. [Google Scholar] [CrossRef]
- Zhuang, S.; Chen, D.; Liao, L.; Zhao, Y.; Xia, N.; Zhang, W.; Wang, C.; Yang, J.; Wu, Z. Hard-sphere random close-packed Au47Cd2(TBBT)31 nanoclusters with a Faradaic efficiency of up to 96% for electrocatalytic CO2 reduction to CO. Angew. Chem. Int. Ed. 2020, 59, 3073–3077. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, L.; Li, L.; Dong, H.; Chen, S.; Zhu, W.; Hu, C.; Deng, W.; Zhao, Z.-J.; Gong, J. Ultrathin Pd-Au shells with controllable alloying degree on Pd nanocubes toward carbon dioxide reduction. J. Am. Chem. Soc. 2019, 141, 4791–4794. [Google Scholar] [CrossRef]
- Sun, K.; Ji, Y.; Liu, Y.; Wang, Z. Synergies between electronic and geometric effects of Mo-doped Au nanoparticles for effective CO2 electrochemical reduction. J. Mater. Chem. A 2020, 8, 12291–12295. [Google Scholar] [CrossRef]
- Gao, D.; Zhang, Y.; Zhou, Z.; Cai, F.; Zhao, X.; Huang, W.; Li, Y.; Zhu, J.; Liu, P.; Yang, F. Enhancing CO2 electroreduction with the metal—Oxide interface. J. Am. Chem. Soc. 2017, 139, 5652–5655. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, D.R.; Alfonso, D.R.; Tafen, D.N.; Wang, C.; Zhou, Y.; Yu, Y.; Lekse, J.W.; Deng, X.; Espinoza, V.; Trindell, J. Selective electrocatalytic reduction of CO2 into CO at small, thiol-capped Au/Cu nanoparticles. J. Phys. Chem. C 2018, 122, 27991–28000. [Google Scholar] [CrossRef]
- Nie, X.; Esopi, M.R.; Janik, M.J.; Asthagiri, A. Selectivity of CO2 Reduction on Copper Electrode: The Role of Kinetics of Elementary Steps. Angew. Chem. Int. Ed. 2013, 52, 2459–2462. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiao, J.; Peng, H.; Hong, X.; Chan, K.; Nørskov, J.K. Understanding trends in electrochemical carbon dioxide reduction rates. Nat. Commun. 2017, 8, 15438. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A Climbing Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfonso, D. High-Performance Ligand-Protected Metal Nanocluster Catalysts for CO2 Conversion through the Exposure of Undercoordinated Sites. Catalysts 2022, 12, 505. https://doi.org/10.3390/catal12050505
Alfonso D. High-Performance Ligand-Protected Metal Nanocluster Catalysts for CO2 Conversion through the Exposure of Undercoordinated Sites. Catalysts. 2022; 12(5):505. https://doi.org/10.3390/catal12050505
Chicago/Turabian StyleAlfonso, Dominic. 2022. "High-Performance Ligand-Protected Metal Nanocluster Catalysts for CO2 Conversion through the Exposure of Undercoordinated Sites" Catalysts 12, no. 5: 505. https://doi.org/10.3390/catal12050505
APA StyleAlfonso, D. (2022). High-Performance Ligand-Protected Metal Nanocluster Catalysts for CO2 Conversion through the Exposure of Undercoordinated Sites. Catalysts, 12(5), 505. https://doi.org/10.3390/catal12050505