
Synthetic Routes to Crystalline Complex Metal Alkyl Carbonates and Hydroxycarbonates via Sol–Gel Chemistry—Perspectives for Advanced Materials in Catalysis

 ,
,  ,
,  ,
,  and
and
Abstract
:
1. Introduction
2. Results and Discussion
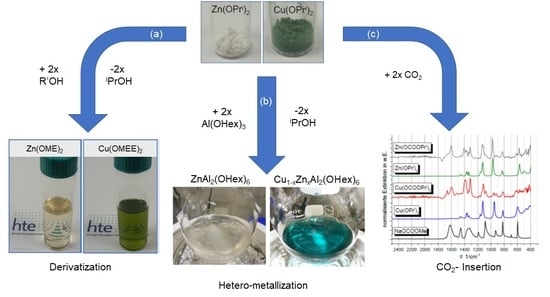
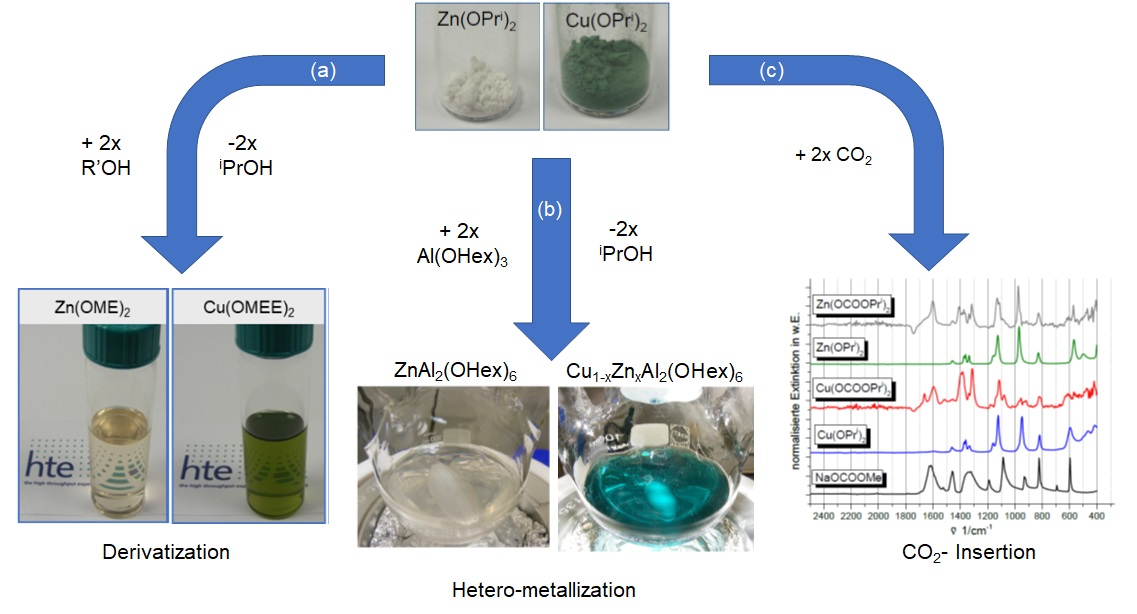
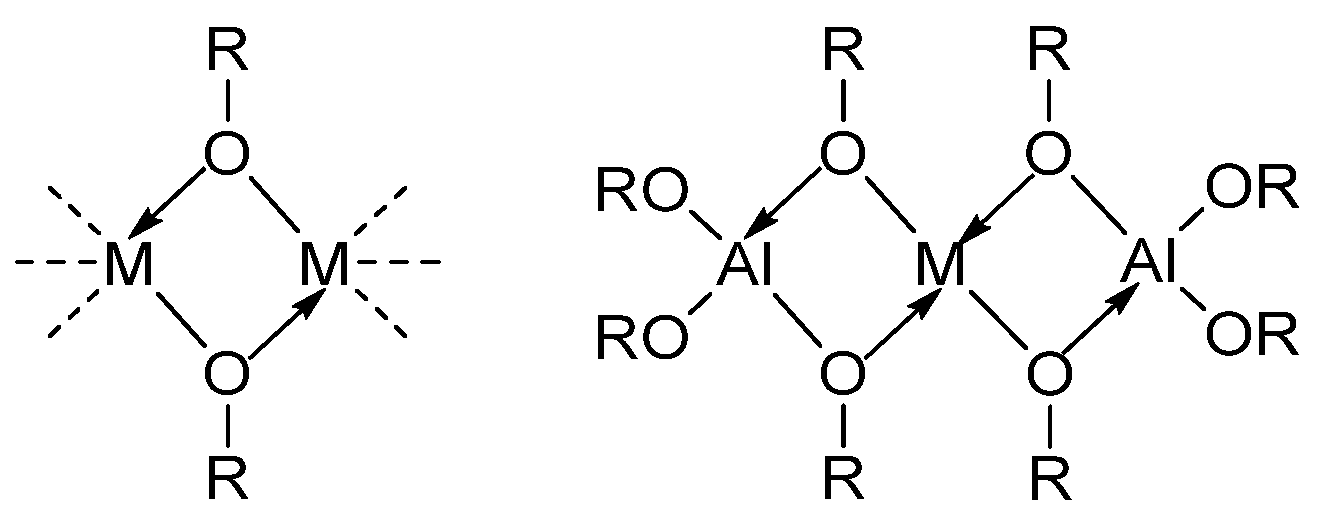
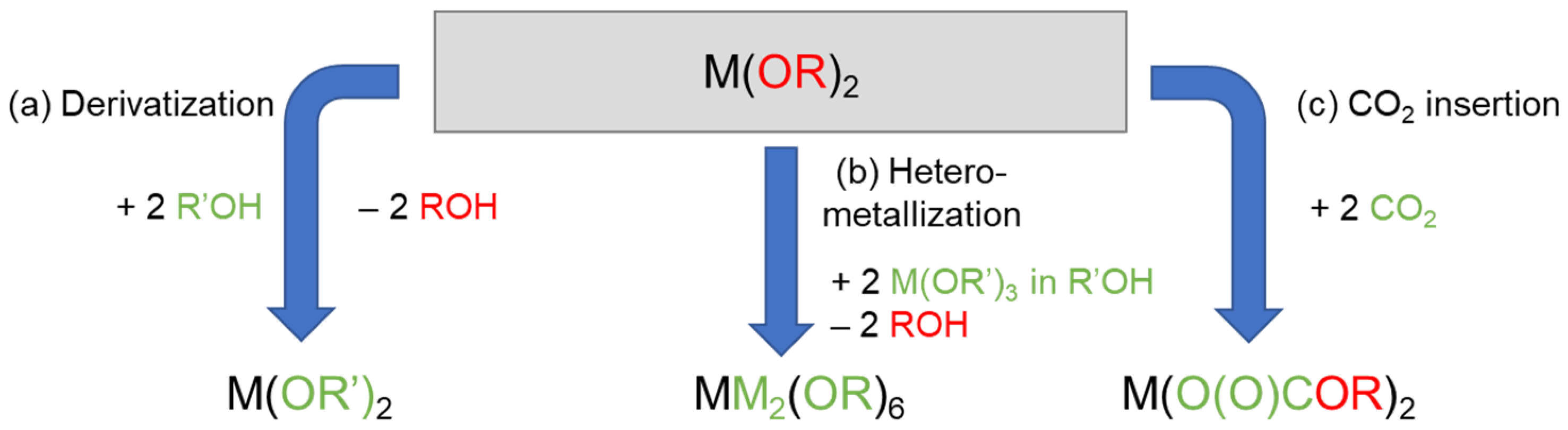
2.1. Solubilization Strategies of Metal Alkoxide Precursors
- (a)
- Derivatization (alcohol exchange)
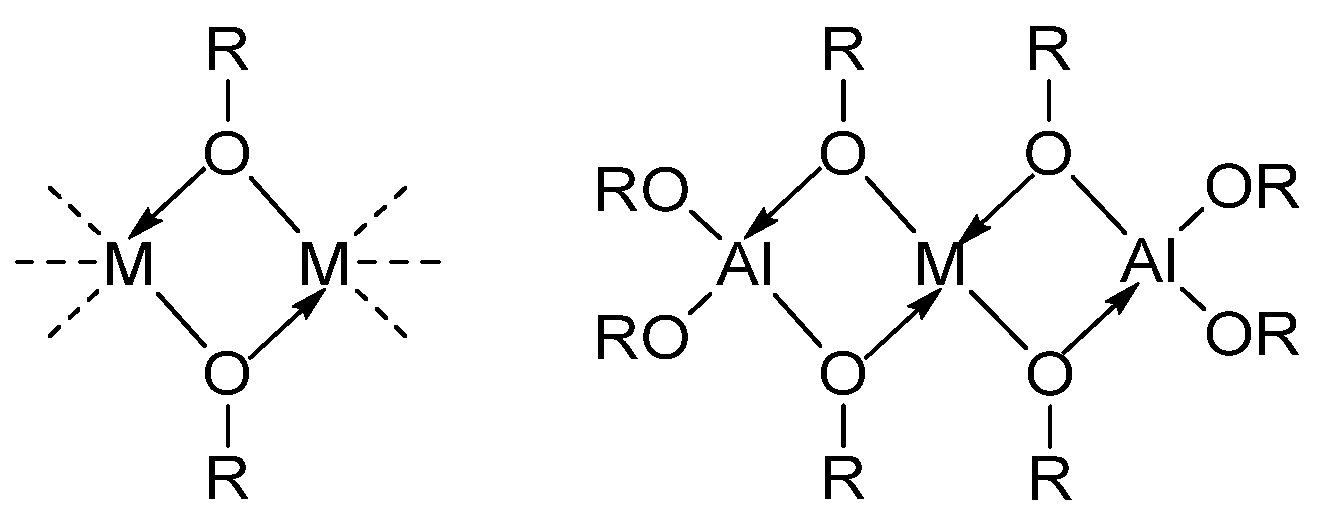
- (b)
- Hetero-metallization
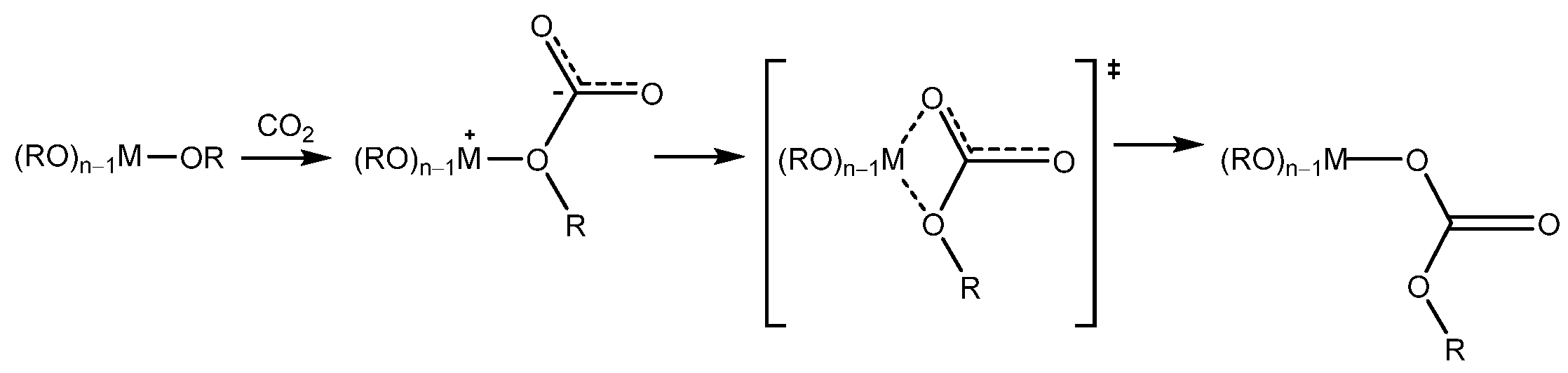
- (c)
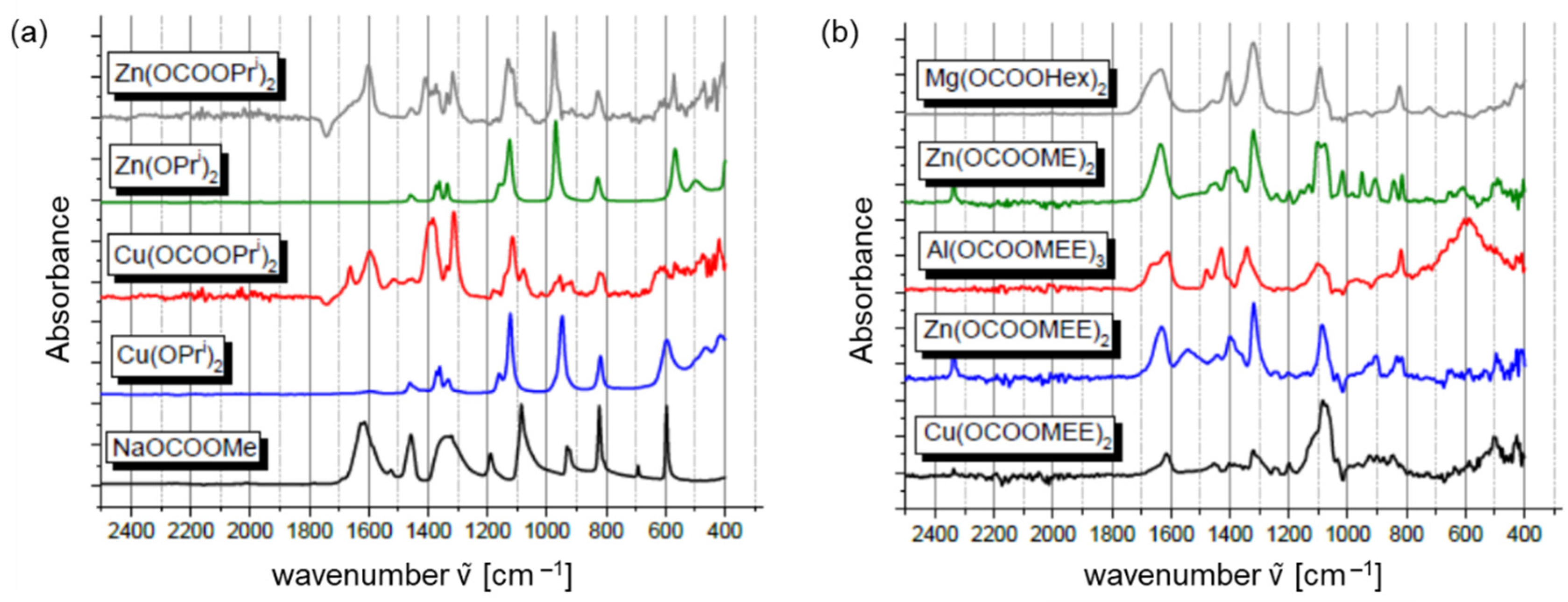
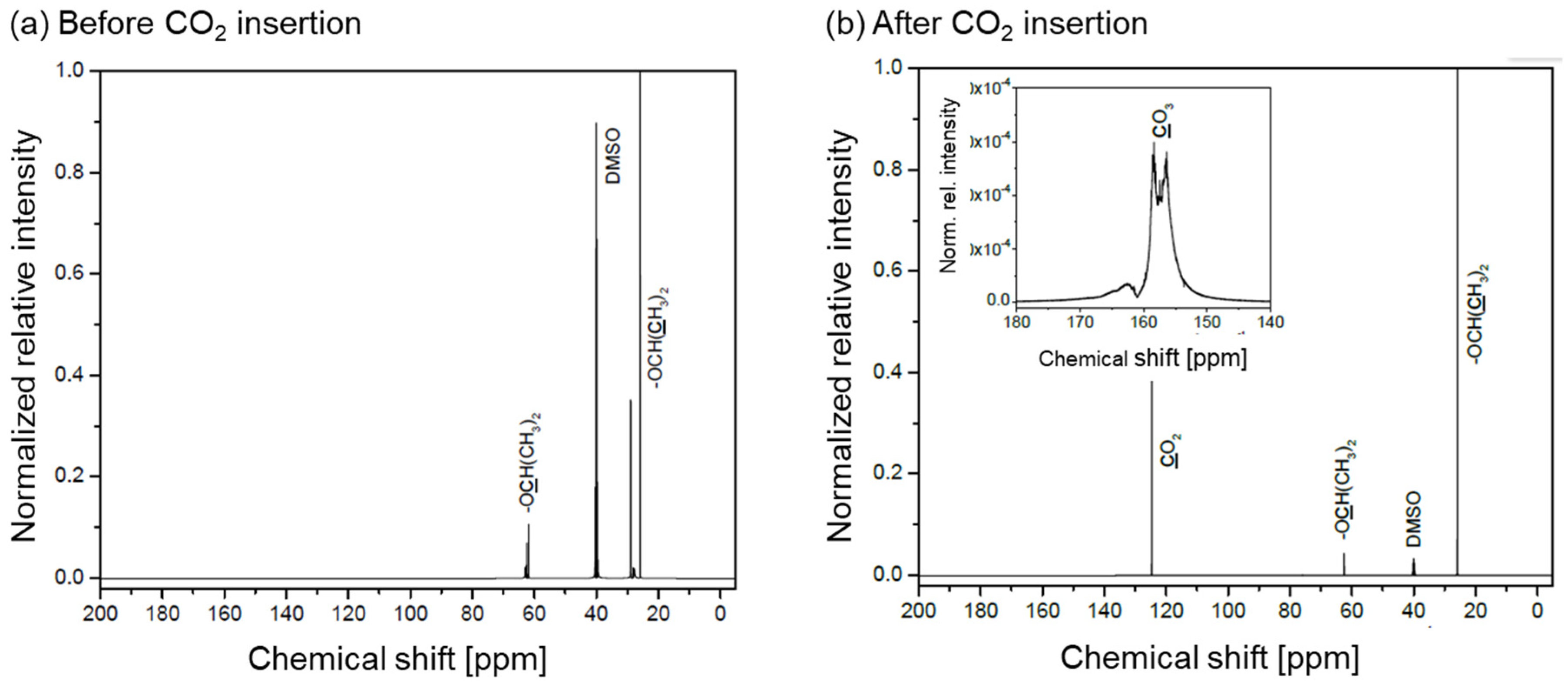
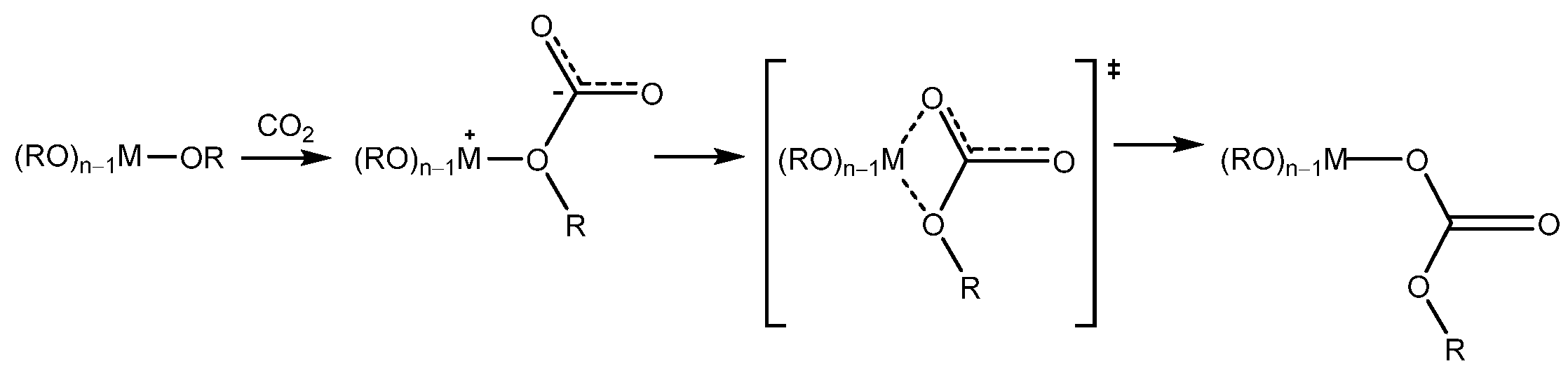
- CO2 insertion
- (a)
- Derivatization
- (b)
- Hetero-metallization
- (c)
- CO2 insertion
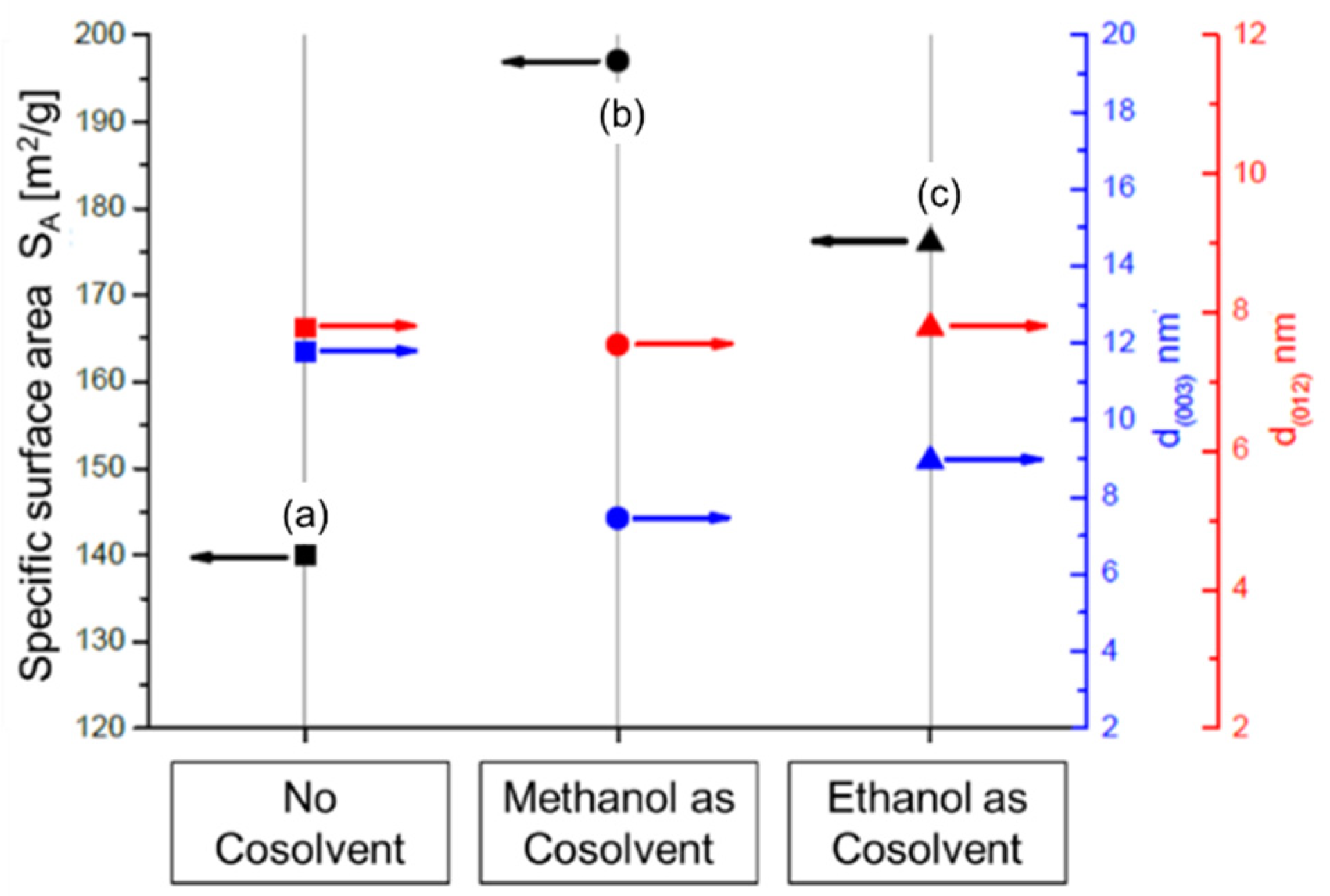
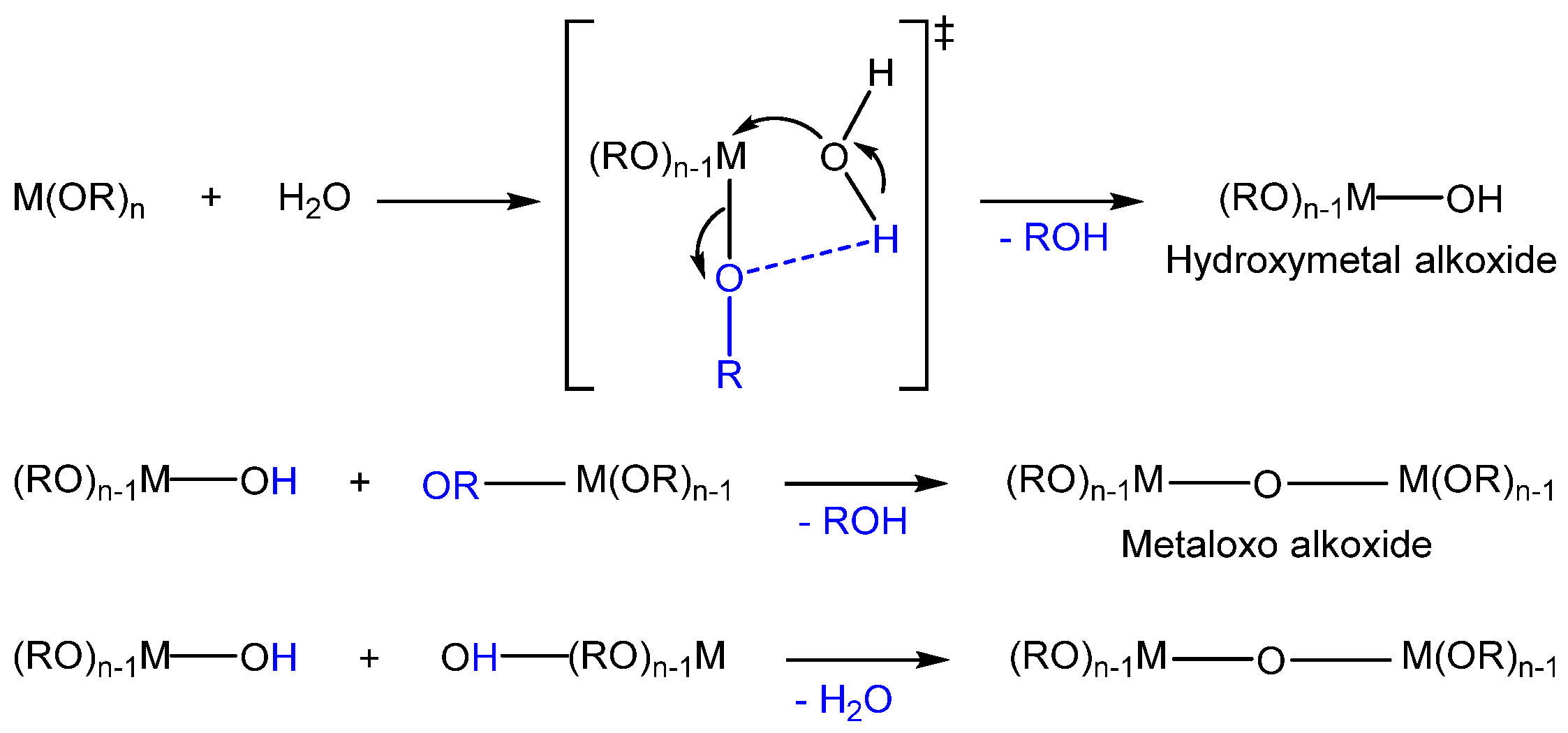
2.2. Hydrolysis of Metal Alkoxides
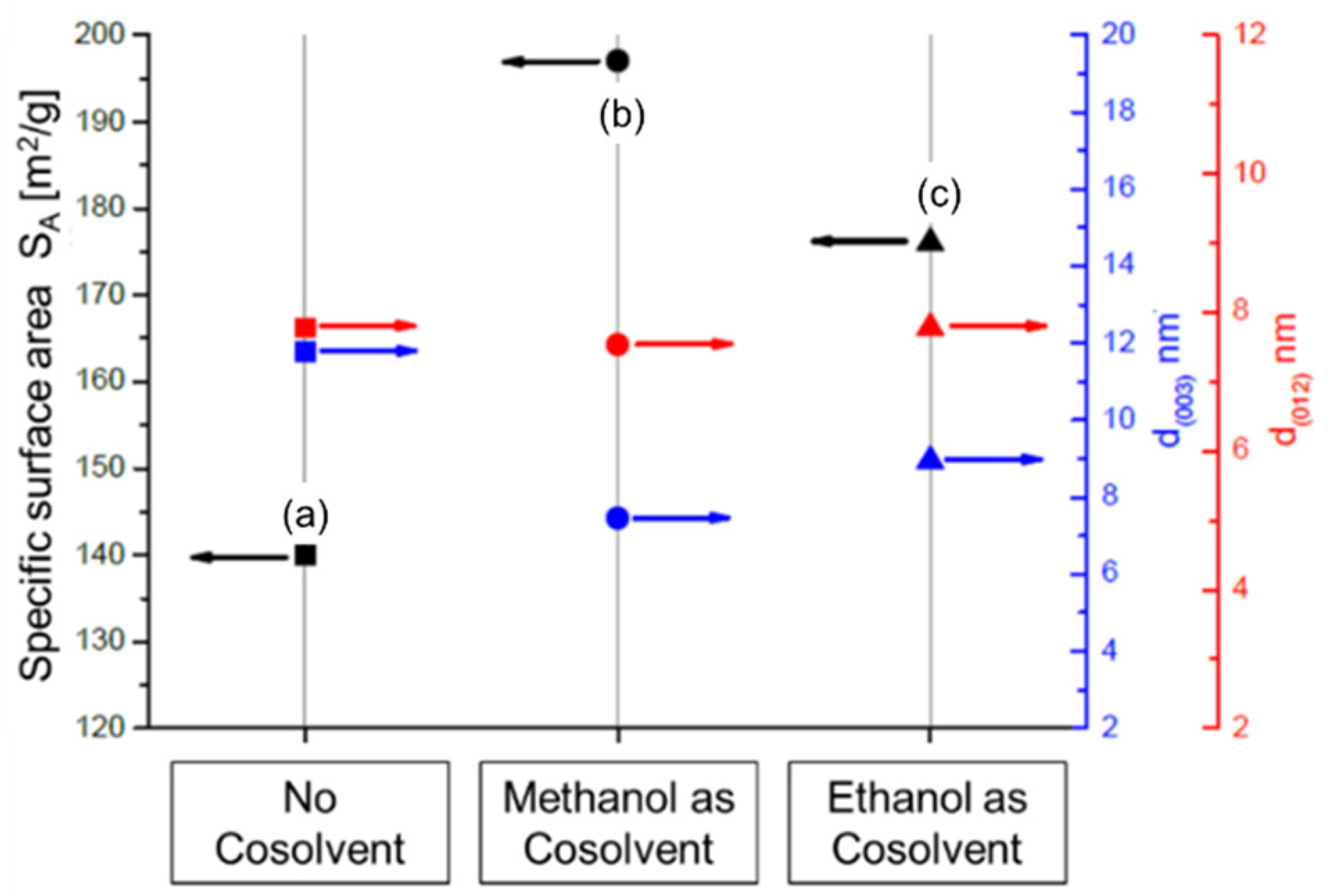
- Classic biphasic system. HTC crystallite appearance almost independent from solvent composition. Aspect ratio ≈ 1
- HTC crystallite formation is strongly dependent on the water content. With decreasing water content, an increased anisotropic character of the HTC crystallites can be observed. x[H2O] = 0.45, aspect ratio ≈ 3; x[H2O] = 0.25, aspect ratio ≈ 7
- Formation of film-like HTC morphologies with a high degree of anisotropy. x[H2O] = 0.05, aspect ratio ≈ 25
- Formation of gel-like structures, which cannot be assigned as HTC structures according to the characteristic [0 0 3] and [0 0 6] reflexes.
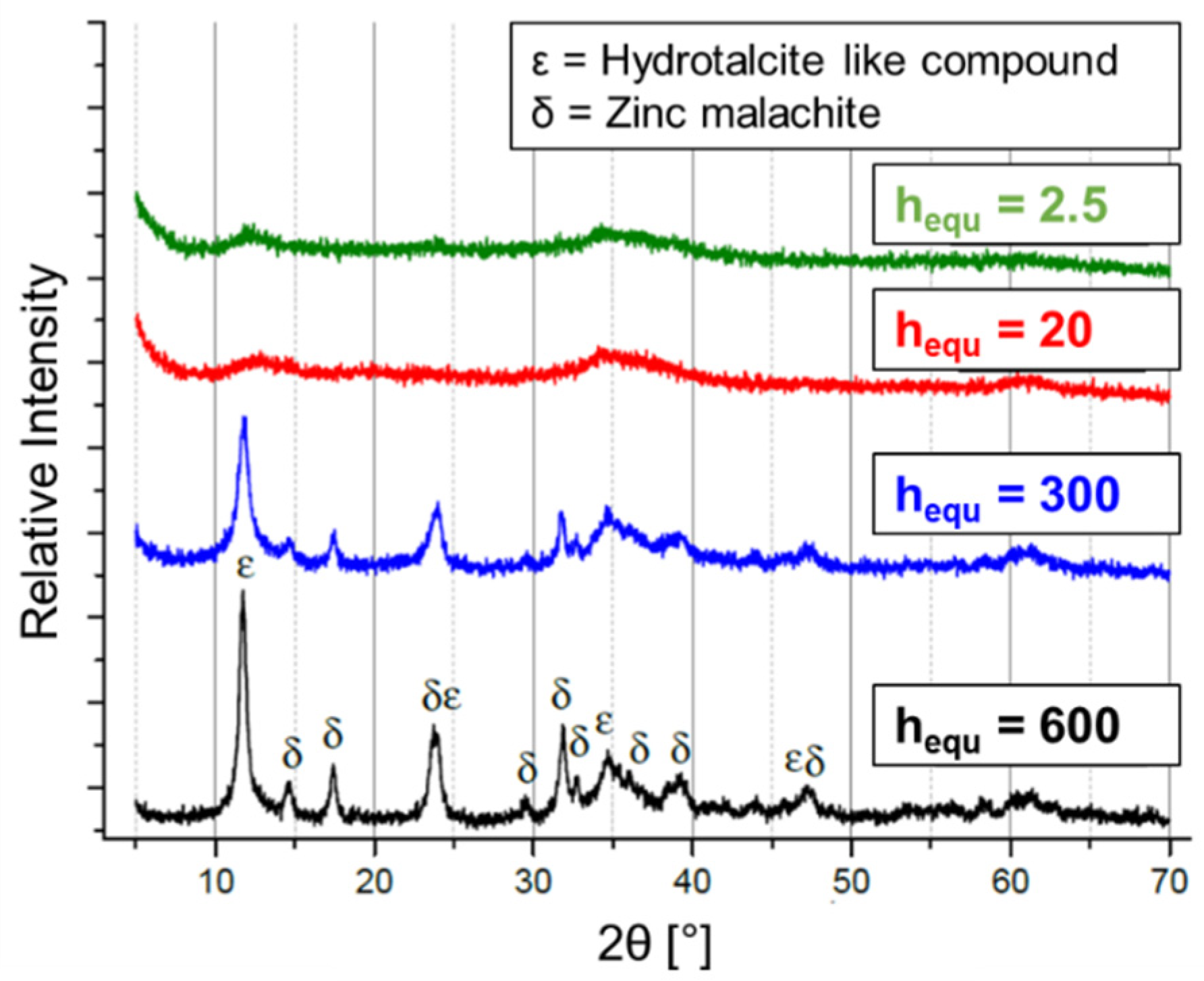
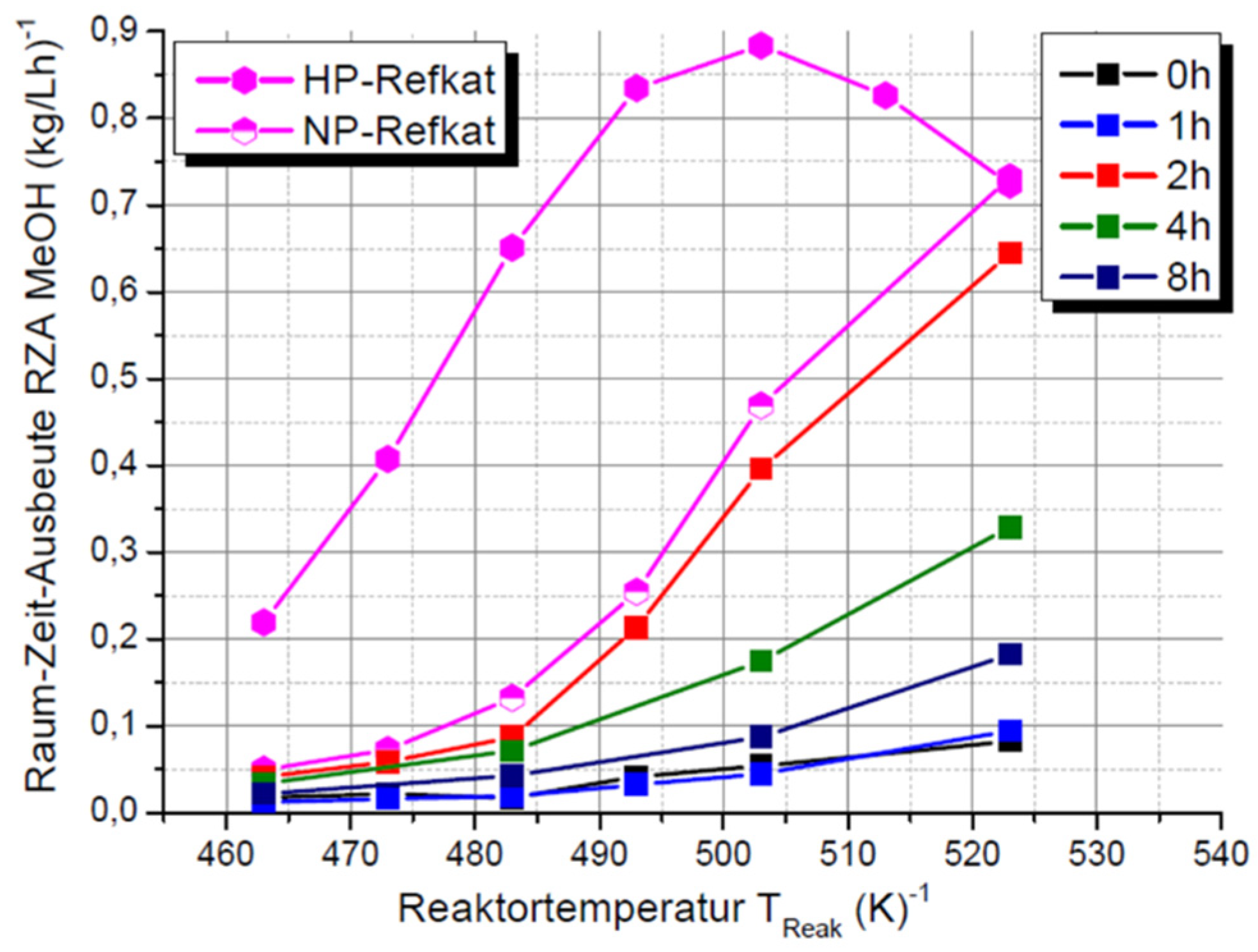
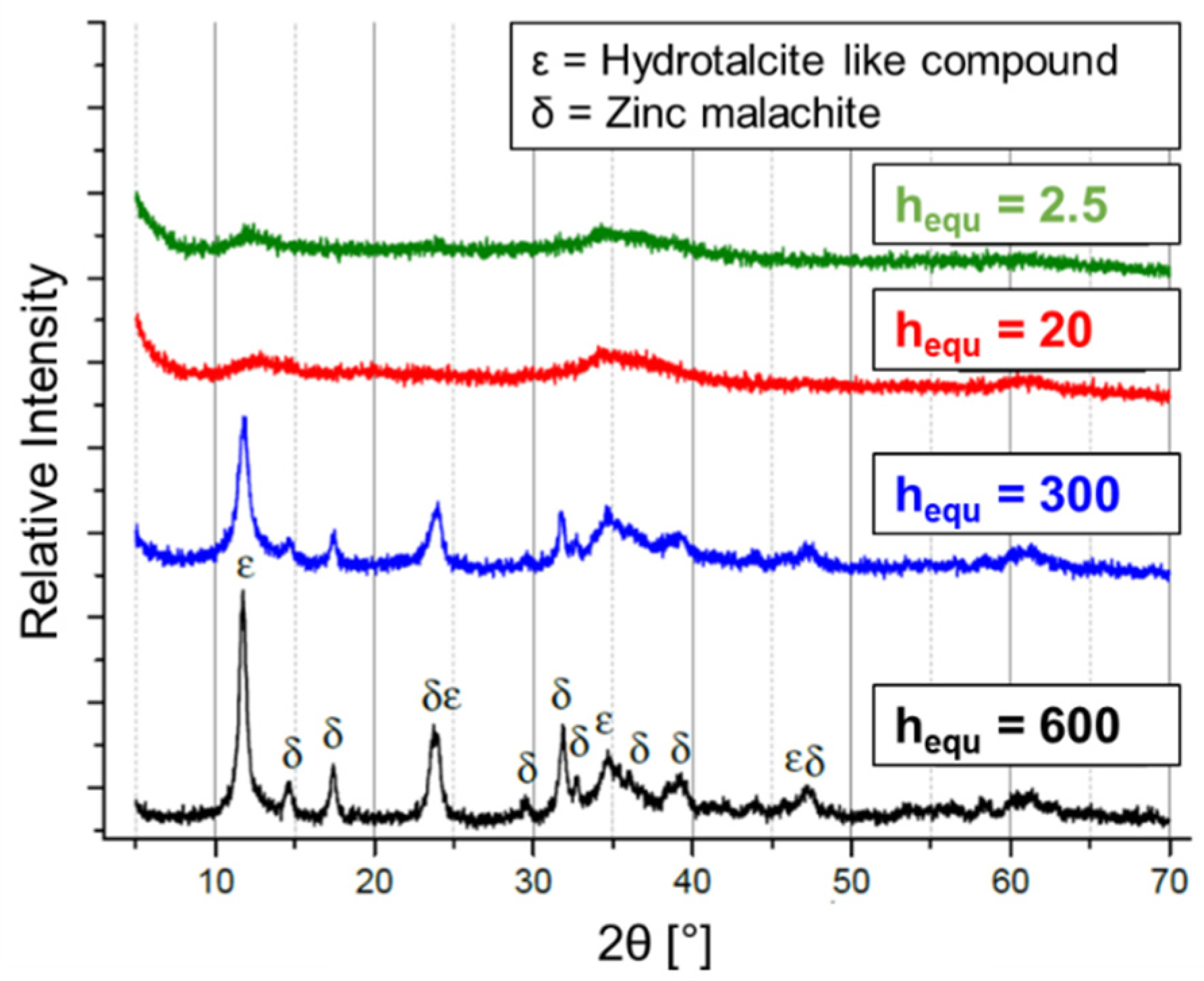
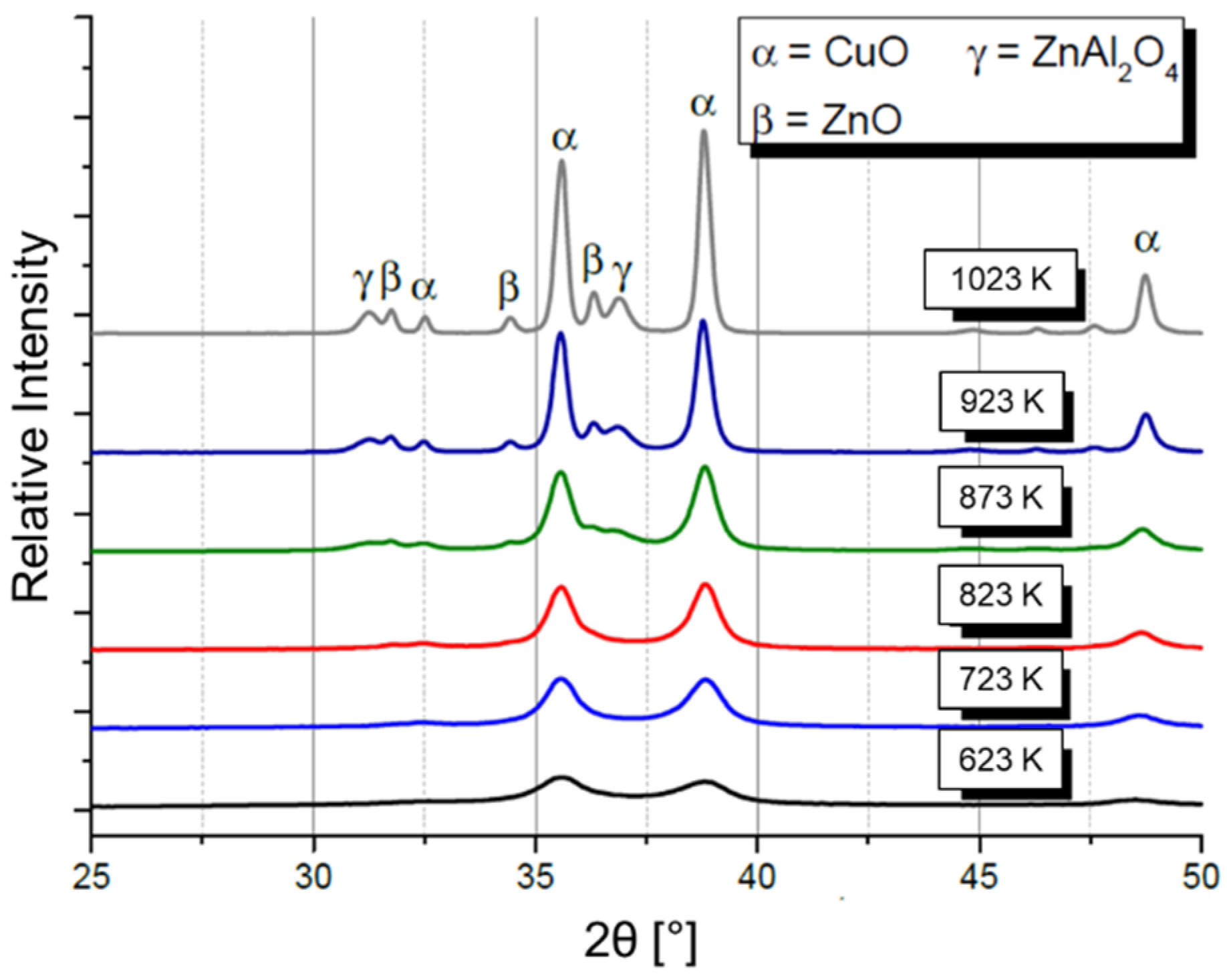
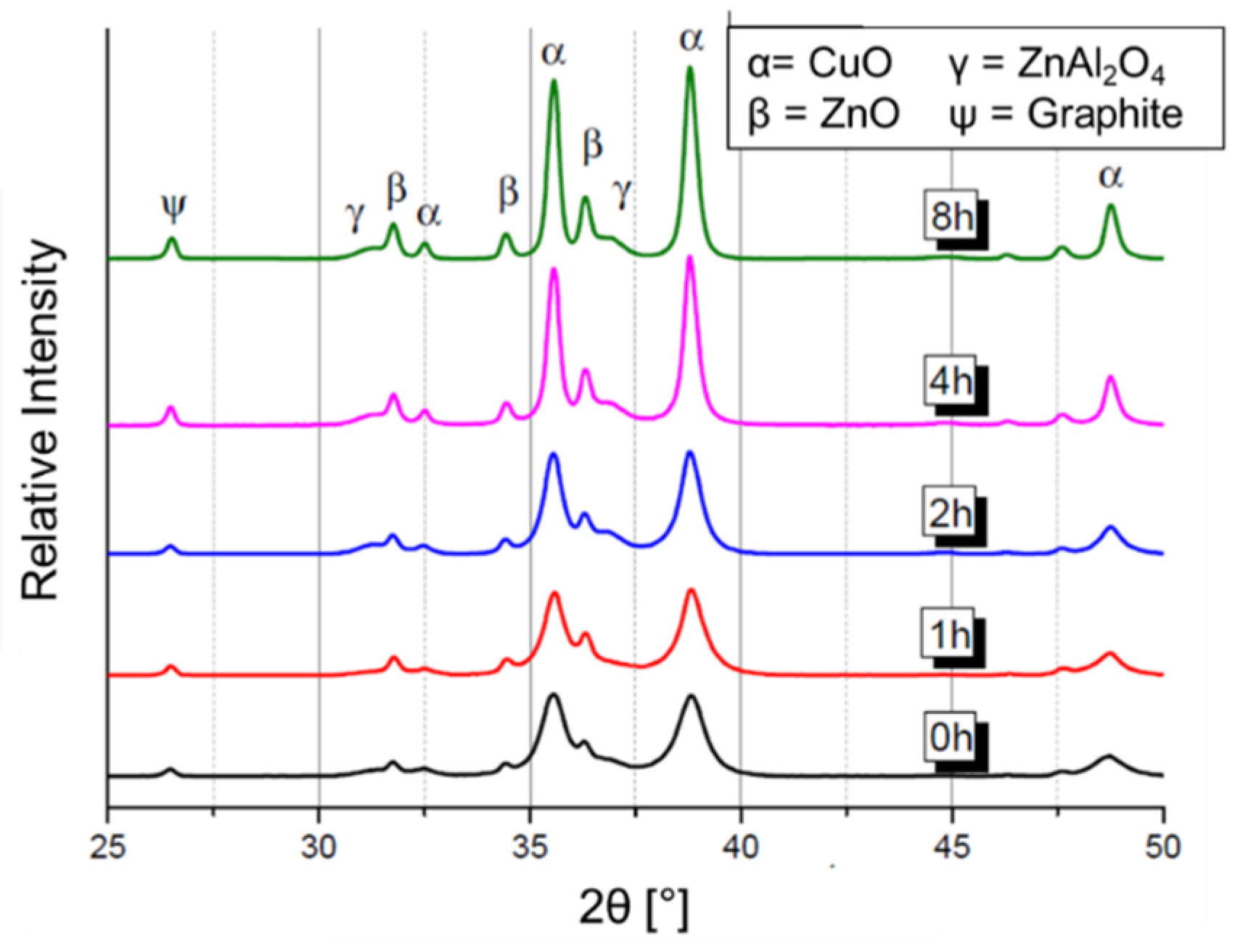
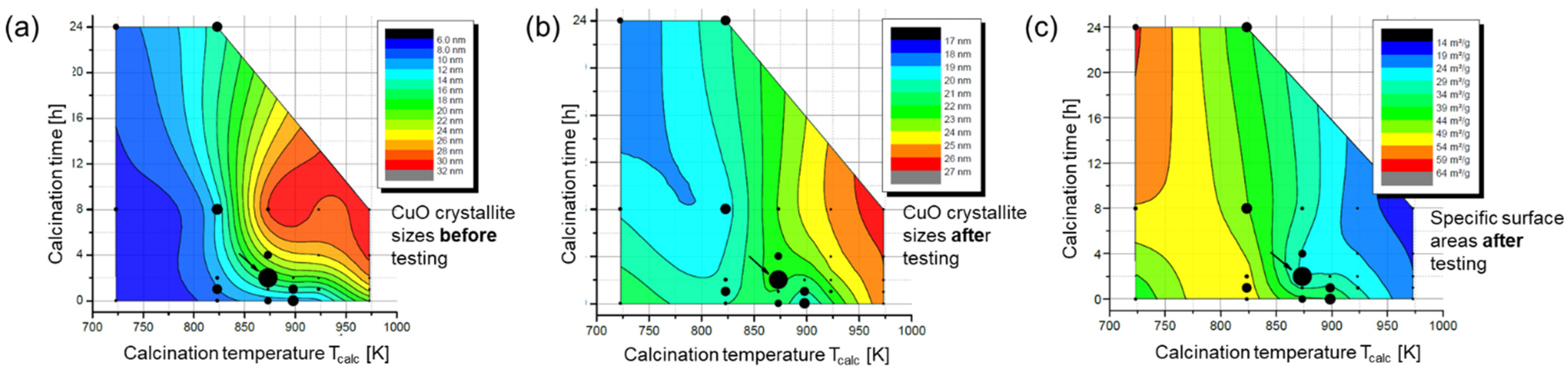
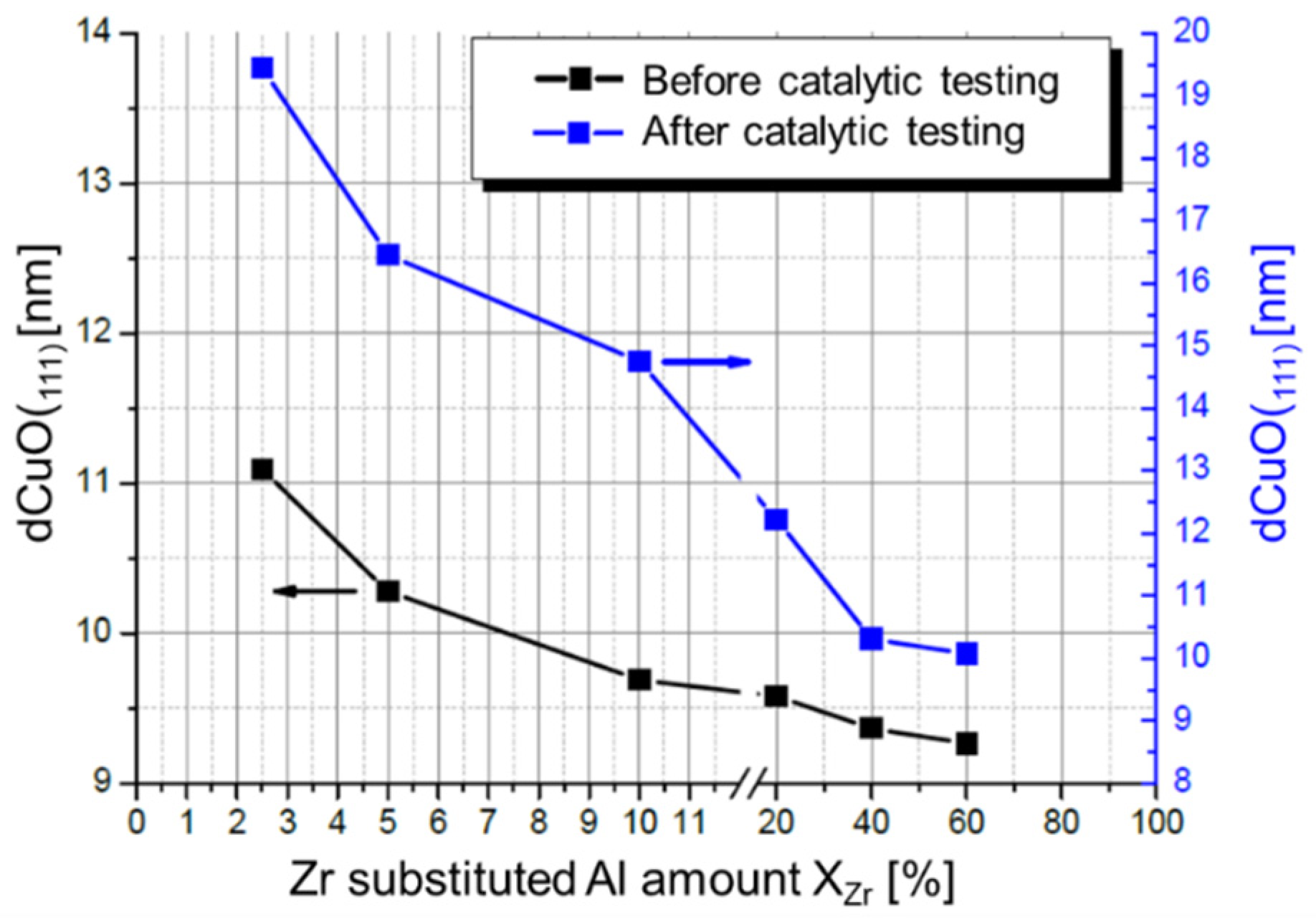
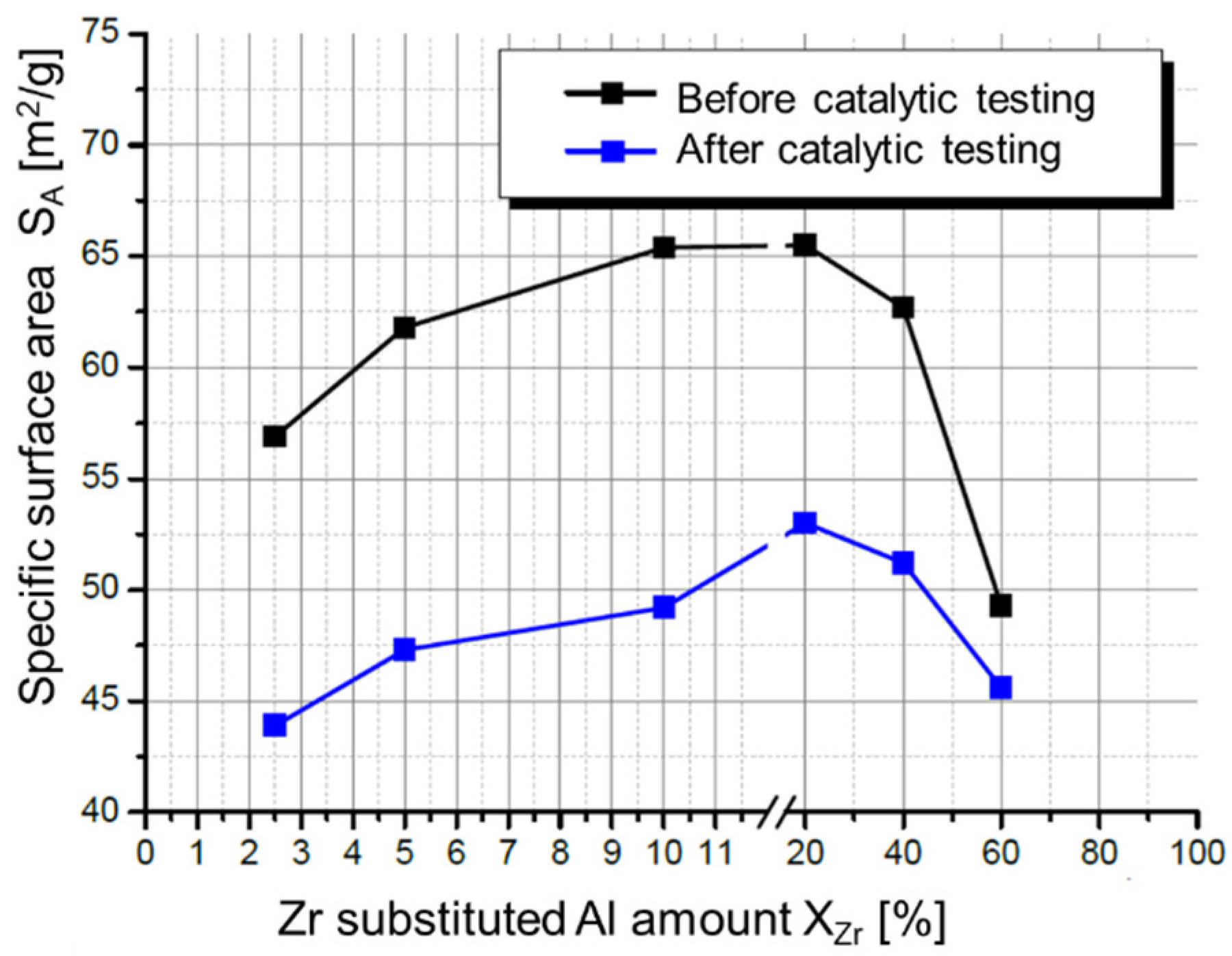
2.3. Hydrolysis of Metal Alkyl Carbonates for Cu/ZnO/Al2O3 Catalyst Synthesis
- (a)
- Cu/Zn-based complex carbonates
- (b)
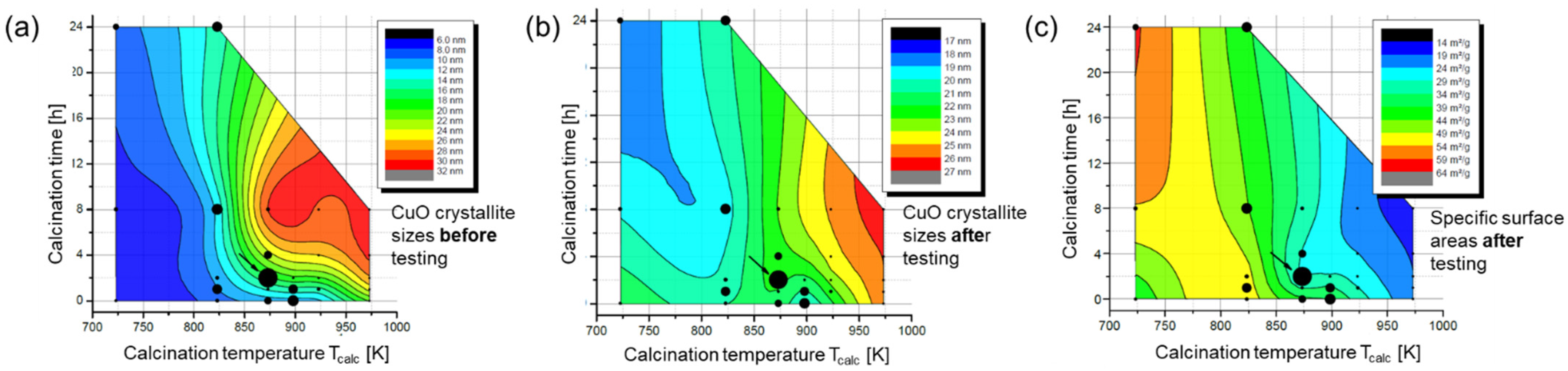
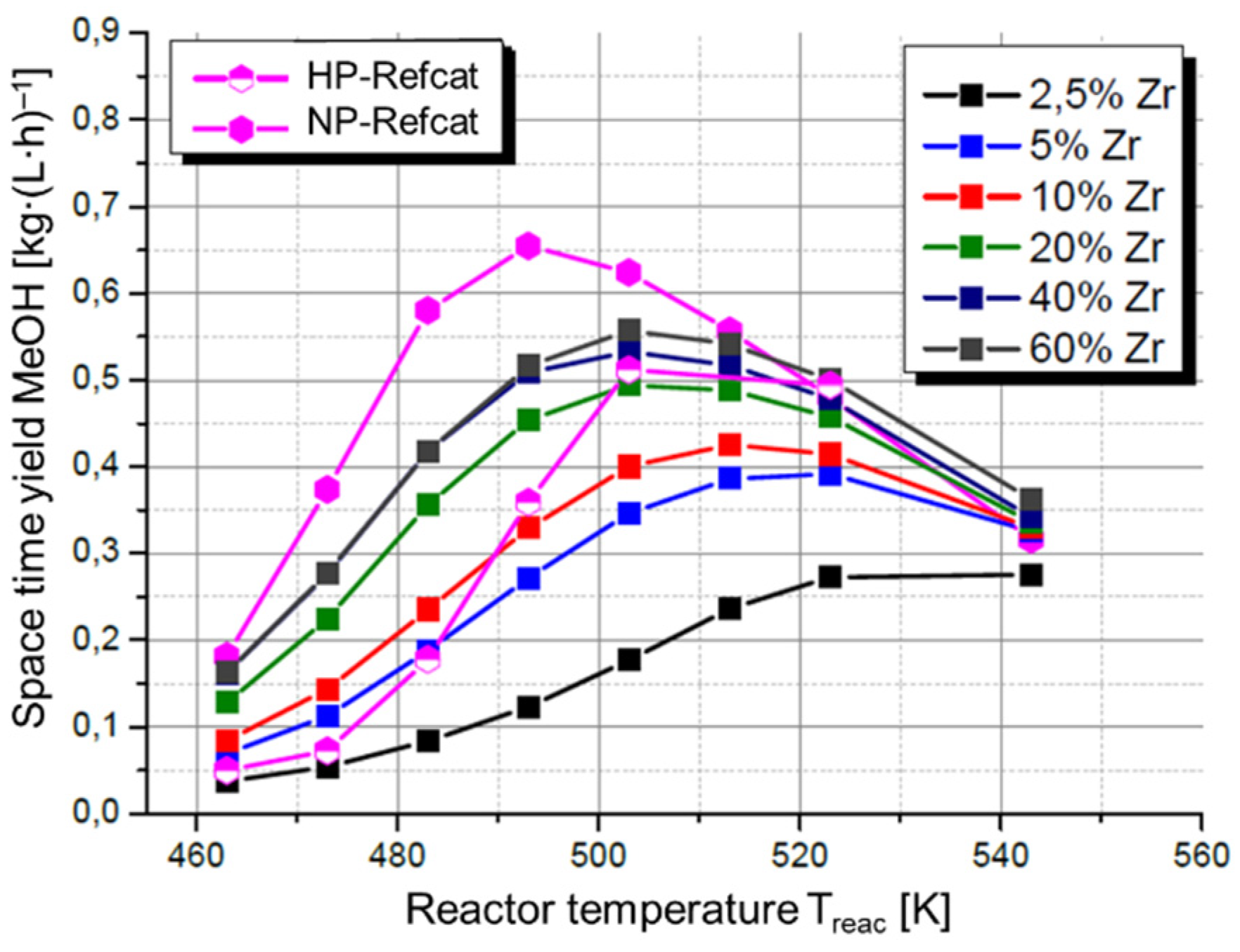
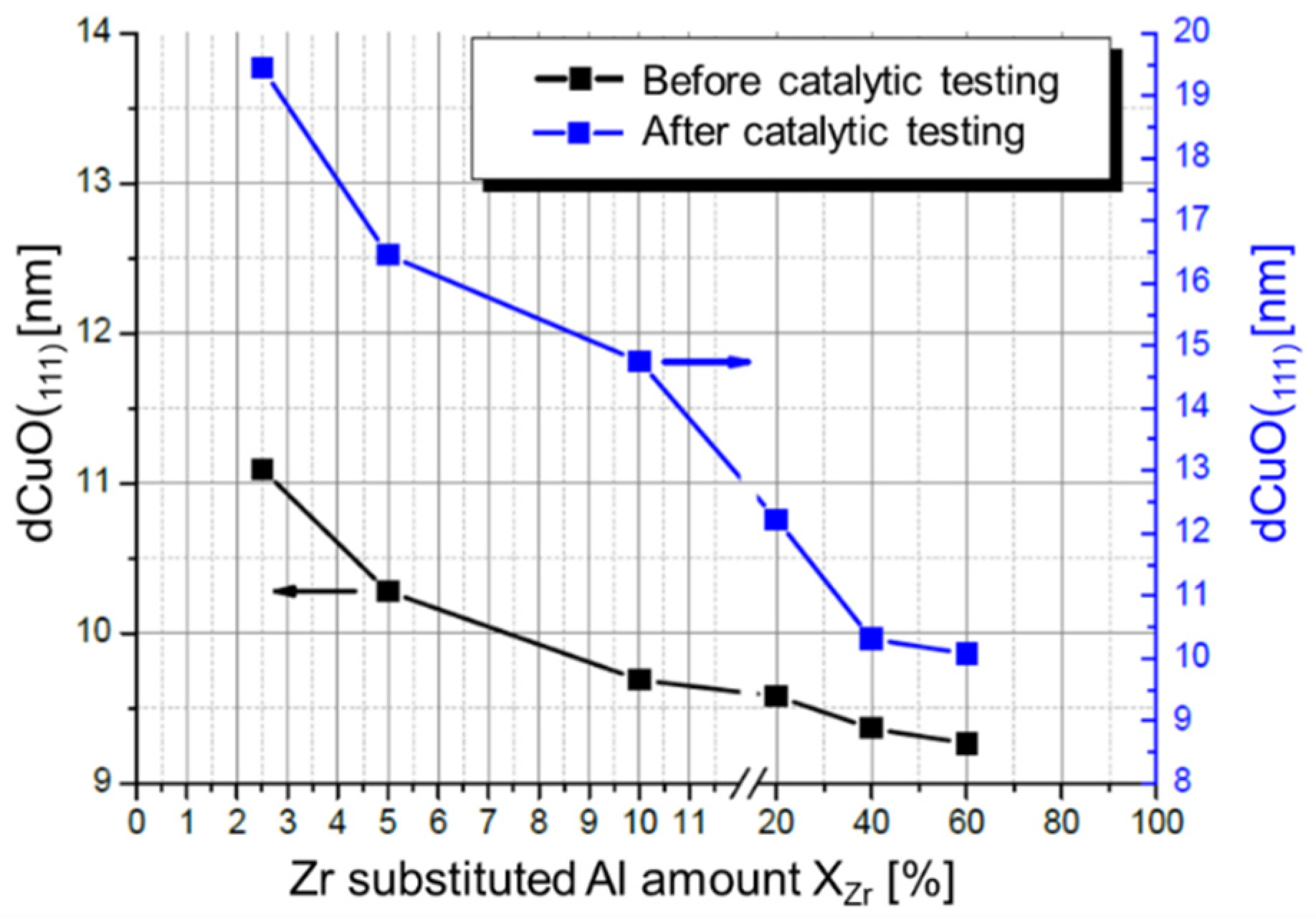
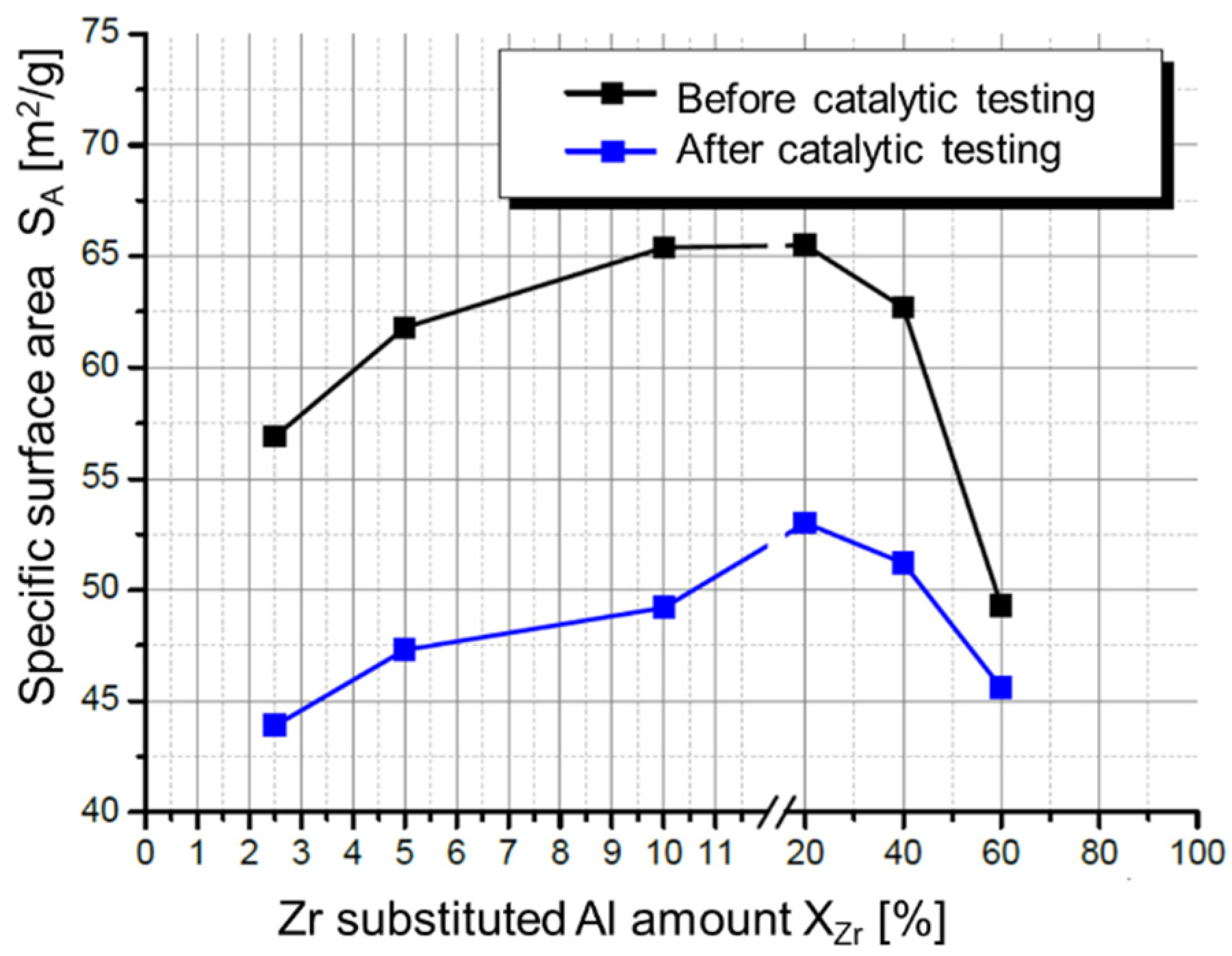
- Zr-promoted Cu/ZnO/Al2O3 catalysts
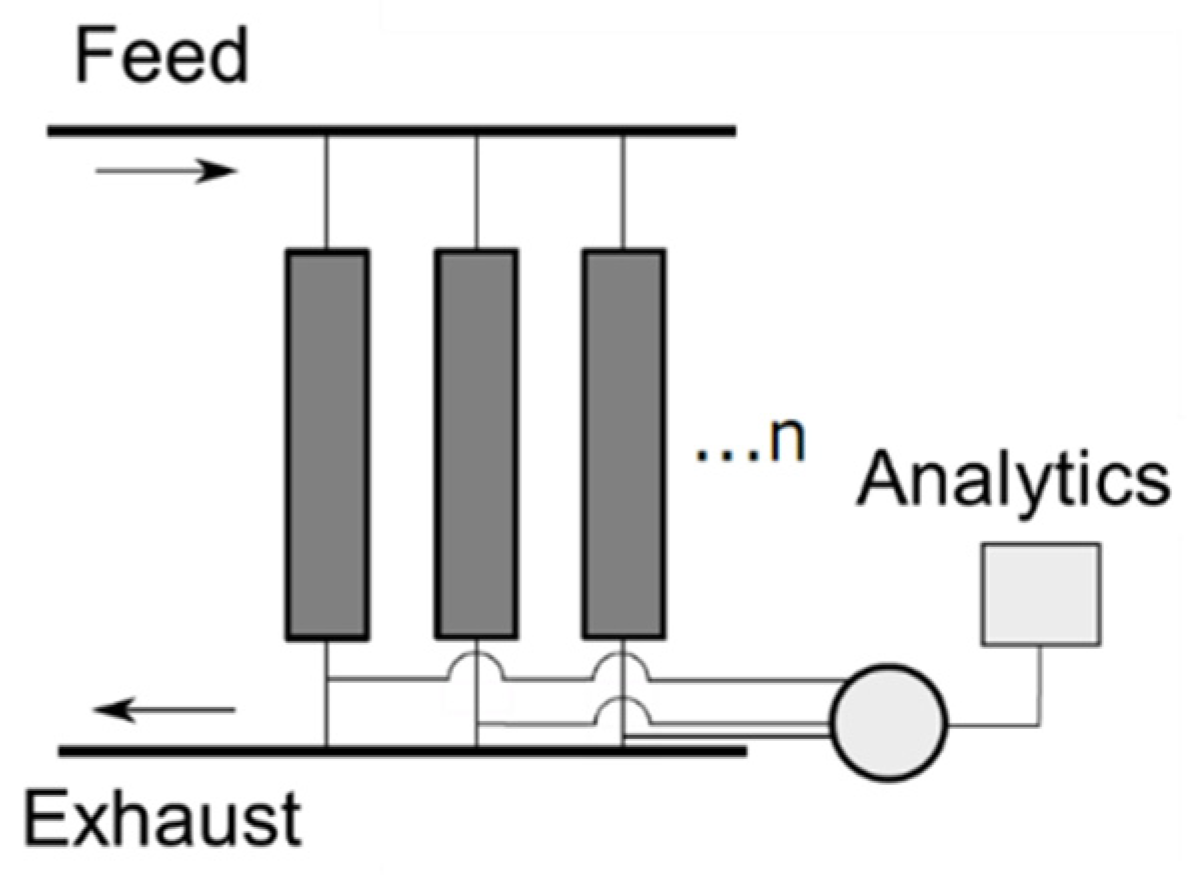
3. Materials and Methods
3.1. Analytical Methods
- Powder X-ray diffraction
- N2 physisorption
- ATR-IR spectroscopy
3.2. Experimental Details
- Preparation of Cu(OiPr)2
- Preparation of Zn(OiPr)2
- Preparation of Co(OiPr)3
- Preparation of Mn(OMe)2
- Preparation of Al(OMEE)3
- Preparation of Zr(OMEE)4
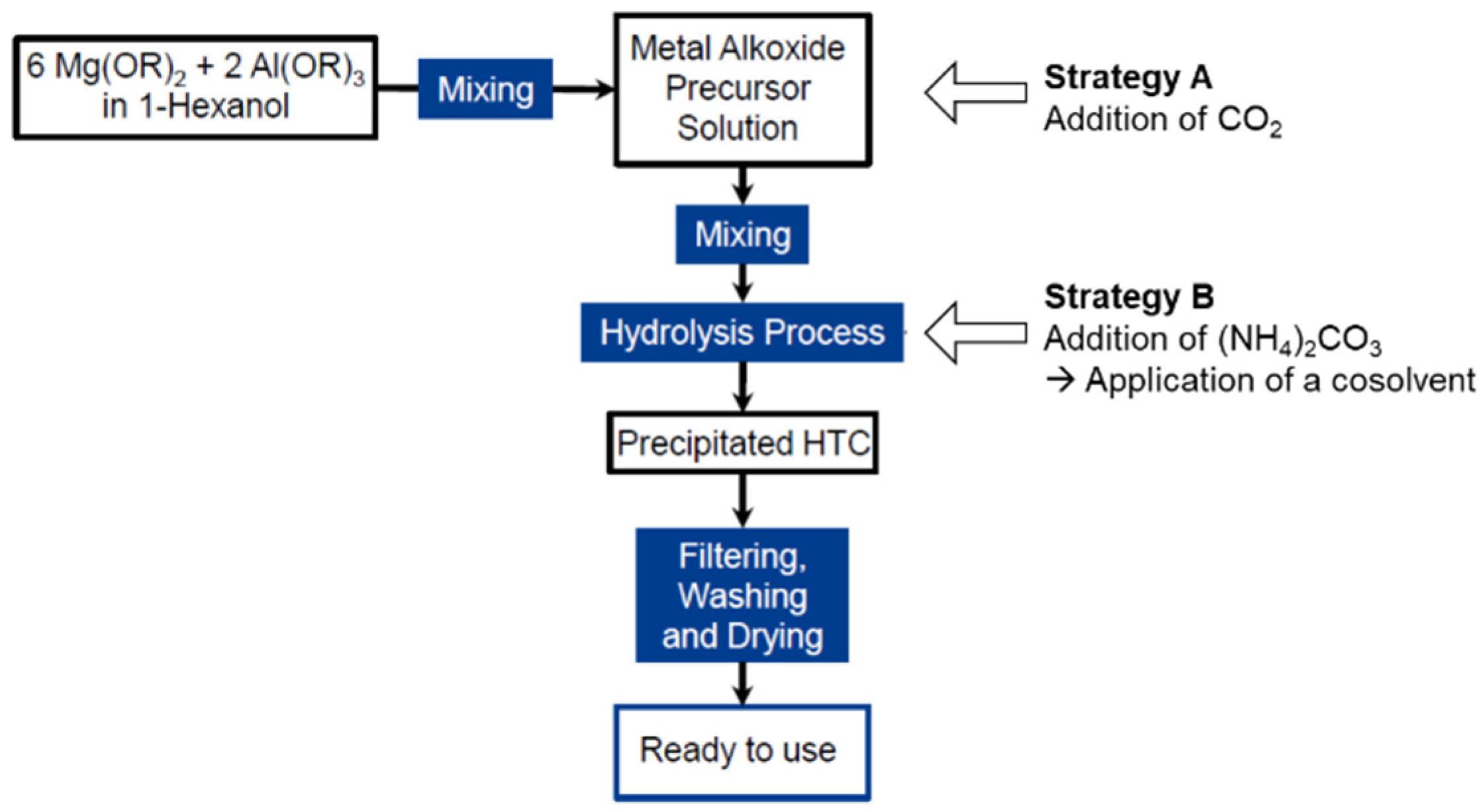
- General preparation of hydrotalcites
- Preparation of Mg6Al2(OH)18∙4 H2O
- Preparation of Mg6Al2(CO3)(OH)16∙4 H2O
- Preparation of Cu/ZnO/Al2O3 bulk catalysts
- Preparation of zirconium-promoted Cu/ZnO/Al2O3 bulk catalysts
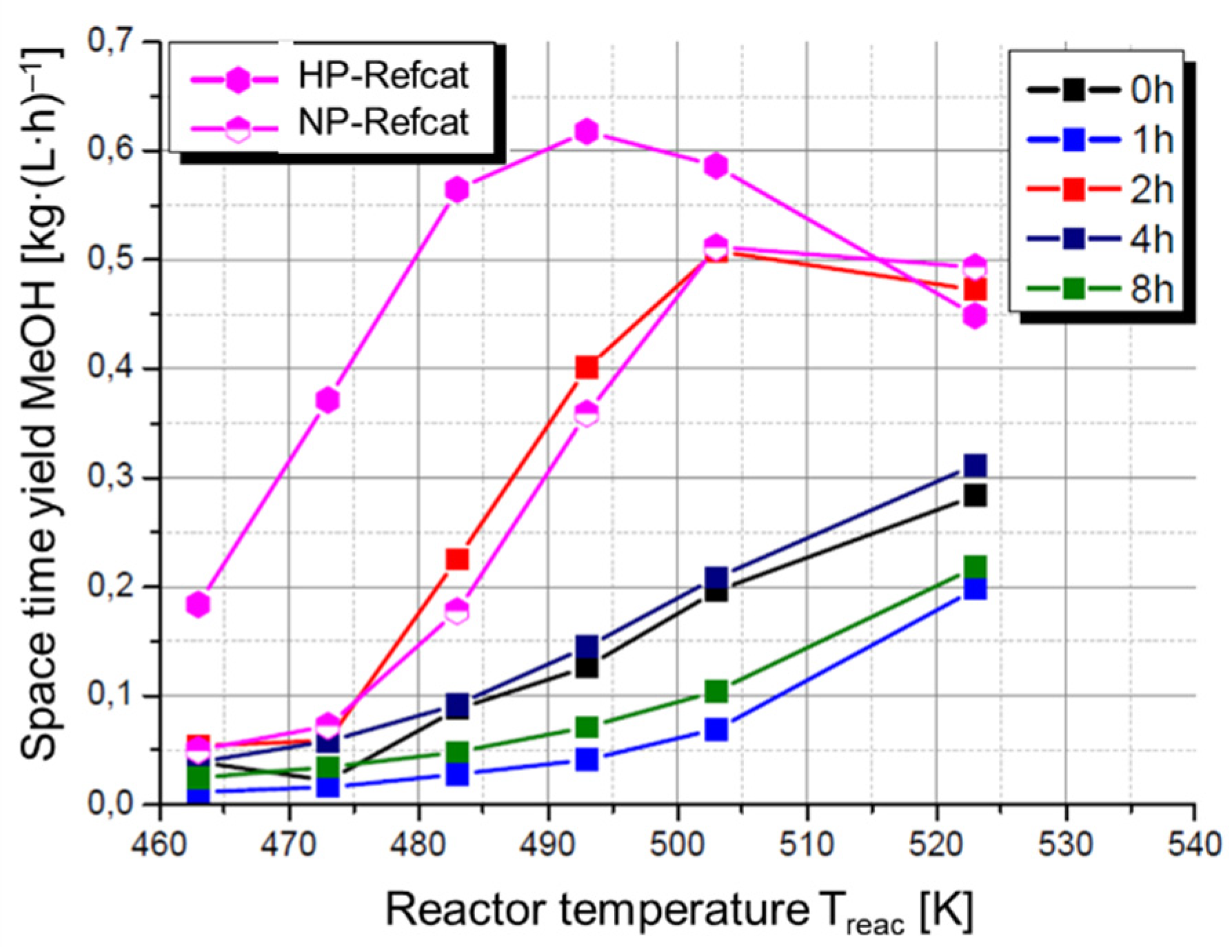
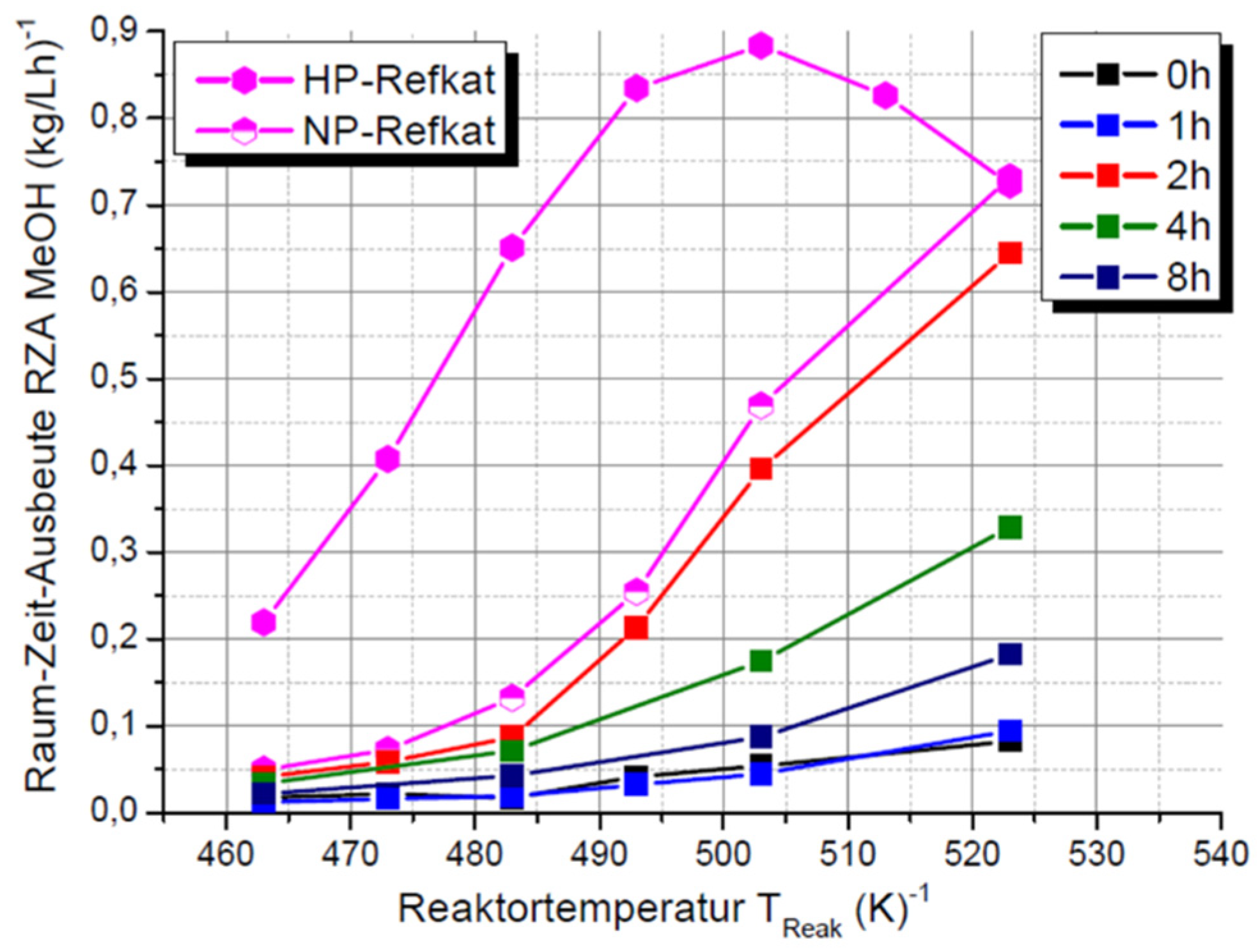
3.3. Catalytic Testing
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lu, H.; Wright, D.S.; Pike, S.D. The use of mixed-metal single source precursors for the synthesis of complex metal oxides. Chem. Commun. 2020, 56, 854–871. [Google Scholar] [CrossRef] [PubMed]
- Hanf, S.; Matthews, P.D.; Li, N.; Luo, H.K.; Wright, D.S. The Influence of Halides in Polyoxotitanate Cages; Dipole Moment, Splitting and Expansion of d-Orbitals and Electron-Electron Repulsion. Dalt. Trans. 2017, 46, 578–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatte, T.; Hussainov, M.; Paalo, M.; Part, M.; Talviste, R.; Kiisk, V.; Mändar, H.; Põhako, K.; Pehk, T.; Reivelt, K.; et al. Alkoxide-based precursors for direct drawing of metal oxide micro- and nanofibres. Sci. Technol. Adv. Mater. 2011, 12, 34412. [Google Scholar] [CrossRef] [PubMed]
- Kessler, V.G. The Synthesis and Solution Stability of Alkoxide Precursors. In Handbook of Sol-Gel Science and Technology: Processing, Characterization and Applications; Springer International Publishing: Berlin/Heidelberg, Germany, 2018; pp. 31–80. [Google Scholar]
- Turova, N.Y.; Turevskaya, E.P.; Kessler, V.G.; Yanovskaya, M.I. The Chemistry of Metal Alkoxides; Kluwer Academic Publishers Group: Drive Norwell, MA, USA, 2002. [Google Scholar]
- Turevskaya, E.P.; Yanovskaya, M.I.; Turova, N.Y. Preparation of Oxide Materials from Metal Alkoxides. In Inorganic Materials; Springer: Berlin/Heidelberg, Germany, 2000; pp. 260–270. [Google Scholar]
- Mishra, S.; Daniele, S. Molecular Engineering of Metal Alkoxides for Solution Phase Synthesis of High-Tech Metal Oxide Nanomaterials. Chem. Eur. J. 2020, 26, 9292–9303. [Google Scholar] [CrossRef]
- Emmert, T.P. Übergangsmetallalkoxide als Precursoren für nano-strukturierte Funktionsmaterialen. Ph.D. Dissertation, Leipzig University, Leipzig, Germany, 2016. [Google Scholar]
- Saini, A.; Jat, S.K.; Shekhawat, D.S.; Kumar, A.; Dhayal, V.; Agarwal, D.C. Oxime-modified aluminium(III) alkoxides: Potential precursors for γ-alumina nano-powders and optically transparent alumina film. Mater. Res. Bull. 2017, 93, 373–380. [Google Scholar] [CrossRef]
- Díez-Sierra, J.; López-Domínguez, P.; Rijckaert, H.; Rikel, M.; Hänisch, J.; Khan, M.Z.; Falter, M.; Bennewitz, J.; Huhtinen, H.; Schäfer, S.; et al. High Critical Current Density and Enhanced Pinning in Superconducting Films of YBa2Cu3O7-ΔNanocomposites with Embedded BaZrO3, BaHfO3, BaTiO3, and SrZrO3Nanocrystals. ACS Appl. Nano Mater. 2020, 3, 5542–5553. [Google Scholar] [CrossRef]
- Bäcker, M.; Bennewitz, J.; de Keukeleere, K.; de Roo, J.; van Driessche, I.; Falter, M.; Dominguez, P.L.; Meyer, A.; Rijackaert, H.; Schäfer, S.; et al. Process for Producing Nanoparticles. WO2020049019A1, 12 March 2020. [Google Scholar]
- Brinker, C.J.; Scherer, G.W. Sol-Gel Science: The Physics and Chemistry of Sol-Gel Processing; Elsevier Inc.: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Danks, A.E.; Hall, S.R.; Schnepp, Z. The Evolution of “sol-Gel” Chemistry as a Technique for Materials Synthesis. Mater. Horiz. 2016, 3, 91–112. [Google Scholar] [CrossRef] [Green Version]
- Clément, S.; Mehdi, A. Sol-Gel Chemistry: From Molecule to Functional Materials. Molecules 2020, 25, 2538. [Google Scholar] [CrossRef]
- Liu, X.; Iocozzia, J.; Wang, Y.; Cui, X.; Chen, Y.; Zhao, S.; Li, Z.; Lin, Z. Noble Metal-Metal Oxide Nanohybrids with Tailored Nanostructures for Efficient Solar Energy Conversion, Photocatalysis and Environmental Remediation. Energy Environ. Sci. 2017, 10, 402–434. [Google Scholar] [CrossRef]
- Ma, C.; Alvarado, J.; Xu, J.; Clément, R.J.; Kodur, M.; Tong, W.; Grey, C.P.; Meng, Y.S. Exploring Oxygen Activity in the High Energy P2-Type Na0.78Ni0.23Mn0.69O2 Cathode Material for Na-Ion Batteries. J. Am. Chem. Soc. 2017, 139, 4835–4845. [Google Scholar] [CrossRef] [Green Version]
- Mensinger, Z.L.; Gatlin, J.T.; Meyers, S.T.; Zakharov, L.N.; Keszler, D.A.; Johnson, D.W. Synthesis of Heterometallic Group 13 Nanoclusters and Inks for Oxide Thin-Film Transistors. Angew. Chem. Int. Ed. 2008, 47, 9484–9486. [Google Scholar] [CrossRef] [PubMed]
- Dubal, D.P.; Gomez-Romero, P. (Eds.) Metal Oxides in Supercapacitors, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Li, W.M.; Zhao, J.F.; Cao, L.P.; Hu, Z.; Huang, Q.Z.; Wang, X.C.; Liu, Y.; Zhao, G.Q.; Zhang, J.; Liu, Q.Q.; et al. Superconductivity in a unique type of copper oxide. Proc. Natl. Acad. Sci. USA 2019, 116, 12156–12160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrotra, R.C. Transition-Metal Alkoxides. Adv. Inorg. Chem. 1983, 26, 269–335. [Google Scholar] [CrossRef]
- Bradley, D. Metal Alkoxides and Dialkylamides. Adv. Inorg. Chem. 1972, 15, 259–322. [Google Scholar] [CrossRef]
- Bradley, D.C.; Mehrotra, R.C.; Wardlaw, W. Structural chemistry of the alkoxides. Part I. Amyloxides of silicon, titanium, and zirconium. J. Chem. Soc. 1952, 2027–2032. [Google Scholar] [CrossRef]
- Bradley, D.C.; Mehrotra, R.C.; Wardlaw, W. Structural chemistry of the alkoxides. Part II. Tertiary alkoxides of silicon, titanium, zirconium, and hafnium. J. Chem. Soc. 1952, 4204–4209. [Google Scholar] [CrossRef]
- Bradley, D.C.; Mehrotra, R.C.; Wardlaw, W. Structural chemistry of the alkoxides. Part III. Secondary alkoxides of silicon, titanium, and zirconium. J. Chem. Soc. 1952, 5020–5023. [Google Scholar] [CrossRef]
- Bradley, D.C.; Mehrotra, R.C.; Swanwick, J.D.; Wardlaw, W. 412. Structural chemistry of the alkoxides. Part IV. Normal alkoxides of silicon, titanium, and zirconium. J. Chem. Soc. 1953, 2025–2030. [Google Scholar] [CrossRef]
- Daniele, S.; Tcheboukov, D.; Pfalzgraf, L.G.H. Functional homo- and heterometallic alkoxides as precursors for sol–gel routes to transparent ZnGa2O4 coatings. J. Mater. Chem. 2002, 12, 2519–2524. [Google Scholar] [CrossRef]
- Caulton, K.G.; Pfalzgraf, L.G.H. Synthesis, Structural Principles, and Reactivity of Heterometailic Alkoxides. Chem. Rev. 1990, 90, 969–995. [Google Scholar] [CrossRef]
- Arkled, B. (Ed.) Double Metal Alkoxides/Heterometallic Alkoxides. In Silicon, Germanium, Tin and Lead Compounds; Metal Alkoxides, Diketonates and Carboxylates; Gelest Inc.: Morrisville, PA, USA, 1995. [Google Scholar]
- Livage, J.; Henry, M.; Sanchez, C. Sol-gel chemistry of transition metal oxides. Prog. Solid State Chem. 1988, 18, 259–341. [Google Scholar] [CrossRef]
- Hazari, N.; Heimann, J.E. Carbon Dioxide Insertion into Group 9 and 10 Metal-Element σ Bonds. Inorg. Chem. 2017, 56, 13655–13678. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, W.; Gattow, G. Über Chalkogenolate. LXII. Untersuchungen Über Halbester Der Kohlensäure 2. Darstellung Und Eigenschaften Der Monomethylkohlensäure. ZAAC 1973, 398, 198–206. [Google Scholar]
- Hidai, M.; Hikita, T.; Uchida, Y. Reactions of carbon dioixde with transition metal alkoxides. Chem. Lett. 1972, 1, 521–522. [Google Scholar] [CrossRef] [Green Version]
- Chisholm, M.H.; Cotton, F.A.; Extine, M.W.; Reichert, W.W. The molybdenum-molybdenum triple bond. 4. Insertion reactions of hexakis(alkoxy)dimolybdenum compounds with carbon dioxide and single-crystal X-ray structural characterization of bis(tert-butylcarbonato)tetrakis(tert-butoxy)dimolybdenum. J. Am. Chem. Soc. 2002, 100, 1727–1734. [Google Scholar] [CrossRef]
- Tsuda, T.; Saegusa, T. Reaction of cupric methoxide and carbon dioxide. Inorg. Chem. 1972, 11, 2561–2563. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Zhou, Z. New generation polymers: The role of metal alkoxides as catalysts in the production of polyoxygenates. J. Mater. Chem. 2004, 14, 3081–3092. [Google Scholar] [CrossRef]
- Polarz, S.; Pueyo, C.L.; Krumm, M. The molecular path to inorganic materials—Zinc oxide and beyond. Inorg. Chim. Acta 2010, 363, 4148–4157. [Google Scholar] [CrossRef]
- Prasad, S.; Kumar, V.; Kirubanandam, S.; Barhoum, A. Engineered Nanomaterials: Nanofabrication and Surface Functionalization. In Emerging Applications of Nanoparticles and Architectural Nanostructures: Current Prospects and Future Trends; Elsevier Inc.: Amsterdam, The Netherlands, 2018; pp. 305–340. [Google Scholar]
- Neto, V.D.O.S.; Freire, T.M.; Saraiva, G.D.; Muniz, C.R.; Cunha, M.S.; Fechine, P.B.A.; Nascimento, R.F.D. Water Treatment Devices Based on Zero-Valent Metal and Metal Oxide Nanomaterials. In Nanomaterials Applications for Environmental Matrices: Water, Soil and Air; Elsevier: Amsterdam, The Netherlands, 2019; pp. 187–225. [Google Scholar]
- Teixeira, G.F.; Lustosa, G.M.; Zanetti, S.M.; Zaghete, M.A. Chemical synthesis and epitaxial growth methods for the preparation of ferroelectric ceramics and thin films. In Magnetic, Ferroelectric, and Multiferroic Metal Oxides; Elsevier: Amsterdam, The Netherlands, 2018; pp. 121–137. [Google Scholar]
- Abou Neel, E.A.; Salih, V.; Knowles, J.C. Phosphate-Based Glasses. In Comprehensive Biomaterials II; Elsevier: Amsterdam, The Netherlands, 2017; pp. 392–405. [Google Scholar]
- Opuchovic, O.; Kareiva, A. Sol–Gel Synthesis and Characterization of Iron-Containing Garnets. In Biocompatible Hybrid Oxide Nanoparticles for Human Health; Elsevier: Amsterdam, The Netherlands, 2019; pp. 233–261. [Google Scholar]
- Lu, Y.; Dong, W.; Ding, J.; Wang, W.; Wang, A. Hydroxyapatite Nanomaterials: Synthesis, Properties, and Functional Applications. In Nanomaterials from Clay Minerals: A New Approach to Green Functional Materials; Elsevier: Amsterdam, The Netherlands, 2019; pp. 485–536. [Google Scholar]
- Hubert-Pfalzgraf, L.G. To What Extent Can Design of Molecular Precursors Control the Preparation of High Tech Oxides? J. Mater. Chem. 2004, 14, 3113–3123. [Google Scholar] [CrossRef]
- Kessler, V.G. Molecular structure design and synthetic approaches to the heterometallic alkoxide complexes (soft chemistry approach to inorganic materials by the eyes of a crystallographer). Chem. Commun. 2003, 1213–1222. [Google Scholar] [CrossRef]
- Hubert-Pfalzgraf, L.G. Some trends in the design of homo- and heterometallic molecular precursors of high-tech oxides. Inorg. Chem. Commun. 2003, 6, 102–120. [Google Scholar] [CrossRef]
- Choi, J.-C.; Sakakura, T.; Sako, T. Reaction of Dialkyltin Methoxide with Carbon Dioxide Relevant to the Mechanism of Catalytic Carbonate Synthesis. J. Am. Chem. Soc. 1999, 121, 3793–3794. [Google Scholar] [CrossRef]
- Vaccari, A. Preparation and catalytic properties of cationic and anionic clays. Catal. Today 1998, 41, 53–71. [Google Scholar] [CrossRef]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-Type Anion Clays: Preparation, Properties and Applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Hochstetter, C. Untersuchung über die Zusammensetzung einiger Mineralien. J. Prakt. Chem. 1842, 27, 375–378. [Google Scholar] [CrossRef]
- Bertau, M.; Offermanns, H.; Plass, L.; Schmidt, F.; Wernicke, H.-J. (Eds.) Methanol: The Basic Chemical and Energy Feedstock of the Future; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Ott, J.; Gronemann, V.; Pontzen, F.; Fiedler, E.; Grossmann, G.; Kersebohm, D.B.; Weiss, G.; Witte, C. “Methanol,” in: Ullmann’s Encyclopedia of Industrial Chemistry Enhanced Reader. Ullmann’s Encycl. Ind. Chem. 2012. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kühl, S.; Hävecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.L.; et al. The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Laudenschleger, D.; Ruland, H.; Muhler, M. Identifying the nature of the active sites in methanol synthesis over Cu/ZnO/Al2O3 catalysts. Nat. Commun. 2020, 11, 3898. [Google Scholar] [CrossRef]
- Grabow, L.C.; Mavrikakis, M. Mechanism of Methanol Synthesis on Cu through CO2 and COHydrogenation. ACS Catal. 2011, 1, 365–384. [Google Scholar] [CrossRef]
- Studt, F.; Behrens, M.; Kunkes, E.L.; Thomas, N.; Zander, S.; Tarasov, A.; Schumann, J.; Frei, E.; Varley, J.B.; Abild-Pedersen, F.; et al. The Mechanism of CO and CO2 Hydrogenation to Methanol over Cu-Based Catalysts. ChemCatChem 2015, 7, 1105–1111. [Google Scholar] [CrossRef] [Green Version]
- Baltes, C.; Vukojevic, S.; Schuth, F. Correlations between synthesis, precursor, and catalyst structure and activity of a large set of CuO/ZnO/Al2O3 catalysts for methanol synthesis. J. Catal. 2008, 258, 334–344. [Google Scholar] [CrossRef]
- Spencer, M. Precursors of copper/zinc oxide catalysts. Catal. Lett. 2000, 66, 255–257. [Google Scholar] [CrossRef]
- Behrens, M.; Brennecke, D.; Girgsdies, F.; Kißner, S.; Trunschke, A.; Nasrudin, N.; Zakaria, S.; Idris, N.F.; Hamid, S.B.A.; Kniep, B.; et al. Understanding the Complexity of a Catalyst Synthesis: Co-Precipitation of Mixed Cu,Zn,Al Hydroxycarbonate Precursors for Cu/ZnO/Al2O3 Catalysts Investigated by Titration Experiments. Appl. Catal. A Gen. 2011, 392, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Behrens, M.; Kasatkin, I.; Kühl, S.; Weinberg, G. Phase-Pure Cu,Zn,Al Hydrotalcite-like Materials as Precursors for Copper Rich Cu/ZnO/Al2O3 Catalysts. Chem. Mater. 2010, 22, 386–397. [Google Scholar] [CrossRef]
- Gordillo, A.; Titlbach, S.; Futter, C.; Lejkowski, M.L.; Prasetyo, E.; Rupflin, L.T.A.; Emmert, T.; Schunk, S.A. High-Throughput Experimentation in Catalysis and Materials Science. Ullmann’s Encycl. Ind. Chem. 2014, 1–19. [Google Scholar] [CrossRef]
- Titlbach, S.; Futter, C.; Lejkowski, M.; de Oliveira, A.L.; Schunk, S.A. High Throughput Technology: Exemplary Highlights of Advanced Technical Tools. Chem. Ing. Tech. 2014, 86, 1013–1028. [Google Scholar] [CrossRef]
- Schunk, S.A.; Böhmer, N.; Futter, C.; Kuschel, A.; Prasetyo, E.; Roussière, T. High throughput technology: Approaches of research in homogeneous and heterogeneous catalysis. Catalysis 2013, 25, 172–215. [Google Scholar] [CrossRef]
- Raudaskoski, R.; Niemelä, M.V.; Keiski, R.L. The effect of ageing time on co-precipitated Cu/ZnO/ZrO2 catalysts used in methanol synthesis from CO2 and H2. In Topics in Catalysis; Springer: Dordrecht, The Netherlands, 2007; Volume 45, pp. 57–60. [Google Scholar]
- Suh, Y.-W.; Moon, S.-H.; Rhee, H.-K. Active sites in Cu/ZnO/ZrO2 catalysts for methanol synthesis from CO/H2. Catal. Today 2000, 63, 447–452. [Google Scholar] [CrossRef]
- Liu, X.-M.; Lu, G.Q.; Yan, Z.-F.; Beltramini, J. Recent Advances in Catalysts for Methanol Synthesis via Hydrogenation of CO and CO2. Ind. Eng. Chem. Res. 2003, 42, 6518–6530. [Google Scholar] [CrossRef]
- Huang, L.; Chu, W.; Long, Y.; Ci, Z.; Luo, S. Influence of Zirconia Promoter on Catalytic Properties of Cu–Cr–Si Catalysts for Methanol Synthesis at High CO Conversion in Slurry Phase. Catal. Lett. 2006, 108, 113–118. [Google Scholar] [CrossRef]
- Raudaskoski, R.; Turpeinen, E.; Lenkkeri, R.; Pongrácz, E.; Keiski, R. Catalytic activation of CO2: Use of secondary CO2 for the production of synthesis gas and for methanol synthesis over copper-based zirconia-containing catalysts. Catal. Today 2009, 144, 318–323. [Google Scholar] [CrossRef]
- Liu, X.-M.; Lu, G.; Yan, Z.-F. Nanocrystalline zirconia as catalyst support in methanol synthesis. Appl. Catal. A Gen. 2005, 279, 241–245. [Google Scholar] [CrossRef]
- Wu, G.-S.; Mao, D.-S.; Lu, G.-Z.; Cao, Y.; Fan, K.-N. The Role of the Promoters in Cu Based Catalysts for Methanol Steam Reforming. Catal. Lett. 2009, 130, 177–184. [Google Scholar] [CrossRef]
- Amenomiya, Y. Methanol synthesis from CO2 + H2 II. Copper-based binary and ternary catalysts. Appl. Catal. 1987, 30, 57–68. [Google Scholar] [CrossRef]
- Denise, B.; Sneeden, R.P.A. Oxide-Supported Copper Catalysts Prepared from Copper Formate: Differences in Behavior in Methanol Synthesis from CO/H2 and CO2/H2 Mixtures. Appl. Catal. 1986, 28, 235–239. [Google Scholar] [CrossRef]
- Jeong, H.; Cho, C.H.; Kim, T.H. Effect of Zr and pH in the preparation of Cu/ZnO catalysts for the methanol synthesis by CO2 hydrogenation. React. Kinet. Mech. Catal. 2012, 106, 435–443. [Google Scholar] [CrossRef]
- Kanoun, N.; Astier, M.P.; Pajonk, G.M. Catalytic properties of new Cu based catalysts containing Zr and/or V for methanol synthesis from a carbon dioxide and hydrogen mixture. Catal. Lett. 1992, 15, 231–235. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]




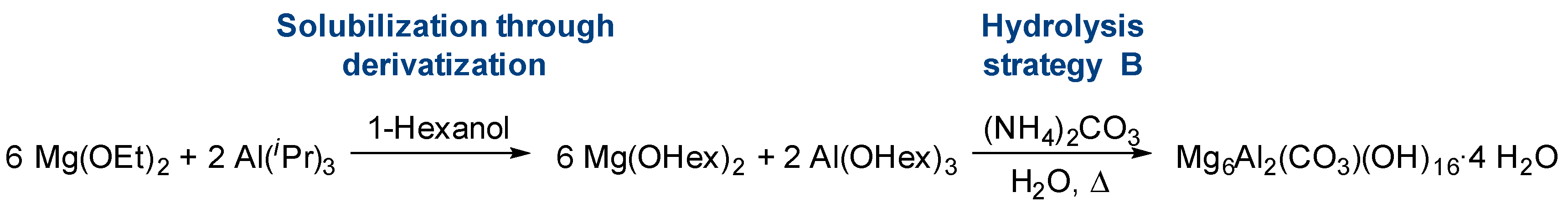






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
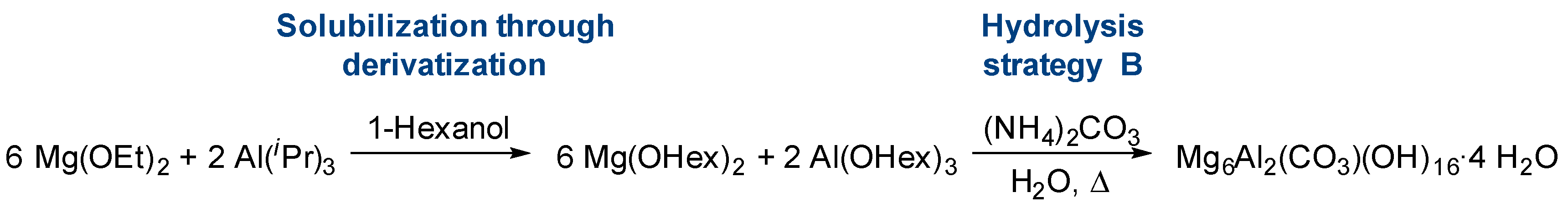{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Derivatized Alkoxide M(OR’)n | Starting Alkoxide M(OR)n | Exch. Alcohol R’OH | |
|---|---|---|---|
| 1 | Al(OHex)3 | Al(OiPr)3 | 1-Hexanol |
| 2 | Al(OMEE)3 | Al(OiPr)3 | OMEE |
| 3 | Mg(OHex)2 | Mg(OEt)2 | 1-Hexanol |
| 4 | Mg(OMEE)2 | Mg(OEt)2 | OMEE |
| 5 | Zr(OMEE)4 | Zr(OnPr)4 | OMEE |
| 6 | Ti(OMEE)4 | Ti(OnPr)4 | OMEE |
| 7 | Cu(OMEE)2 | Cu(OiPr)2 | OMEE |
| 8 | Zn(OMEE)2 | Zn(OiPr)2 | OMEE |
| 9 | Zn(OME)2 | Zn(OiPr)2 | OME |
| 10 | Co(OMEE)2 | Co(OiPr)2 | OMEE |
| 11 | Mn(OMEE)2 | Mn(OMe)2 | OMEE |
| Starting Alkoxide M(OR)n | Heterometallic Alkoxide M[Al(OHex)4]z | |
|---|---|---|
| 1 | Mg(OEt)2 | Mg[Al(OHex)4]2 |
| 2 | Cu(OiPr)2 | Cu[Al(OHex)4]2 |
| 3 | Zn(OiPr)2 | Zn[Al(OHex)4]2 |
| 4 | Co(OiPr)2 | Co[Al(OHex)4]2 |
| 5 | Mn(OMe)2 | Mn[Al(OHex)4]2 |
| Alkoxide | Solvent | Time until Solubilized |
|---|---|---|
| Cu(OiPr)2 | nHexane | Not solubilized |
| Cu(OiPr)2 | Pyridine | 0.2 h |
| Cu(OtBu)2 | tButanol | 1.3 h |
| Cu(OiPr)2 | THF | 1.8 h |
| Cu(OiPr)2 | iPropanol | 2.1 h |
| Cu(OMe)2 | Methanol | 3.5 h |
| Starting Alkoxide M(OR)n | Solvent | tCO2 (h) | IF | Formulae of Compound Based on CO2 Adsorption Experiment | |
|---|---|---|---|---|---|
| 1 | Cu(OiPr)2 | iPrOH | 6 | 0.93 | Cu(O(O)COiPr)1.86(OiPr)0.14 |
| 2 | Zn(OiPr)2 | iPrOH | 6 | 0.89 | Zn(O(O)COiPr)1.78(OiPr)0.22 |
| 3 | Al(OiPr)3 | iPrOH | 6 | 0.65 | Al(O(O)COiPr)1.95(OiPr)1.05 |
| 4 | Zr(OPr)4 | nPrOH | 6 | 0.94 | Zr(O(O)COPr)3.76(OPr)0.24 |
| 5 | Cu(OMEE)2 | OMEE | 1 | 0.88 | Cu(O(O)COMEE)1.76(OMEE)0.24 |
| 6 | Zn(OMEE)2 | OMEE | 1 | 0.84 | Zn(O(O)COMEE)1.68(OMEE)0.32 |
| 7 | Zn(OME)2 | OME | 1 | 0.96 | Zn(O(O)COME)1.92(OME)0.08 |
| 8 | Al(OMEE)3 | OMEE | 1 | 0.82 | Al(O(O)COMEE)2.46(OMEE)0.54 |
| 9 | Zr(OMEE)4 | OMEE | 1 | 0.90 | Zr(O(O)COMEE)3.6(OMEE)0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanf, S.; Lizandara-Pueyo, C.; Emmert, T.P.; Jevtovikj, I.; Gläser, R.; Schunk, S.A. Synthetic Routes to Crystalline Complex Metal Alkyl Carbonates and Hydroxycarbonates via Sol–Gel Chemistry—Perspectives for Advanced Materials in Catalysis. Catalysts 2022, 12, 554. https://doi.org/10.3390/catal12050554
Hanf S, Lizandara-Pueyo C, Emmert TP, Jevtovikj I, Gläser R, Schunk SA. Synthetic Routes to Crystalline Complex Metal Alkyl Carbonates and Hydroxycarbonates via Sol–Gel Chemistry—Perspectives for Advanced Materials in Catalysis. Catalysts. 2022; 12(5):554. https://doi.org/10.3390/catal12050554
Chicago/Turabian StyleHanf, Schirin, Carlos Lizandara-Pueyo, Timo Philipp Emmert, Ivana Jevtovikj, Roger Gläser, and Stephan Andreas Schunk. 2022. "Synthetic Routes to Crystalline Complex Metal Alkyl Carbonates and Hydroxycarbonates via Sol–Gel Chemistry—Perspectives for Advanced Materials in Catalysis" Catalysts 12, no. 5: 554. https://doi.org/10.3390/catal12050554
APA StyleHanf, S., Lizandara-Pueyo, C., Emmert, T. P., Jevtovikj, I., Gläser, R., & Schunk, S. A. (2022). Synthetic Routes to Crystalline Complex Metal Alkyl Carbonates and Hydroxycarbonates via Sol–Gel Chemistry—Perspectives for Advanced Materials in Catalysis. Catalysts, 12(5), 554. https://doi.org/10.3390/catal12050554