
Poisoning and Reuse of Supported Precious Metal Catalysts in the Hydrogenation of N-Heterocycles, Part II: Hydrogenation of 1-Methylpyrrole over Rhodium

 ,
,  , ,
, ,
Abstract
:
1. Introduction
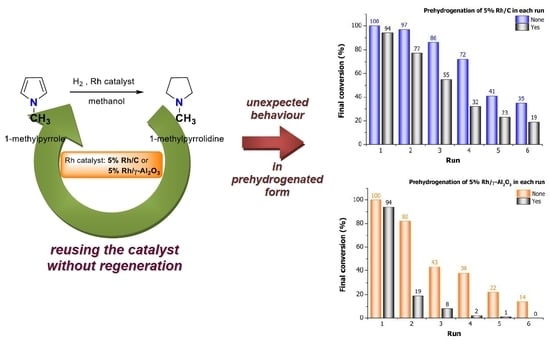
2. Results and Discussion
2.1. Rhodium-Catalysed Reference Hydrogenations of 1-Methylpyrrole
2.2. Effect of Temperature
2.3. NH3-TPD Examinations
2.4. Influence of the Amount of Carbon-Supported Rhodium
2.5. Controlling the Loss of 5% Rh/C
2.6. Acidic Aftertreatment of Catalysts
2.7. Effects of Catalyst Prehydrogenation
3. Materials and Methods
3.1. Materials
3.2. Hydrogenations
3.3. Catalyst Recycling
3.4. Catalyst Pretreatment
3.5. Correction of Catalyst Loss
3.6. Catalyst Characterisation
3.7. Analysis
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hegedűs, L.; Szőke-Molnár, K.; Sajó, I.E.; Srankó, D.F.; Schay, Z. Poisoning and Reuse of Supported Precious Metal Catalysts in the Hydrogenation of N-heterocycles Part I: Ruthenium-Catalysed Hydrogenation of 1-Methylpyrrole. Catal. Lett. 2018, 148, 1939–1950. [Google Scholar] [CrossRef]
- Hegedus, L.L.; McCabe, R.W. Catalyst Poisoning; Marcel Dekker: New York, NY, USA, 1984. [Google Scholar]
- Forzatti, P.; Lietti, L. Catalyst deactivation. Catal. Today 1999, 52, 165–181. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Besson, M.; Gallezot, P. Deactivation of metal catalysts in liquid phase organic reactions. Catal. Today 2003, 81, 547–559. [Google Scholar] [CrossRef]
- Moulijn, J.A.; van Diepen, A.E.; Kapteijn, F. Deactivation and Regeneration. In Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2008; pp. 1829–1845. [Google Scholar]
- Guisnet, M.; Ribeiro, F.R. Deactivation and regeneration of solid catalysts. In Deactivation and Regeneration of Zeolite Catalysts; Guisnet, M., Ribeiro, F.R., Eds.; Catalytic Science Series—Volume 9; Imperial College Press: London, UK, 2011; pp. 3–18. [Google Scholar]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous Catalyst Deactivation and Regeneration: A Review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef] [Green Version]
- Lange, J.-P. Renewable Feedstocks: The Problem of Catalyst Deactivation and its Mitigation. Angew. Chem. Int. Ed. 2015, 54, 13186–13197. [Google Scholar] [CrossRef]
- Hamilton, T.S.; Adams, R. Reduction of pyridine hydrochloride and pyridonium salts by means of hydrogen and platinum-oxide platinum black. XVIII. J. Am. Chem. Soc. 1928, 50, 2260–2263. [Google Scholar] [CrossRef]
- Maxted, E.B.; Walker, A.G. Studies in the detoxication of catalyst poisons. Part VII. The self-poisoning effect in the hydrogenation of pyridine. J. Chem. Soc. 1948, 1093–1097. [Google Scholar] [CrossRef]
- Maxted, E.B. The Poisoning of Metallic Catalysts. Adv. Catal. 1951, 3, 129–178. [Google Scholar]
- Devereux, J.M.; Payne, K.R.; Peeling, E.R.A. Catalytic hydrogenation. Part I. The hydrogenation of unsaturated amines over platinic oxide. J. Chem. Soc. 1957, 2845–2851. [Google Scholar] [CrossRef]
- Maxted, E.B.; Briggs, M.S. The catalytic toxicity of nitrogen compounds. Part I. Toxicity of ammonia and of amines. J. Chem. Soc. 1957, 3844–3847. [Google Scholar] [CrossRef]
- Freifelder, M. Practical Catalytic Hydrogenation; John Wiley: New York, NY, USA, 1971. [Google Scholar]
- Petró, J. Catalyst poisons, catalyst poisoning. In Contact Catalysis; Szabó, Z.G., Kalló, D., Eds.; Elsevier: Amsterdam, The Netherlands, 1976; Volume 2, pp. 65–75. [Google Scholar]
- Hegedűs, L. Katalizátorok mérgeződése, katalizátorméreg típusú vegyületek hidrogénezése. Magy. Kém. Folyóirat 2007, 113, 139–144. [Google Scholar]
- Xi, Y.; Huang, L.; Cheng, H. Mechanisms of Pyrrole Hydrogenation on Ru(0001) and Hydrogen Molybdenum Bronze Surfaces. J. Phys. Chem. C 2015, 119, 22477–22485. [Google Scholar] [CrossRef]
- He, T.; Liu, L.; Wu, G.; Chen, P. Covalent triazine framework-supported palladium nanoparticles for catalytic hydrogenation of N-heterocycles. J. Mater. Chem. A 2015, 3, 16235–16241. [Google Scholar] [CrossRef]
- Kim, T.W.; Oh, J.; Suh, Y.-W. Hydrogenation of 2-benzylpyridine over alumina-supported Ru catalysts: Use of Ru3(CO)12 as a Ru precursor. Appl. Catal. A Gen. 2017, 547, 183–190. [Google Scholar] [CrossRef]
- Moghaddam, A.A.; Krewer, U. Poisoning of Ammonia Synthesis Catalyst Considering Off-Design Feed Compositions. Catalysts 2020, 10, 1225. [Google Scholar] [CrossRef]
- Choi, J.; Cho, A.; Cho, J.H.; Kim, B.M. Bimetallic PdRh-Fe3O4 nanoparticle-catalyzed highly selective quinoline hydrogenation using ammonia borane. Appl. Catal. A Gen. 2022, 642, 118709. [Google Scholar] [CrossRef]
- Hegedűs, L.; Máthé, T.; Tungler, A. Hydrogenation of pyrrole derivatives I. Hydrogenations over palladium. Appl. Catal. A Gen. 1996, 143, 309–316. [Google Scholar] [CrossRef]
- Hegedűs, L.; Máthé, T.; Tungler, A. Hydrogenation of pyrrole derivatives. II. Hydrogenations over supported nobel metal catalysts. Appl. Catal. A Gen. 1996, 147, 407–414. [Google Scholar] [CrossRef]
- Hegedűs, L.; Máthé, T.; Tungler, A. Hydrogenation of pyrrole derivatives. Part IV. Hydrogenation of 1-methylpyrrole. Appl. Catal. A Gen. 1997, 152, 143–151. [Google Scholar] [CrossRef]
- Hegedűs, L.; Máthé, T.; Tungler, A. Hydrogenation of pyrrole derivatives Part III. Hydrogenation of methyl 1-methyl-2-pyrroleacetate. Appl. Catal. A Gen. 1997, 153, 133–139. [Google Scholar] [CrossRef]
- Nikiforov, T.; Stanchev, S.; Milenkov, B.; Dimitrov, V. Asymmetric synthesis of 1-methyl-2-(2-hydroxyethyl)pyrrolidine. Heterocycles 1986, 24, 1825–1829. [Google Scholar] [CrossRef]
- Fournier, A.M.; Brown, R.A.; Farnaby, W.; Miyatake-Ondozabal, H.; Clayden, J. Synthesis of (–)-(S,S)-clemastine by Invertive N → C Aryl Migration in a Lithiated Carbamate. Org. Lett. 2010, 12, 2222–2225. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Jung, J.W.; Kim, T.-H.; Kim, H.-D. Asymmetric synthesis of H1 receptor antagonist (R,R)-clemastine. Arch. Pharm. Res. 2015, 38, 2131–2136. [Google Scholar] [CrossRef] [PubMed]
- Leete, E. 1-Methylpyrrolidine-2-acetic Acid, a Plausible Intermediate in the Biosynthesis of Cocaine. Heterocycles 1989, 28, 481–487. [Google Scholar] [CrossRef]
- Hegedűs, L.; Máthé, T. Hydrogenation of pyrrole derivatives Part V. Poisoning effect of nitrogen on precious metal on carbon catalysts. Appl. Catal. A Gen. 2002, 226, 319–322. [Google Scholar] [CrossRef]
- Tungler, A.; Hegedűs, L.; Háda, V.; Máthé, T.; Szepesy, L. Diastereoselective Heterogeneous Catalytic Hydrogenation of Chiral Aromatic N-Heterocyclic Compounds. Chem. Ind. Cat. Org. React. 2001, 82, 425–437. [Google Scholar]
- Háda, V.; Tungler, A.; Szepesy, L. Diastereoselective heterogeneous catalytic hydrogenation of N-heterocycles: Part II. Hydrogenation of pyrroles. Appl. Catal. A Gen. 2001, 210, 165–171. [Google Scholar] [CrossRef]
- Eblagon, K.M.; Tam, K.; Tsang, S.C.E. Comparison of catalytic performance of supported ruthenium and rhodium for hydrogenation of 9-ethylcarbazole for hydrogen storage applications. Energy Environ. Sci. 2012, 5, 8621–8630. [Google Scholar] [CrossRef]
- Song, S.; Fung Kin Yuen, V.; Di, L.; Sun, Q.; Zhou, K.; Yan, N. Integrating Biomass into the Organonitrogen Chemical Supply Chain: Production of Pyrrole and d-Proline from Furfural. Angew. Chem. Int. Ed. 2020, 59, 19846–19850. [Google Scholar] [CrossRef]
- Overberger, C.G.; Palmer, L.C.; Marks, B.S.; Byrd, N.R. Azo Compounds. Biradical Sources. The Synthesis of Some 1,1-Disubstituted Hydrazines. J. Am. Chem. Soc. 1955, 77, 4100–4104. [Google Scholar] [CrossRef]
- Adams, R.; Miyano, S.; Nair, M.D. Synthesis of Substituted Pyrrolidines and Pyrrolizidines. J. Am. Chem. Soc. 1961, 83, 3323–3327. [Google Scholar] [CrossRef]
- Ortiz, C.; Greenhouse, R. The total synthesis of (+)-isoretronecanol from pyrrole. Tetrahedron Lett. 1985, 26, 2831–2832. [Google Scholar] [CrossRef]
- Jiang, C.; Frontier, A.J. Stereoselective Synthesis of Pyrrolidine Derivatives via Reduction of Substituted Pyrroles. Org. Lett. 2007, 9, 4939–4942. [Google Scholar] [CrossRef]
- Kotthaus, A.F.; Ballaschk, F.; Stakaj, V.; Mohr, F.; Kirsch, S.F. Synthesis and Resolution of a Chiral Diamine: 2,2′-(Propane-2,2-diyl)dipyrrolidine. Synthesis 2017, 49, 3107–3111. [Google Scholar]
- Cincinelli, R.; Beretta, G.; Dallavalle, S. Total synthesis of tetracyclic kynurenic acid analogues isolated from chestnut honey. Tetrahedron Lett. 2018, 59, 163–166. [Google Scholar] [CrossRef]
- Journot, G.; Neier, R.; Gualandi, A. Hydrogenation of Calix [4]pyrrole: From the Formation to the Synthesis of Calix [4]pyrrolidine. Eur. J. Org. Chem. 2021, 2021, 4444–4464. [Google Scholar] [CrossRef]
- Huang, W.; Kuhn, J.N.; Tsung, C.-K.; Zhang, Y.; Habas, S.E.; Yang, P.; Somorjai, G.A. Dendrimer Templated Synthesis of One Nanometer Rh and Pt Particles Supported on Mesoporous Silica: Catalytic Activity for Ethylene and Pyrrole Hydrogenation. Nano Lett. 2008, 8, 2027–2034. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, C.J.; Bieri, M.; Somorjai, G.A. Pyrrole Hydrogenation over Rh(111) and Pt(111) Single-Crystal Surfaces and Hydrogenation Promotion Mediated by 1-Methylpyrrole: A Kinetic and Sum-Frequency Generation Vibrational Spectroscopy Study. J. Phys. Chem. C 2008, 112, 11373–11378. [Google Scholar] [CrossRef] [Green Version]
- Hagelüken, C. Precious metals process catalysts—Material flows and recycling. Chim. Oggi/Chem. Today 2006, 24, 14–17. [Google Scholar]
- Hagelüken, C. Recycling of spent catalysts containing precious metals. In Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2008; pp. 1846–1863. [Google Scholar]
- Axon, S.A.; Casci, J.L. Recycling of spent catalysts containing base metals. In Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2008; pp. 1863–1871. [Google Scholar]
- Trimm, D.L. The regeneration or disposal of deactivated heterogeneous catalysts. Appl. Catal. A Gen. 2001, 212, 153–160. [Google Scholar] [CrossRef]
- Grumett, P. Precious Metal Recovery from Spent Catalysts. Platinum Metals Rev. 2003, 47, 163–166. [Google Scholar]
- Marafi, M.; Stanislaus, A. Options and processes for spent catalyst handling and utilization. J. Hazard. Mater. 2003, B101, 123–132. [Google Scholar] [CrossRef]
- Jackson, S.D. Processes occurring during deactivation and regeneration of metal and metal oxide catalysts. Chem. Eng. J. 2006, 120, 119–125. [Google Scholar]
- Dufresne, P. Hydroprocessing catalysts regeneration and recycling. Appl. Catal. A Gen. 2007, 322, 67–75. [Google Scholar] [CrossRef]
- Al-Sheeha, H.; Marafi, M.; Raghavan, V.; Rana, M.S. Recycling and Recovery Routes for Spent Hydroprocessing Catalyst Waste. Ind. Eng. Chem. Res. 2013, 52, 12794–12801. [Google Scholar] [CrossRef]
- Molnár, Á.; Papp, A. Catalyst recycling—A survey of recent progress and current status. Coord. Chem. Rev. 2017, 349, 1–65. [Google Scholar] [CrossRef]
- Miceli, M.; Frontera, P.; Macario, A.; Malara, A. Recovery/Reuse of Heterogeneous Supported Spent Catalysts. Catalysts 2021, 11, 591. [Google Scholar] [CrossRef]
- EudraLex—Volume 4, Good Manufacturing Practice (GMP) Guidelines. Available online: https://ec.europa.eu/health/medicinal-products/eudralex/eudralex-volume-4_hu (accessed on 5 May 2022).
- Busca, G. The surface of transitional aluminas: A critical review. Catal. Today 2014, 226, 2–13. [Google Scholar] [CrossRef]
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom, P.J.; Mallard, W.G. (Eds.) National Institute of Standards and Technology: Gaithersburg, MD, USA, 2017. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | Reusing the Catalyst | Reaction Time (h) | Conversion (%) | v0 (nL H2 ⋅ gRh−1 ⋅ h−1) | |||
|---|---|---|---|---|---|---|---|
| 5% Rh/C | 5% Rh/γ-Al2O3 | 5% Rh/C | 5% Rh/γ-Al2O3 | 5% Rh/C | 5% Rh/γ-Al2O3 | ||
| 1 | — (Fresh) | 3.5 | 4.0 | 100 | 100 | 121.8 | 77.5 |
| 2 | 1st | 5.0 | 5.0 | 97 | 82 | 99.6 | 66.4 |
| 3 | 2nd | 5.0 | 5.0 | 86 | 43 | 83.0 | 44.3 |
| 4 | 3rd | 6.0 | 5.0 | 72 | 38 | 72.0 | 33.2 |
| 5 | 4th | 6.5 | 5.5 | 41 | 22 | 33.2 | 22.2 |
| 6 | 5th | 6.5 | 5.5 | 35 | 14 | 22.1 | 11.1 |
| Entry | Catalysts/Supports | SBET (m2 · g−1) | NH3 Desorption (norm. Peak Area·gcat−1) |
|---|---|---|---|
| 1 | γ-Al2O3 (Alfa Aesar) | 135 | 23.9 * |
| 2 | 5% Rh/γ-Al2O3, fresh (Degussa G214 R/D) | 288 | 39.0 |
| 3 | 5% Rh/γ-Al2O3, used (Degussa G214 R/D) | n.m. | 11.2 |
| 4 | Activated C (Carbopal P3) | 940 | 10.0 |
| 5 | 5% Rh/C, fresh (own-prepared) | 880 | 8.8 |
| 6 | 5% Rh/C, used (own-prepared) | n.m. | 5.2 |
| Run | Reusing the Catalyst | Constant 0.1 g · g−1 Catalyst/Substrate Ratio | Final Conversion (%) | ||
|---|---|---|---|---|---|
| Amount of Catalyst (g) | Amount of Substrate (g) | Reference | |||
| 1 | — (Fresh) | 0.20 | 2.0 | 100 | 100 |
| 2 | 1st | 0.18 | 1.8 | 96 | 97 |
| 3 | 2nd | 0.15 | 1.5 | 88 | 86 |
| 4 | 3rd | 0.13 | 1.3 | 81 | 72 |
| 5 | 4th | 0.11 | 1.1 | 80 | 41 |
| 6 | 5th | 0.07 | 0.7 | 79 | 35 |
| Entry | Form of 5% Rh/γ-Al2O3 | Dispersion (–) a |
|---|---|---|
| 1 | Untreated (fresh) | 0.38 |
| 2 | Prehydrogenated b (fresh) | 0.38 |
| 3 | Prehydrogenated b (6× used) | 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hegedűs, L.; Nguyen, T.T.T.; Lévay, K.; László, K.; Sáfrán, G.; Beck, A. Poisoning and Reuse of Supported Precious Metal Catalysts in the Hydrogenation of N-Heterocycles, Part II: Hydrogenation of 1-Methylpyrrole over Rhodium. Catalysts 2022, 12, 730. https://doi.org/10.3390/catal12070730
Hegedűs L, Nguyen TTT, Lévay K, László K, Sáfrán G, Beck A. Poisoning and Reuse of Supported Precious Metal Catalysts in the Hydrogenation of N-Heterocycles, Part II: Hydrogenation of 1-Methylpyrrole over Rhodium. Catalysts. 2022; 12(7):730. https://doi.org/10.3390/catal12070730
Chicago/Turabian StyleHegedűs, László, Tien Thuy Thanh Nguyen, Krisztina Lévay, Krisztina László, György Sáfrán, and Andrea Beck. 2022. "Poisoning and Reuse of Supported Precious Metal Catalysts in the Hydrogenation of N-Heterocycles, Part II: Hydrogenation of 1-Methylpyrrole over Rhodium" Catalysts 12, no. 7: 730. https://doi.org/10.3390/catal12070730