
Competitive Adsorption of NOx and Ozone on the Catalyst Surface of Ozone Converters

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
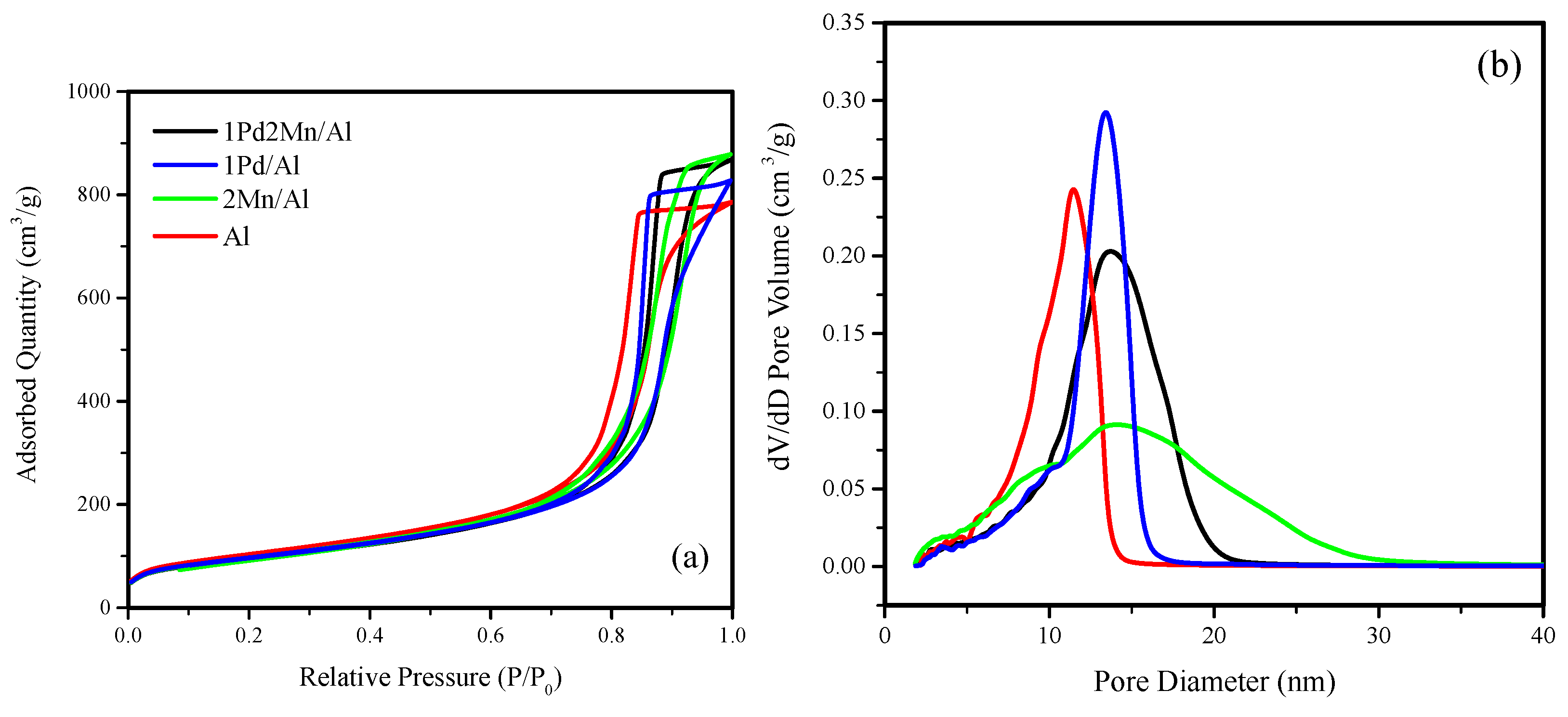
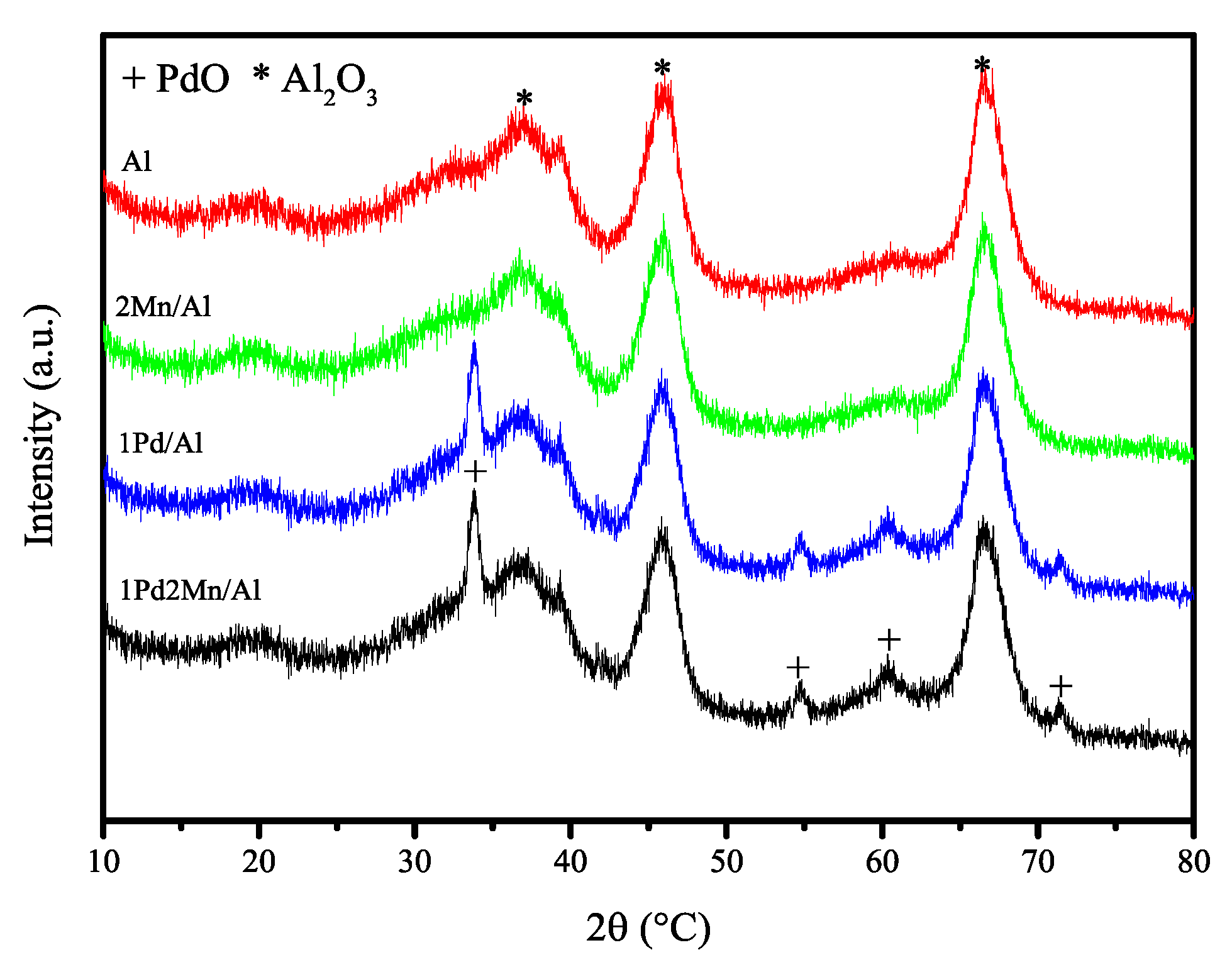
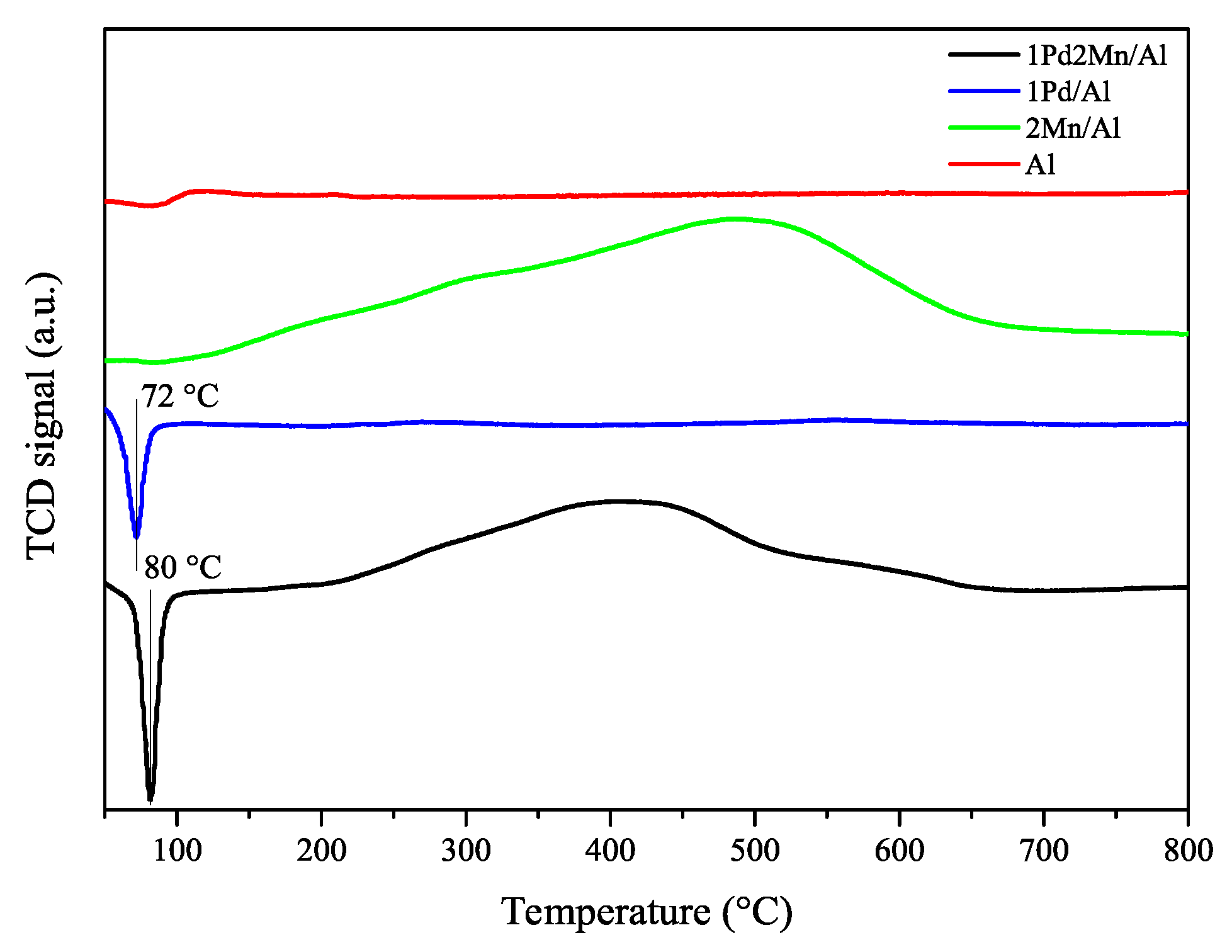
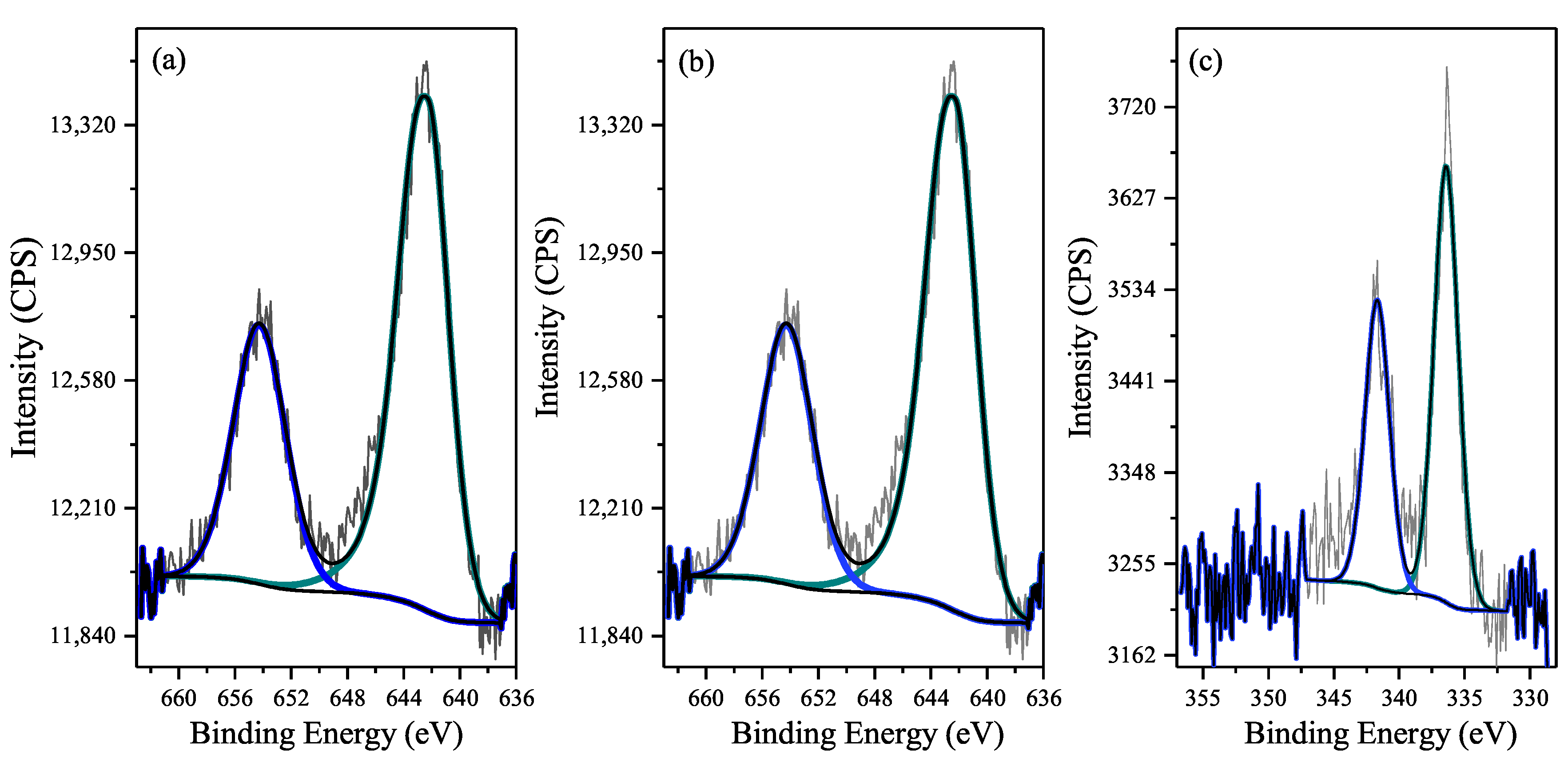
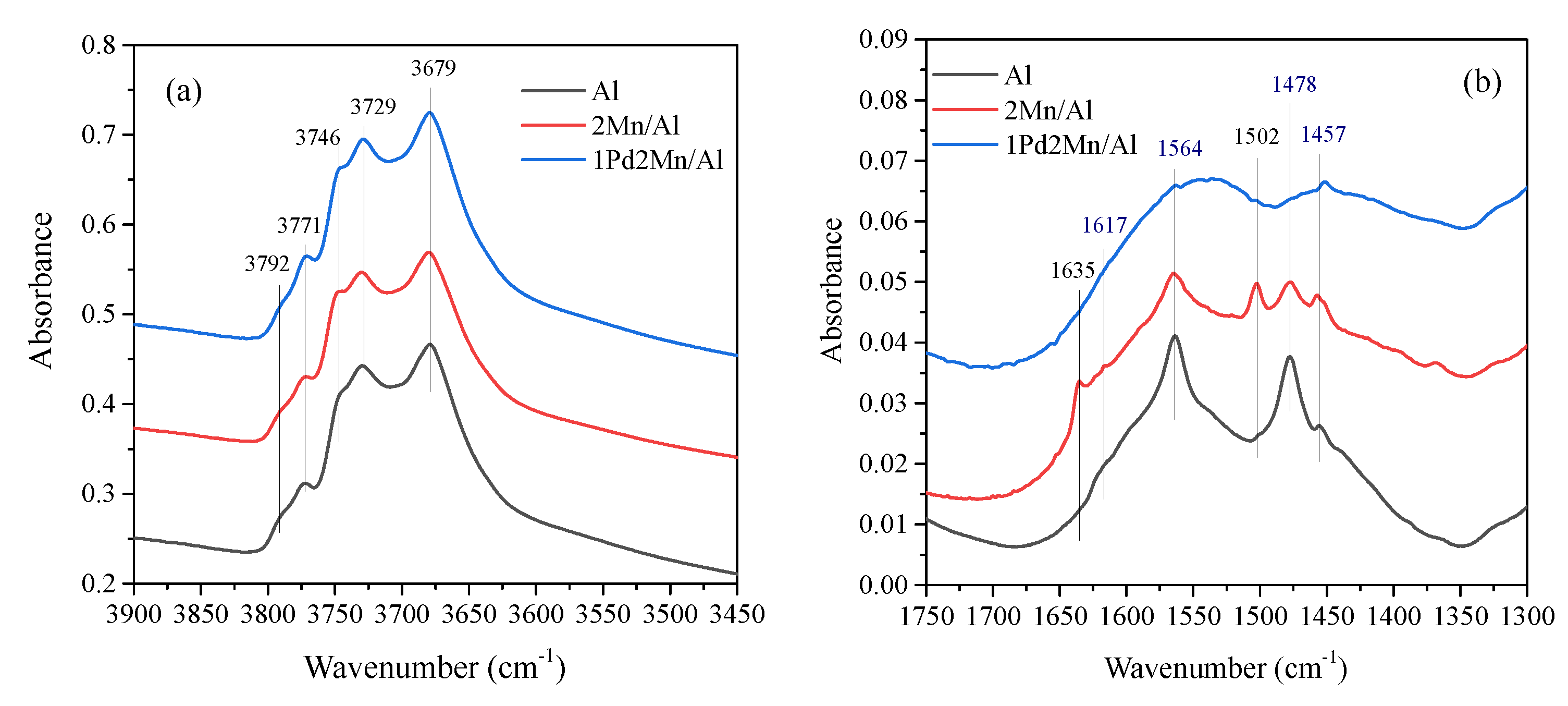
2.1. Catalyst Characterization Results
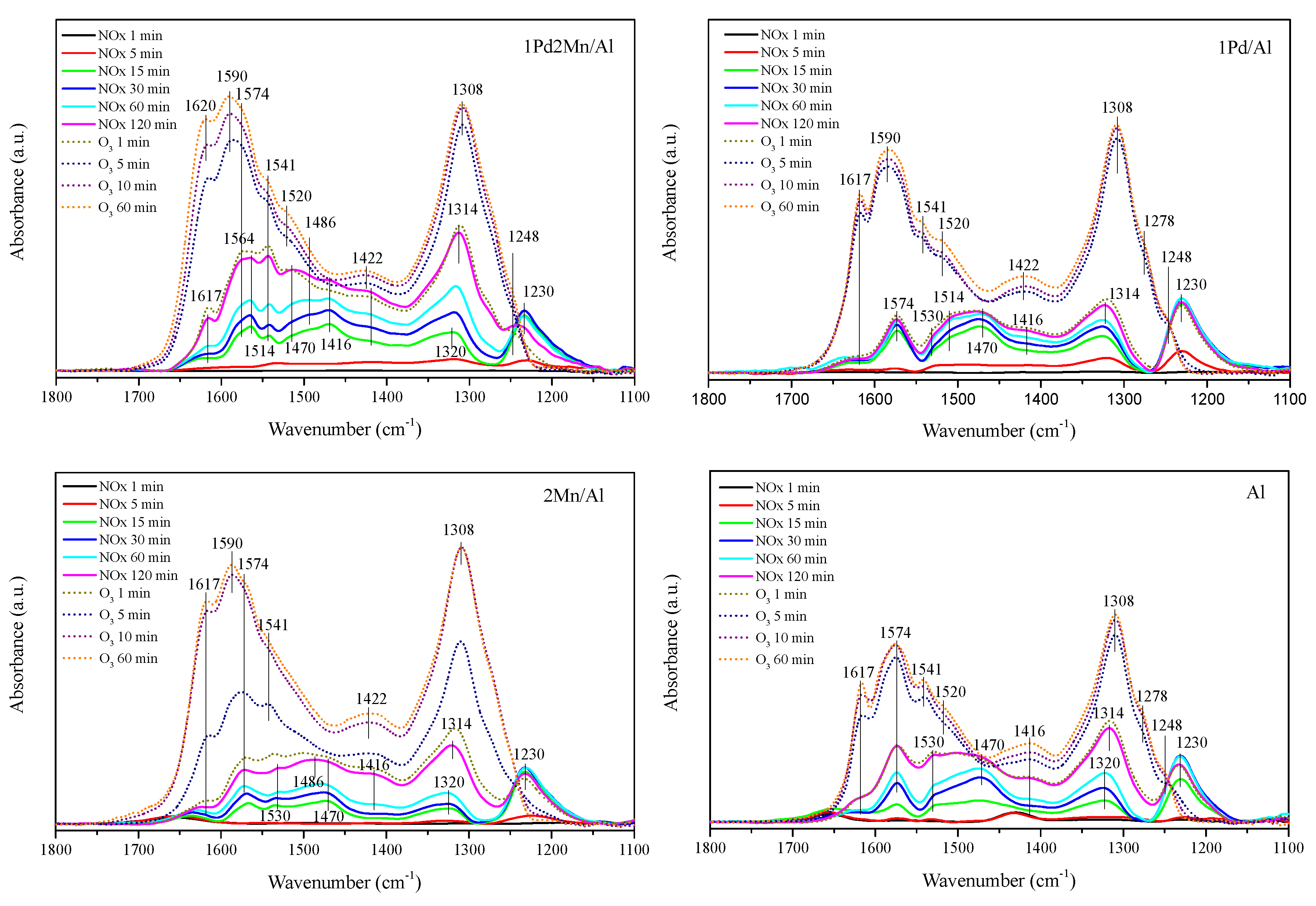
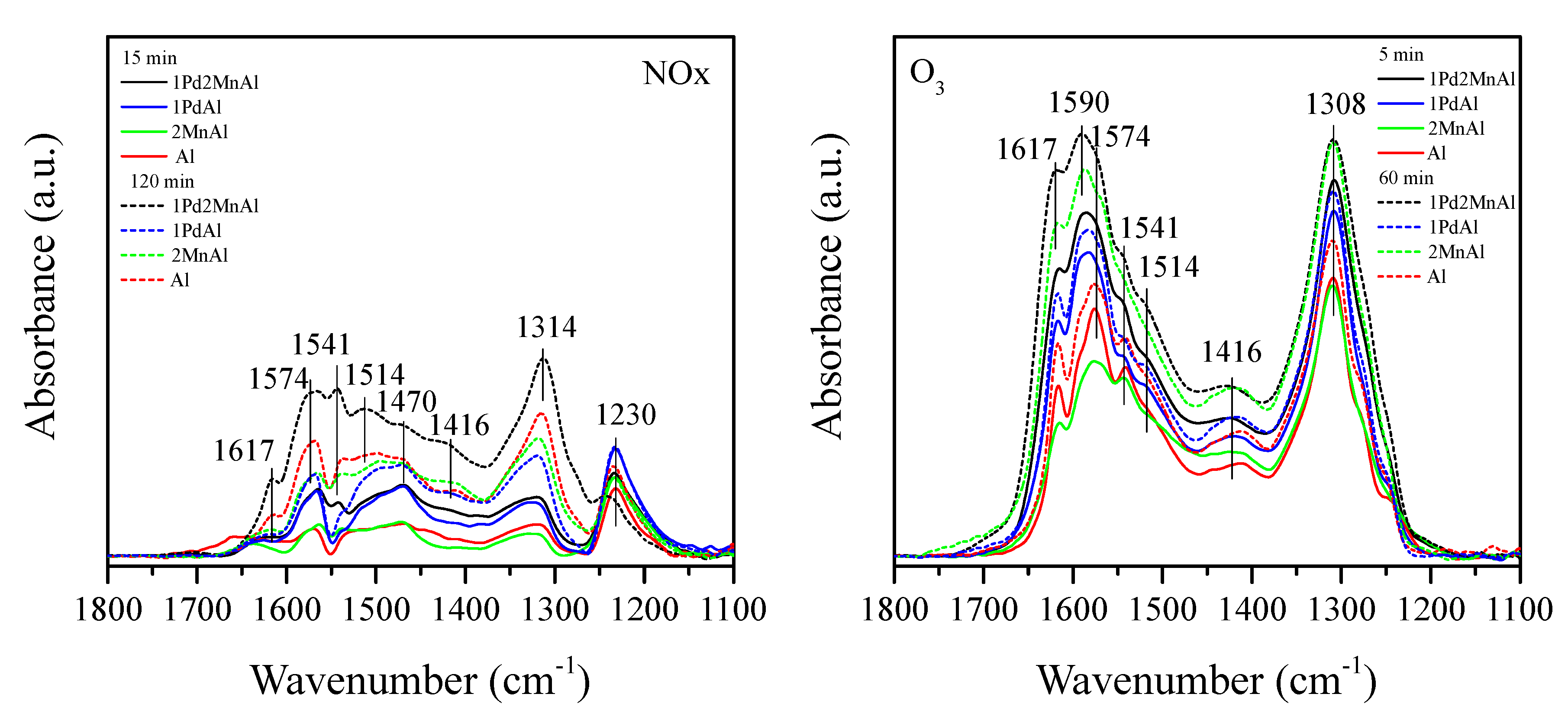
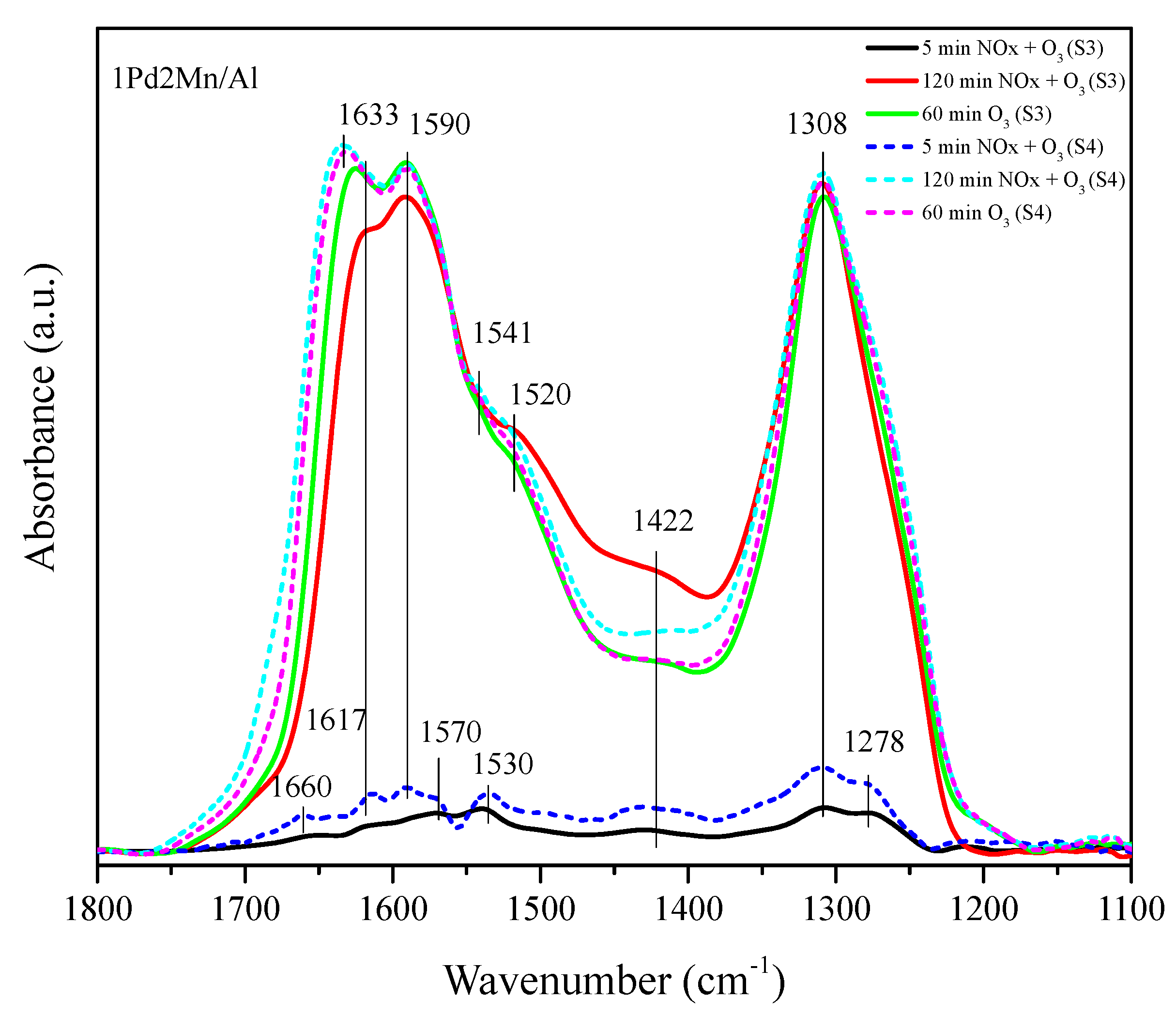
2.2. In Situ DRIFTS Measurements
3. Experimental Section
3.1. Catalyst Preparation
3.2. Catalyst Characterization
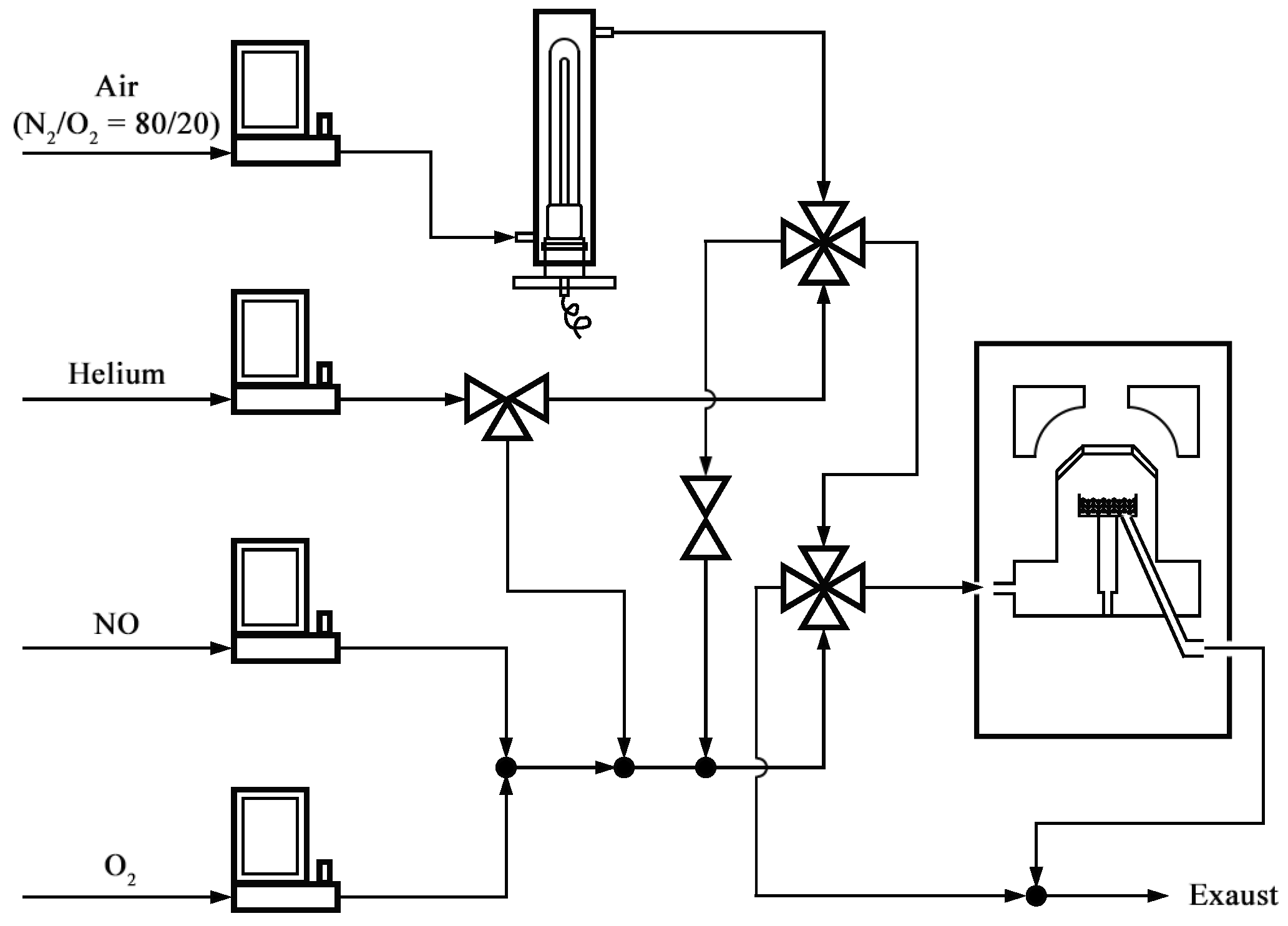
3.3. DRIFTS Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pudasainee, D.; Sapkota, B.; Shrestha, M.L.; Kaga, A.; Kondo, A.; Inoue, Y. Ground Level Ozone Concentrations and Its Association with NOx and Meteorological Parameters in Kathmandu Valley, Nepal. Atmos. Environ. 2006, 40, 8081–8087. [Google Scholar] [CrossRef]
- Hagenbjörk, A.; Malmqvist, E.; Mattisson, K.; Sommar, N.J.; Modig, L. The Spatial Variation of O3, NO, NO2 and NOx and the Relation between Them in Two Swedish Cities. Environ. Monit. Assess. 2017, 189, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhangar, S.; Cowlin, S.C.; Singer, B.C.; Sextro, R.G.; Nazaroff, W.W. Ozone Levels in Passenger Cabins of Commercial Aircraft on North American and Transoceanic Routes. Environ. Sci. Technol. 2008, 42, 3938–3943. [Google Scholar] [CrossRef]
- Jia, J.; Zhang, P.; Chen, L. Catalytic Decomposition of Gaseous Ozone over Manganese Dioxides with Different Crystal Structures. Appl. Catal. B Environ. 2016, 189, 210–218. [Google Scholar] [CrossRef]
- Jiang, C.; Zhang, P.; Zhang, B.; Li, J.; Wang, M. Facile Synthesis of Activated Carbon-Supported Porous Manganese Oxide via in Situ Reduction of Permanganate for Ozone Decomposition. Ozone Sci. Eng. 2013, 35, 308–315. [Google Scholar] [CrossRef]
- Batakliev, T.; Georgiev, V.; Anachkov, M.; Rakovsky, S.; Rakovsky, S. Ozone decomposition. Interdiscip. Toxicol. 2014, 7, 47–59. [Google Scholar] [CrossRef] [Green Version]
- İm, U.; Tayanç, M.; Yenigün, O. Analysis of Major Photochemical Pollutants with Meteorological Factors for High Ozone Days in Istanbul, Turkey. Water Air Soil Pollut. 2006, 175, 335–359. [Google Scholar] [CrossRef]
- Derwent, R.G.; Davies, T.J. Modelling the Impact of NOx or Hydrocarbon Control on Photochemical Ozone in Europe. Atmos. Environ. 1994, 28, 2039–2052. [Google Scholar] [CrossRef]
- Itano, Y.; Bandow, H.; Takenaka, N.; Saitoh, Y.; Asayama, A.; Fukuyama, J. Impact of NOx Reduction on Long-Term Ozone Trends in an Urban Atmosphere. Sci. Total Environ. 2007, 379, 46–55. [Google Scholar] [CrossRef]
- Lin, F.; Wang, Z.; Ma, Q.; He, Y.; Whiddon, R.; Zhu, Y.; Liu, J. N2O5 Formation Mechanism during the Ozone-Based Low-Temperature Oxidation deNOx Process. Energy Fuels 2016, 30, 5101–5107. [Google Scholar] [CrossRef]
- Liu, L.; Shen, B.; Si, M.; Lu, F. Performance and Mechanism of MnOx/γ-Al2O3 for Low-Temperature NO Catalytic Oxidation with O3/NO Ratio of 0.5. Fuel Process. Technol. 2021, 222, 106979. [Google Scholar] [CrossRef]
- Subrahmanyam, C.; Bulushev, D.A.; Kiwi-Minsker, L. Dynamic Behaviour of Activated Carbon Catalysts during Ozone Decomposition at Room Temperature. Appl. Catal. B Environ. 2005, 61, 98–106. [Google Scholar] [CrossRef]
- Mehandjiev, D.; Naydenov, A.; Ivanov, G. Ozone Decomposition, Benzene and CO Oxidation over NiMnO3-ilmenite and NiMn2O4-spinel Catalysts. Appl. Catal. A Gen. 2001, 206, 13–18. [Google Scholar] [CrossRef]
- Spasova, I.; Nikolov, P.; Mehandjiev, D. Ozone Decomposition over Alumina-Supported Copper, Manganese and Copper-Manganese Catalysts. Ozone Sci. Eng. 2007, 29, 41–45. [Google Scholar] [CrossRef]
- Mehandjiev, D.; Naidenov, A. Ozone Decomposition on α-Fe2O3 Catalyst. Ozone Sci. Eng. 1992, 14, 277–282. [Google Scholar] [CrossRef]
- Touati, H.; Guerin, A.; Swesi, Y.; Batiot-Dupeyrat, C.; Philippe, R.; Meille, V.; Clacens, J.M. Unexpected Role of NOx during Catalytic Ozone Abatement at Low Temperature. Catal. Commun. 2021, 148, 106163. [Google Scholar] [CrossRef]
- Sotomayor, F.; Cychosz, K.A.; Thommes, M. Characterization of Micro/Mesoporous Materials byPhysisorption: Concepts and Case Studies. Acc. Mater. Surf. Res. 2018, 3, 34–50. [Google Scholar]
- Daroughegi, R.; Meshkani, F.; Rezaei, M. Characterization and Evaluation of Mesoporous High Surface Area Promoted Ni-Al2O3 Catalysts in CO2 Methanation. J. Energy Inst. 2020, 93, 482–495. [Google Scholar] [CrossRef]
- Habibi, N.; Arandiyan, H.; Rezaei, M. Mesoporous MgO·Al2O3 Nanopowder-Supported Meso–Macroporous Nickel Catalysts: A New Path to High-Performance Biogas Reforming for Syngas. RSC Adv. 2016, 6, 29576–29585. [Google Scholar] [CrossRef]
- Chandra Shekar, S.; Krishna Murthy, J.; Kanta Rao, P.; Rama Rao, K.S. Selective Hydrogenolysis of Dichlorodifluoromethane on Carbon Covered Alumina Supported Palladium Catalyst. J. Mol. Catal. A Chem. 2003, 191, 45–59. [Google Scholar] [CrossRef]
- Kapteijn, F.; Singoredjo, L.; Andreini, A.; Moulijn, J.A. Activity and Selectivity of Pure Manganese Oxides in the Selective Catalytic Reduction of Nitric Oxide with Ammonia. Appl. Catal. B Environ. 1994, 3, 173–189. [Google Scholar] [CrossRef]
- Ardizzone, S.; Bianchi, C.L.; Tirelli, D. Mn3O4 and γ-MnOOH Powders, Preparation, Phase Composition and XPS Characterisation. Colloids Surfaces A Physicochem. Eng. Asp. 1998, 134, 305–312. [Google Scholar] [CrossRef]
- Hadjiivanov, K. Chapter Two—Identification and Characterization of Surface Hydroxyl Groups by Infrared Spectroscopy. In Advances in Catalysis; Jentoft, F.C., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 57, pp. 99–318. [Google Scholar] [CrossRef]
- Knözinger, H.; Ratnasamy, P. Catalytic Aluminas: Surface Models and Characterization of Surface Sites. Catal. Rev. 2007, 17, 31–70. [Google Scholar] [CrossRef]
- Vimont, A.; Lavalley, J.C.; Sahibed-Dine, A.; Otero Areán, C.; Rodríguez Delgado, M.; Daturi, M. Infrared Spectroscopic Study on the Surface Properties of γ-Gallium Oxide as Compared to Those of γ-Alumina. J. Phys. Chem. B 2005, 109, 9656–9664. [Google Scholar] [CrossRef] [PubMed]
- Haneda, M.; Joubert, E.; Ménézo, J.C.; Duprez, D.; Barbier, J.; Bion, N.; Daturi, M.; Saussey, J.; Lavalley, J.C.; Hamada, H. Surface Characterization of Alumina-Supported Catalysts Prepared by Sol–Gel Method. Part I.—Acid–Base Properties. Phys. Chem. Chem. Phys. 2001, 3, 1366–1370. [Google Scholar] [CrossRef]
- Dhandapani, B.; Oyama, S.T. Kinetics and Mechanism of Ozone Decomposition on a Manganese Oxide Catalyst. Chem. Lett. 1995, 24, 413–414. [Google Scholar] [CrossRef]
- Castoldi, L.; Matarrese, R.; Morandi, S.; Righini, L.; Lietti, L. New insights on the adsorption, thermal decomposition and reduction of NOx over Pt- and Ba-based catalysts. Appl. Catal. B Environ. 2018, 224, 249–263. [Google Scholar] [CrossRef]
- Su, Y.; Amiridis, M.D. In situ FTIR studies of the mechanism of NOx storage and reduction on Pt/Ba/Al2O3 catalysts. Catal. Today 2004, 96, 31–41. [Google Scholar] [CrossRef]
- Hadjiivanov, K.I. Identification of Neutral and Charged NxOy Surface Species by IR Spectroscopy. Catal. Rev. 2000, 42, 71–144. [Google Scholar] [CrossRef]
- Huang, S.J.; Walters, A.; Vannice, M. TPD, TPR and DRIFTS studies of adsorption and reduction of NO on La2O3 dispersed on Al2O3. Appl. Catal. B Environ. 2000, 26, 101–118. [Google Scholar] [CrossRef]
- Koukiou, S.; Konsolakis, M.; Lambert, R.; Yentekakis, I. Spectroscopic evidence for the mode of action of alkali promoters in Pt-catalyzed de-NOx chemistry. Appl. Catal. B Environ. 2007, 76, 101–106. [Google Scholar] [CrossRef]
- Granger, P.; Praliaud, H.; Billy, J.; Leclercq, L.; Leclercq, G. Infrared Investigation of the Transformation of NO over Supported Pt- and Rh-based Three-Way Catalysts. Surf. Interface Anal. 2002, 34, 92–96. [Google Scholar] [CrossRef]
- Westerberg, B.; Fridell, E. A transient FTIR study of species formed during NOx storage in the Pt/BaO/Al2O3 system. J. Mol. Catal. A Chem. 2001, 165, 249–263. [Google Scholar] [CrossRef]
- Bourane, A.; Dulaurent, O.; Salasc, S.; Sarda, C.; Bouly, C.; Bianchi, D. Heats of Adsorption of Linear NO Species on a Pt/Al2O3 Catalyst Using In Situ Infrared Spectroscopy under Adsorption Equilibrium. J. Catal. 2001, 204, 77–88. [Google Scholar] [CrossRef]
- Wu, F.; Wang, M.; Lu, Y.; Zhang, X.; Yang, C. Catalytic removal of ozone and design of an ozone converter for the bleeding air purification of aircraft cabin. Build. Environ. 2017, 115, 25–33. [Google Scholar] [CrossRef]
- Yan, Z.; Zuo, Z.; Li, Z.; Zhang, J. A Cluster DFT Study of NH3 and NO Adsorption on the MoO22+/HZSM-5 Surface: Lewis versus Brønsted Acid Sites. Appl. Surf. Sci. 2014, 321, 339–347. [Google Scholar] [CrossRef]
- Oton, L.F.; Oliveira, A.C.; de Araujo, J.C.S.; Araujo, R.S.; de Sousa, F.F.; Saraiva, G.D.; Lang, R.; Otubo, L.; Carlos da Silva Duarte, G.; Campos, A. Selective Catalytic Reduction of NOx by CO (CO-SCR) over Metal-Supported Nanoparticles Dispersed on Porous Alumina. Adv. Powder Technol. 2020, 31, 464–476. [Google Scholar] [CrossRef]
- Lin, F.; Wang, Z.; Ma, Q.; Yang, Y.; Whiddon, R.; Zhu, Y.; Cen, K. Catalytic deep oxidation of NO by ozone over MnOx loaded spherical alumina catalyst. Appl. Catal. B Environ. 2016, 198, 100–111. [Google Scholar] [CrossRef]
- Irfan, M.F.; Goo, J.H.; Kim, S.D.; Hong, S.C. Effect of CO on NO Oxidation over Platinum Based Catalysts for Hybrid Fast SCR Process. Chemosphere 2007, 66, 54–59. [Google Scholar] [CrossRef]
- Irfan, M.F.; Goo, J.H.; Kim, S.D. Co3O4 Based Catalysts for NO Oxidation and NOx Reduction in Fast SCR Process. Appl. Catal. B Environ. 2008, 78, 267–274. [Google Scholar] [CrossRef]
- Roscoe, J.M.; Abbatt, J.P.D. Diffuse Reflectance FTIR Study of the Interaction of Alumina Surfaces with Ozone and Water Vapor. J. Phys. Chem. A 2005, 109, 9028–9034. [Google Scholar] [CrossRef] [PubMed]
- Touati, H.; Mehri, A.; Karouia, F.; Richard, F.; Batiot-Dupeyrat, C.; Daniele, S.; Clacens, J.M. Low-Temperature O3 Decomposition over Pd−TiO2 Hybrid Catalysts. Catalysts 2022, 12, 448. [Google Scholar] [CrossRef]
- Abdallah, G.; Bitar, R.; Kaliya Perumal Veerapandian, S.; Giraudon, J.M.; De Geyter, N.; Morent, R.; Lamonier, J.F. Acid Treated Ce Modified Birnessite–Type MnO2 for Ozone Decomposition at Low Temperature: Effect of Nitrogen Containing Co-Pollutants and Water. Appl. Surf. Sci. 2022, 571, 151240. [Google Scholar] [CrossRef]
- Bleta, R.; Alphonse, P.; Pin, L.; Gressier, M.; Menu, M.J. An efficient route to aqueous phase synthesis of nanocrystalline γ-Al2O3 with high porosity: From stable boehmite colloids to large pore mesoporous alumina. J. Colloid Interface Sci. 2012, 367, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Wulf, O.R.; Tolman, R.C. The Thermal Decomposition of Ozone. Proc. Natl. Acad. Sci. USA 1927, 13, 272–275. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | BET Surface Area (m/g) | Total Pore Volume (cm/g) | Average Pore Diameter (nm) |
|---|---|---|---|
| 1Pd2Mn/Al | 341 | 1.33 | 14 |
| 1Pd/Al | 340 | 1.27 | 14 |
| 2Mn/Al | 349 | 1.35 | 14 |
| Al | 372 | 1.21 | 12 |
| Element | 1Pd2Mn/Al | 2Mn/Al | Al |
|---|---|---|---|
| C 1s | 1.2 | 1.3 | 1.9 |
| O 1s | 59.6 | 59.3 | 60 |
| Al 2p | 38.6 | 38.8 | 38.1 |
| Mn 2p | 0.7 (1.8 wt %) | 0.6 (1.6 wt %) | |
| Pd 3d | 0.06 (0.3 wt %) |
| Strong LAS | Weak LAS | Total | |
|---|---|---|---|
| Al | 106 | 278 | 384 |
| 2Mn/Al | 31 | 317 | 348 |
| 1Pd2Mn/Al | 47 | 376 | 423 |
| Surface Species | Peak Position (cm) | References |
|---|---|---|
| Ionic nitrites NO2− | 1215–1260 | [28] |
| Monodentate nitrites | 1375–1470 | [29] |
 | 1065–1206 | [30] |
| Bridged nitrites | 1266–1390 | [31] |
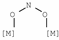 | 1230–1276 | [32] |
| Monodentate nitrates | 1415–1520 | [30] |
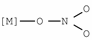 | 1250–1330 | [28] |
| Bidentate nitrates | 1550–1611 | [29,33,34,35] |
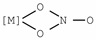 | 1200–1300 | |
| Bridging nitrates | 1600–1660 | [29,33,34,35] |
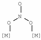 | 1200–1300 | |
| Atomic oxygen | 1318–1380 | [36] |
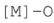 | ||
| Chemically adsorbed ozone | 1100 | [36] |
 |
| Seq. | Step 1 | Step 2 | Step 3 |
|---|---|---|---|
| 1 | 200 ppm NO, 20% O2 (He balance, 40 mL/min) 2 h | 500 ppm O3 (Air balance, 40 mL/min) 1 h | |
| 2 | 500 ppm O3 (Air balance, 40 mL/min) 1 h | 200 ppm NO, 20% O2 (He balance, 40 mL/min) 2 h | 500 ppm O3 (Air balance, 40 mL/min) 1 h |
| 3 | 500 ppm O3 (Air balance, 20 mL/min) + 200 ppm NO, 20% O2 (He balance, 20 mL/min) 2 h | 500 ppm O3 (Air balance, 40 mL/min) 1 h | |
| 4 | 500 ppm O3 (Air balance, 40 mL/min) 1 h | 500 ppm O3 (Air balance, 20 mL/min) + 200 ppm NO, 20% O2 (He balance, 20 mL/min) 2 h | 500 ppm O3 (Air balance, 40 mL/min) 1 h |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, X.; Clacens, J.-M.; Can, F.; Boréave, A.; Veyre, L.; Gil, S.; Meille, V. Competitive Adsorption of NOx and Ozone on the Catalyst Surface of Ozone Converters. Catalysts 2022, 12, 738. https://doi.org/10.3390/catal12070738
Ji X, Clacens J-M, Can F, Boréave A, Veyre L, Gil S, Meille V. Competitive Adsorption of NOx and Ozone on the Catalyst Surface of Ozone Converters. Catalysts. 2022; 12(7):738. https://doi.org/10.3390/catal12070738
Chicago/Turabian StyleJi, Xiaolong, Jean-Marc Clacens, Fabien Can, Antoinette Boréave, Laurent Veyre, Sonia Gil, and Valérie Meille. 2022. "Competitive Adsorption of NOx and Ozone on the Catalyst Surface of Ozone Converters" Catalysts 12, no. 7: 738. https://doi.org/10.3390/catal12070738