
Electron Paramagnetic Resonance Spin Trapping (EPR–ST) Technique in Photopolymerization Processes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
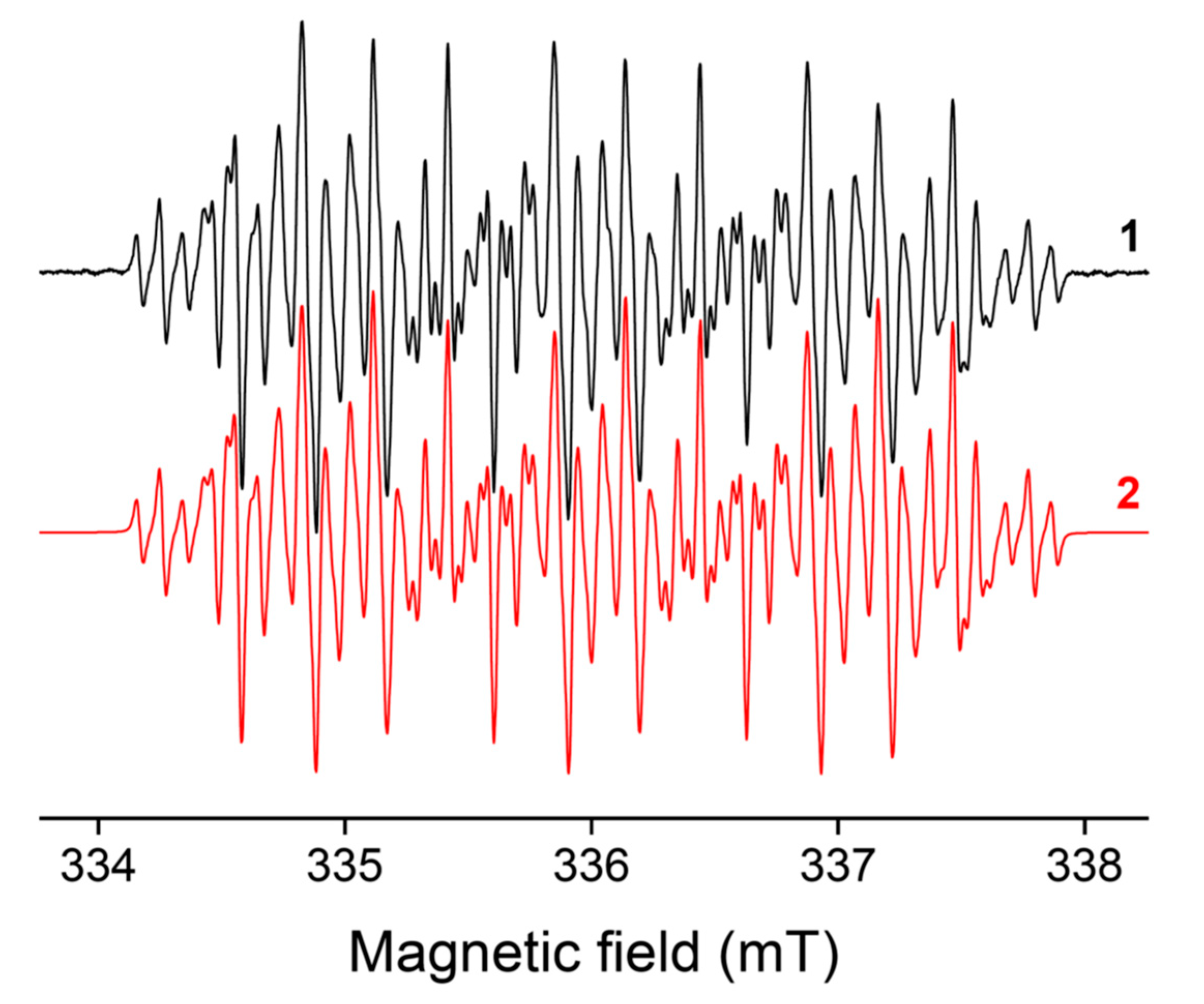{kind=link}
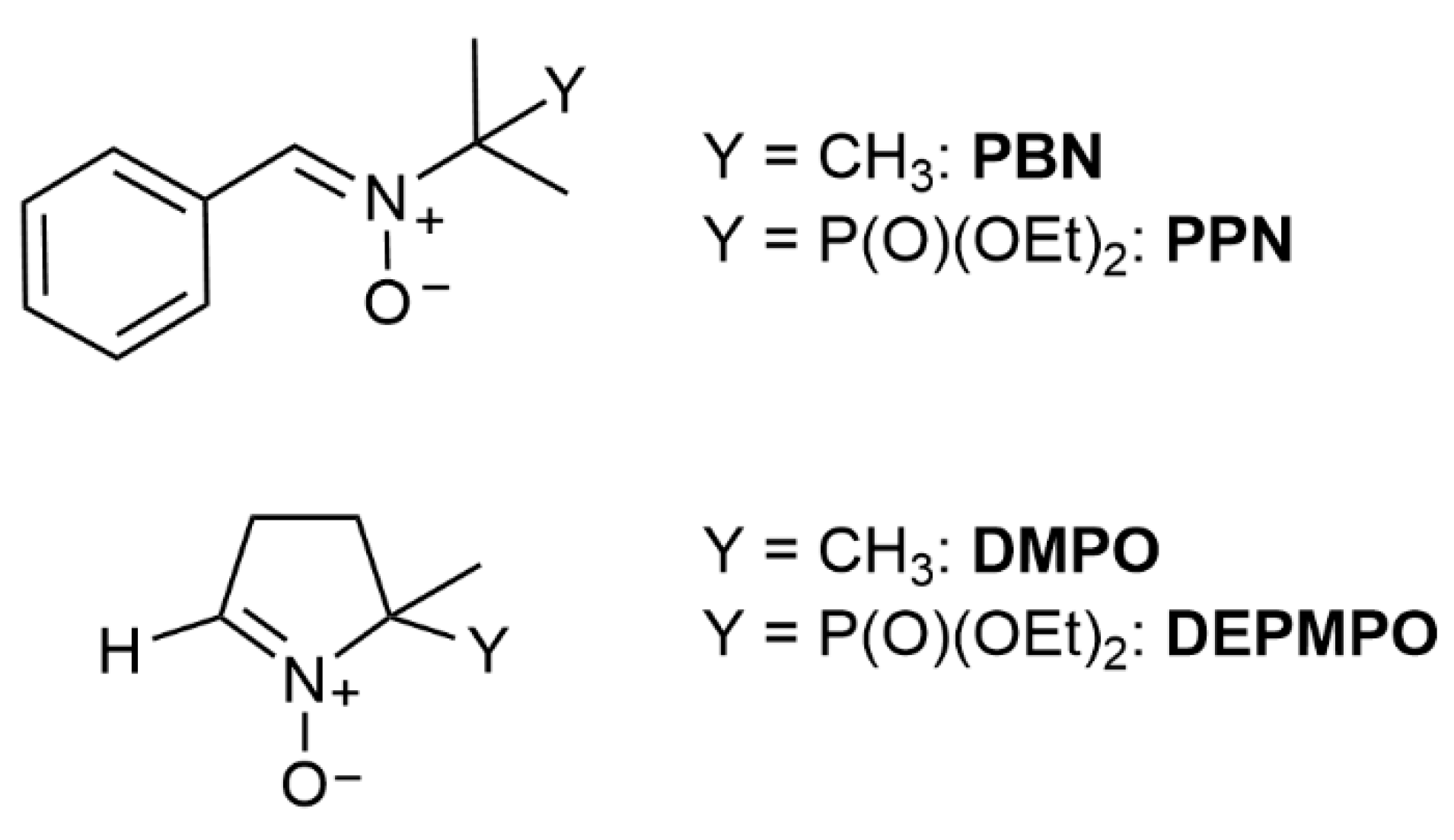{kind=link}
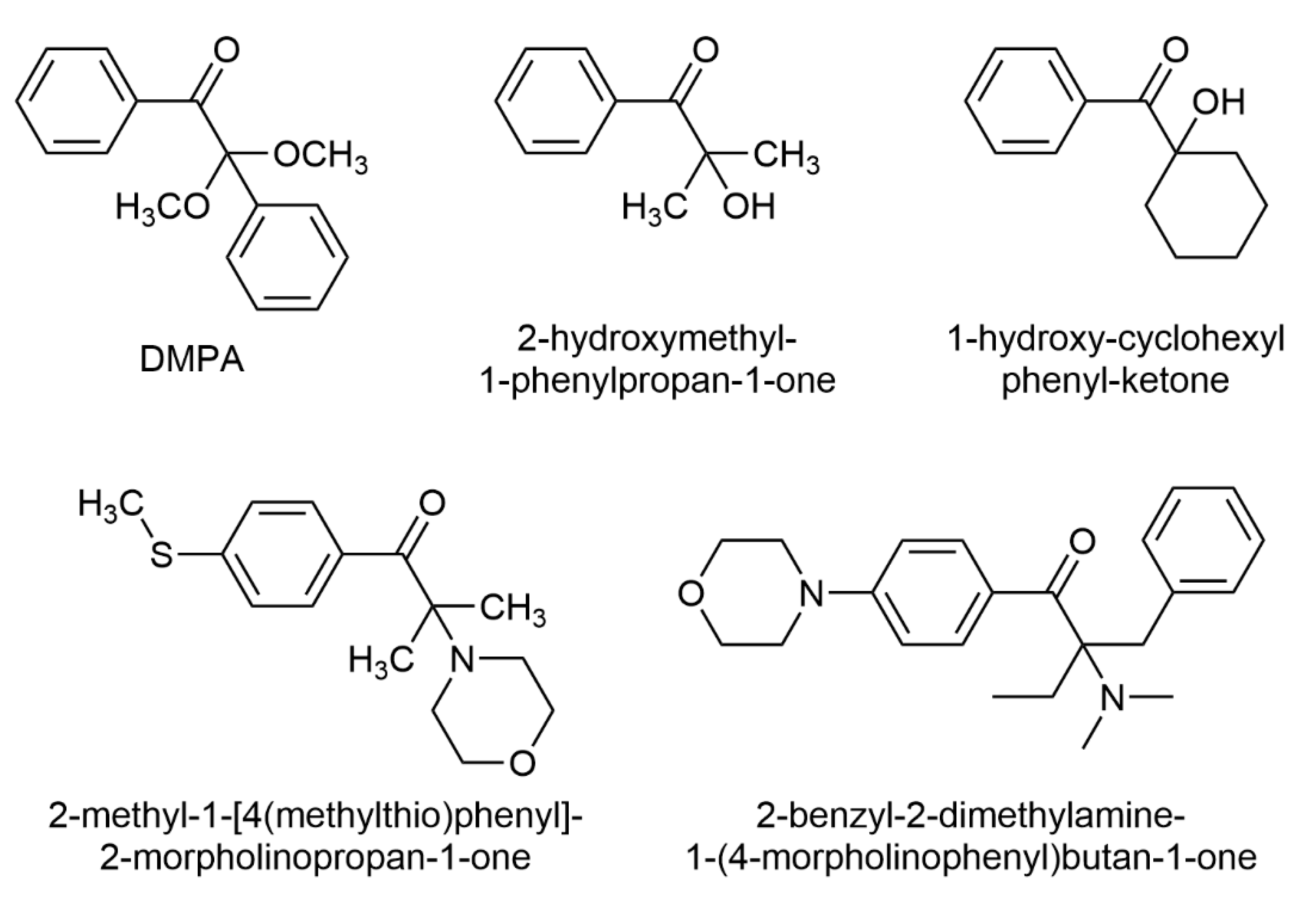{kind=link}
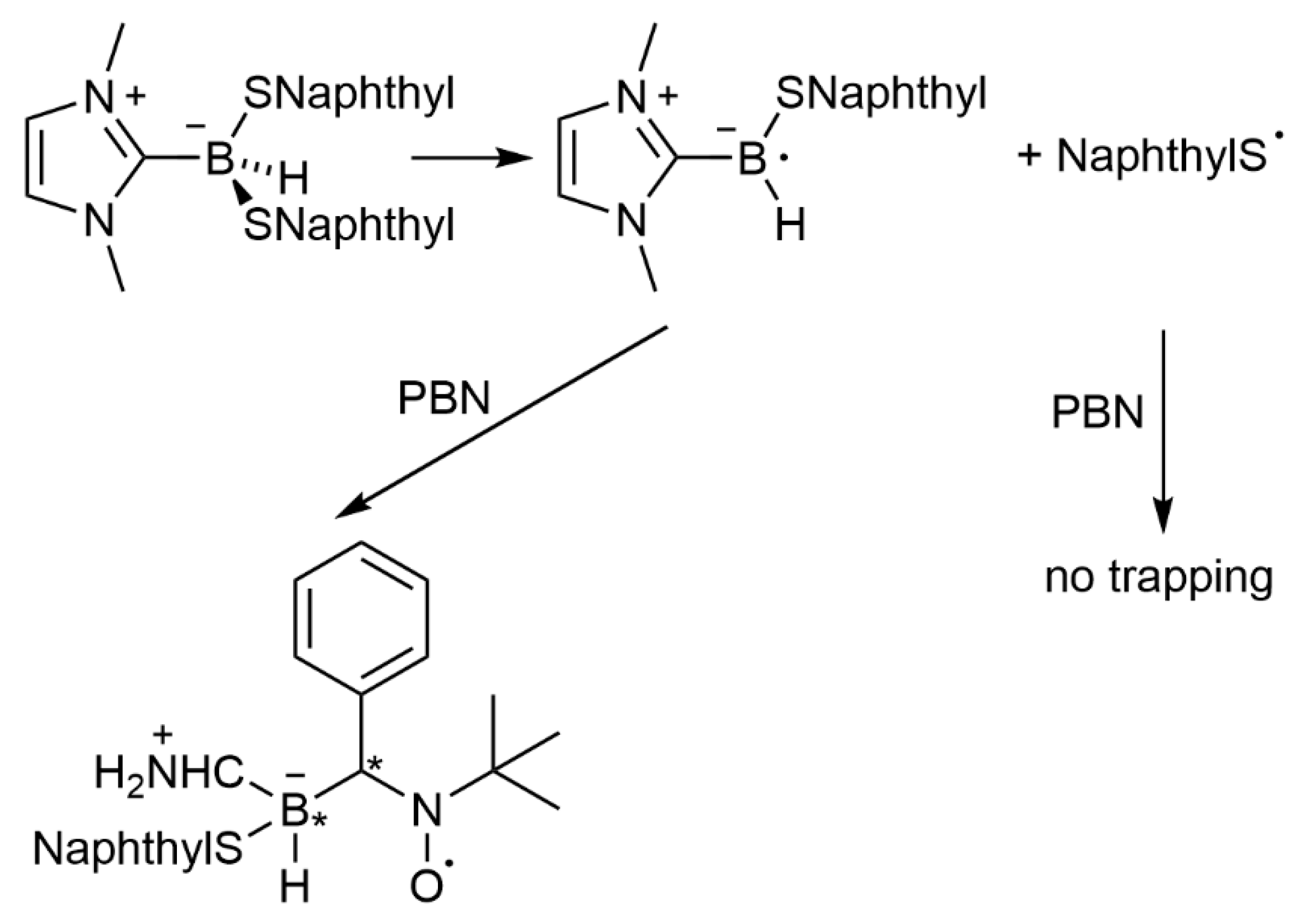{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
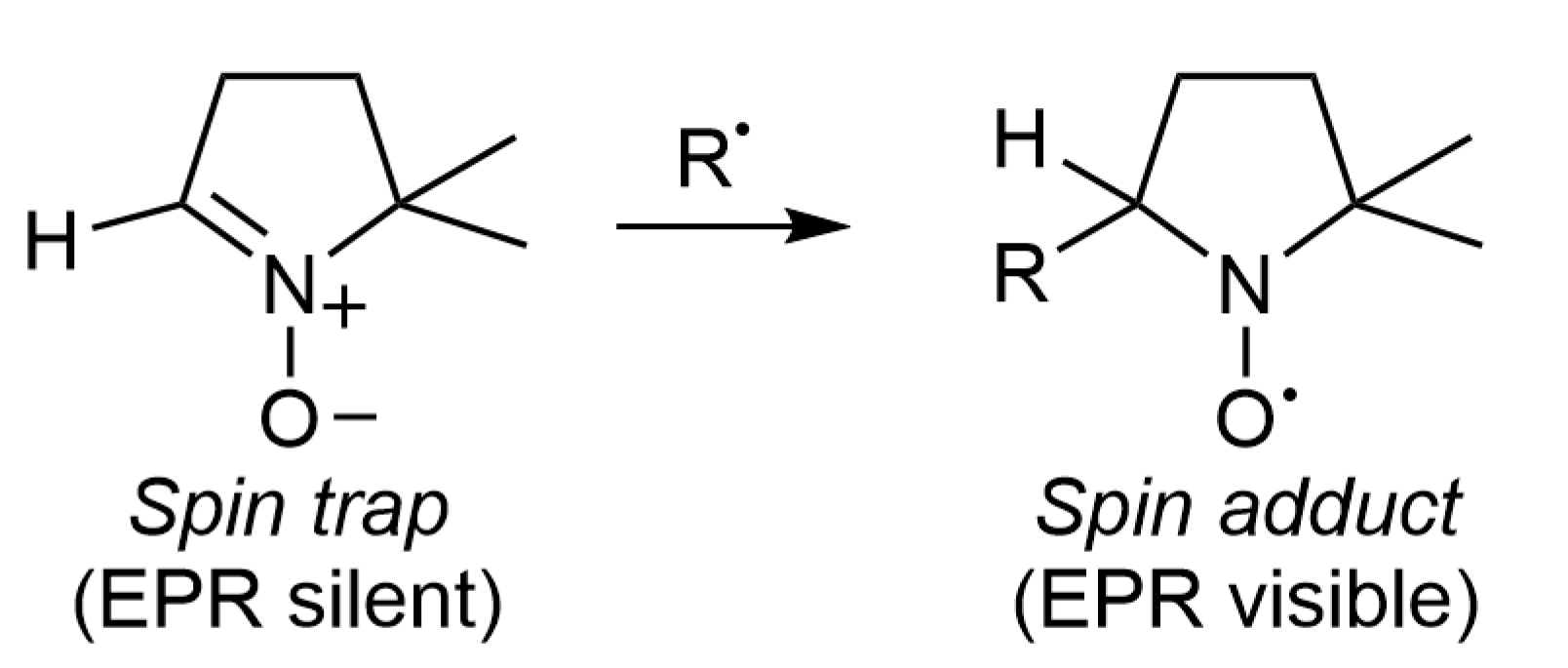
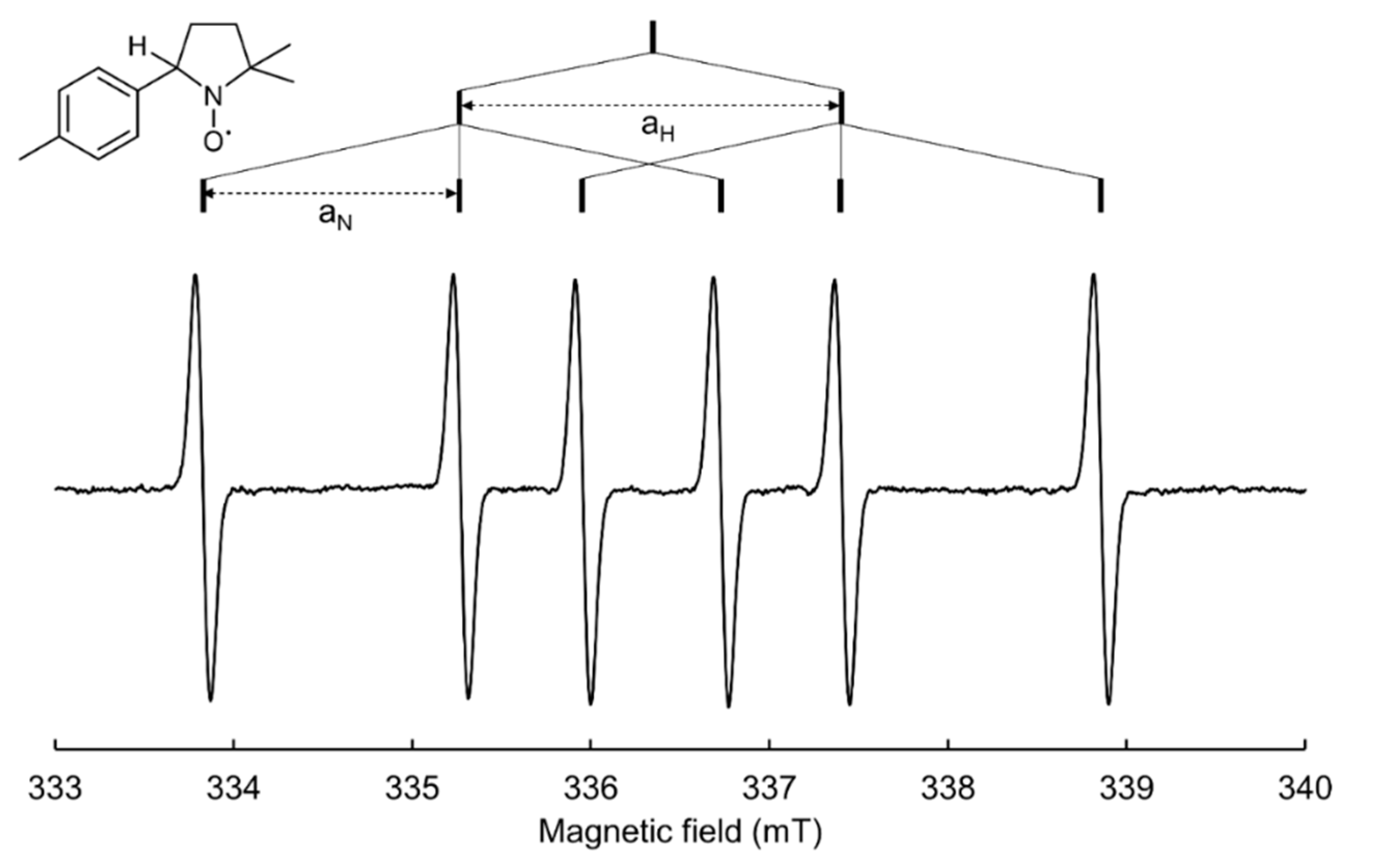
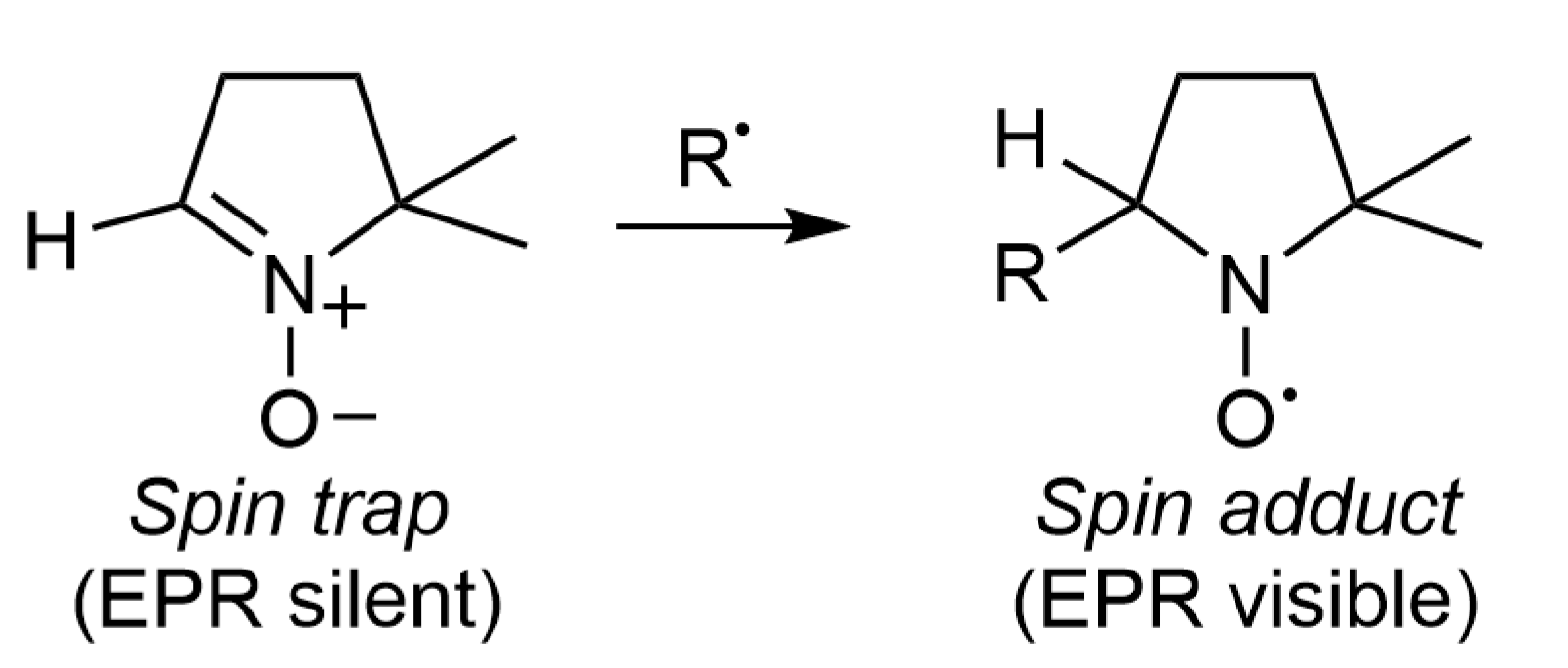
2. Spin Trapping Principle and Brief Description of EPR Spectra
3. Three Classes of Spin Traps
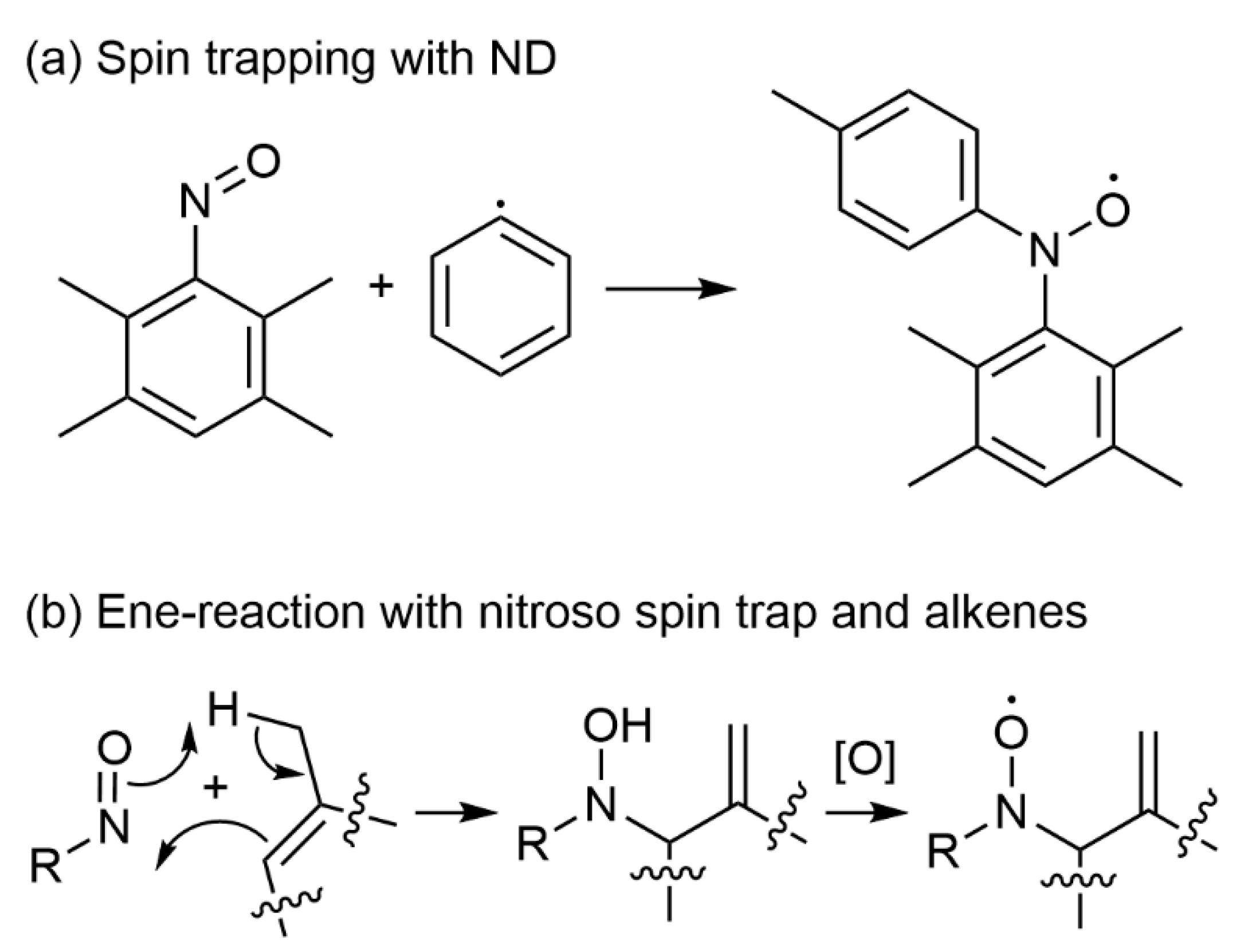
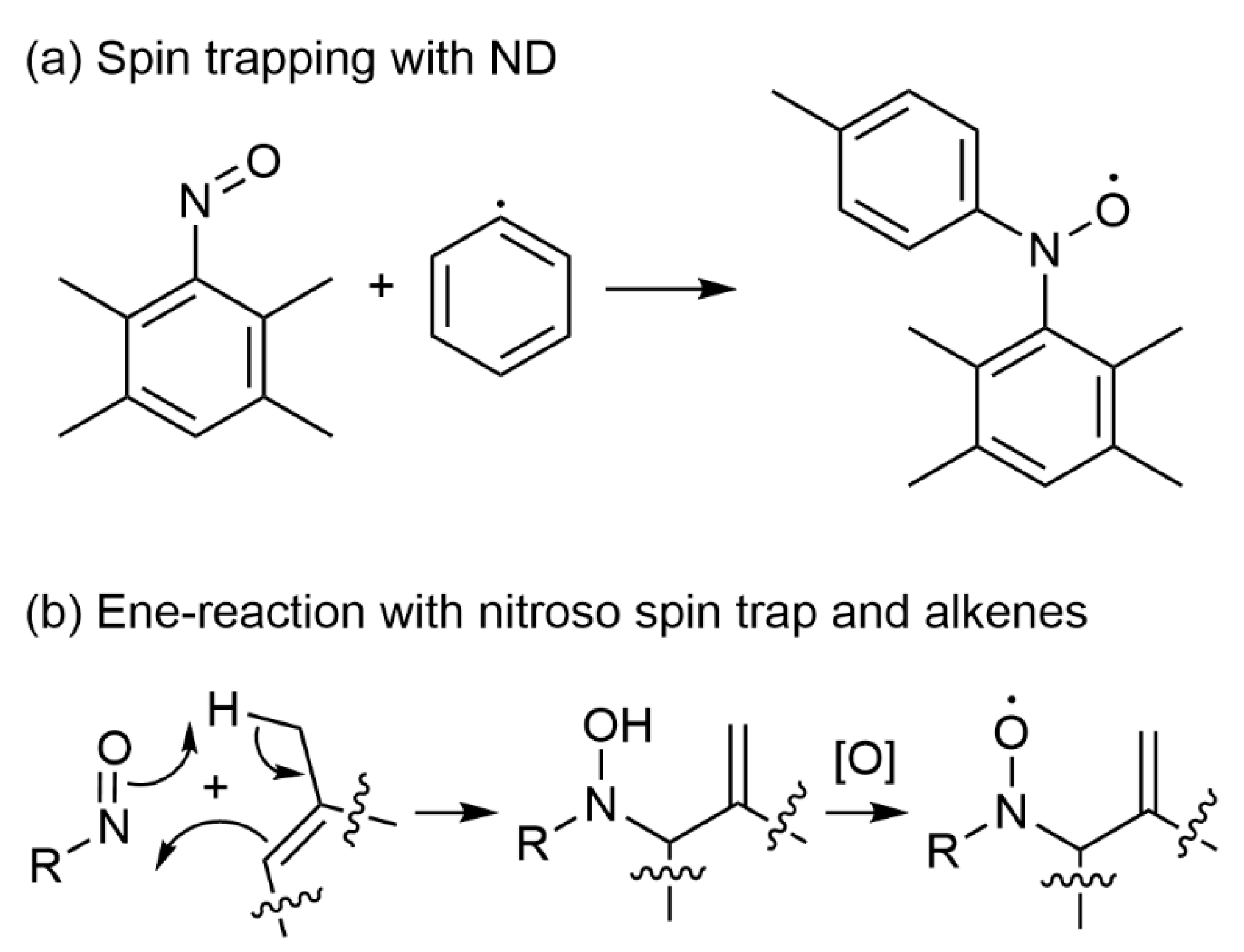
3.1. Nitroso Spin Traps
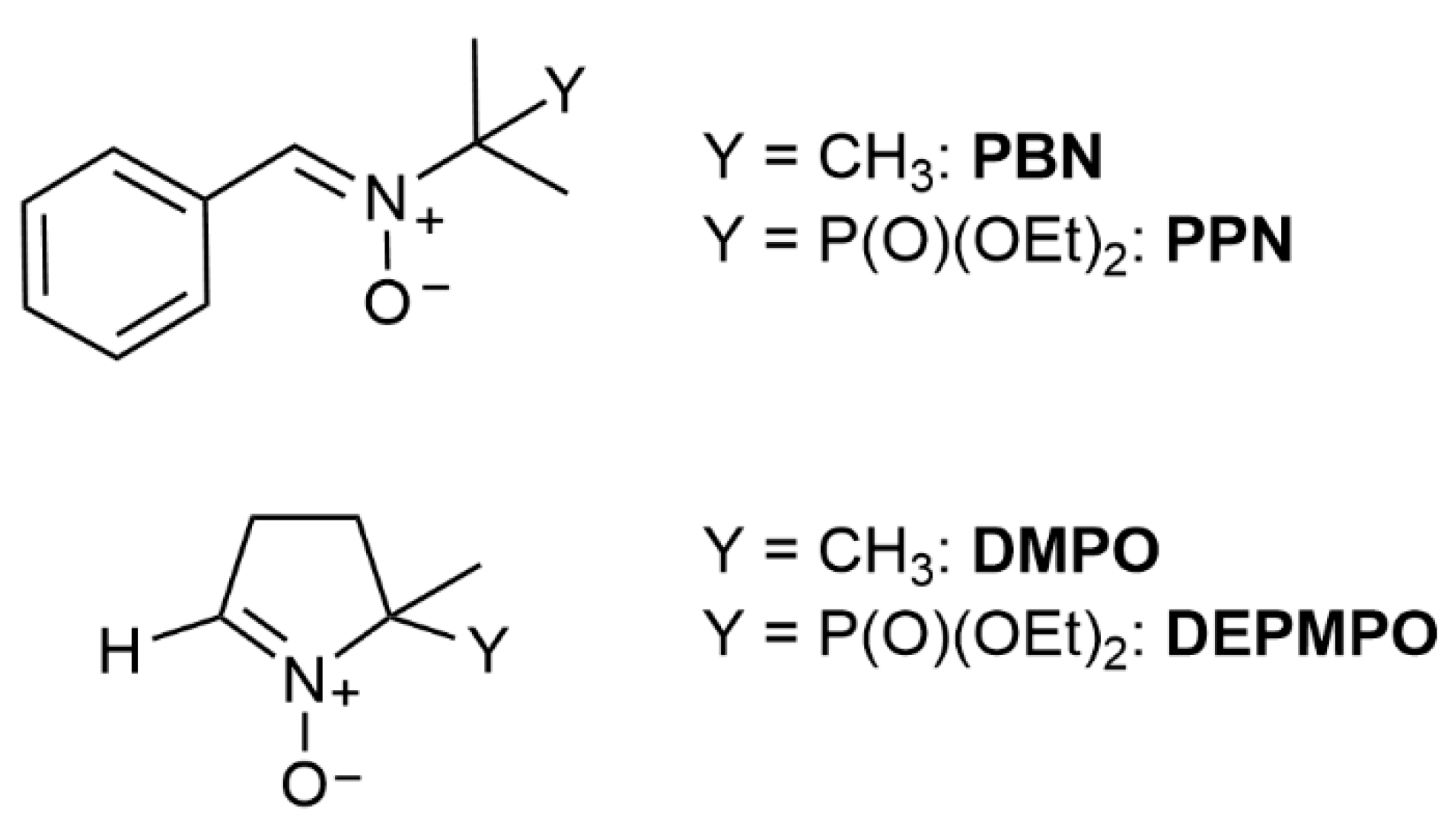
3.2. Linear Nitrone Spin Traps
3.2.1. Carbon-Centred Radical Spin Adducts of PBN
3.2.2. Silyl Radical Spin Adducts of PBN
3.2.3. Boryl Radical Spin Adducts of PBN in FRP
3.2.4. Phosphinoyl Radical Spin Adducts of PBN
3.2.5. Other PBN-Radical Adducts
3.3. Cyclic Nitrone Spin Traps
3.3.1. α-Aminoalkyl Radical Spin Adducts of DMPO
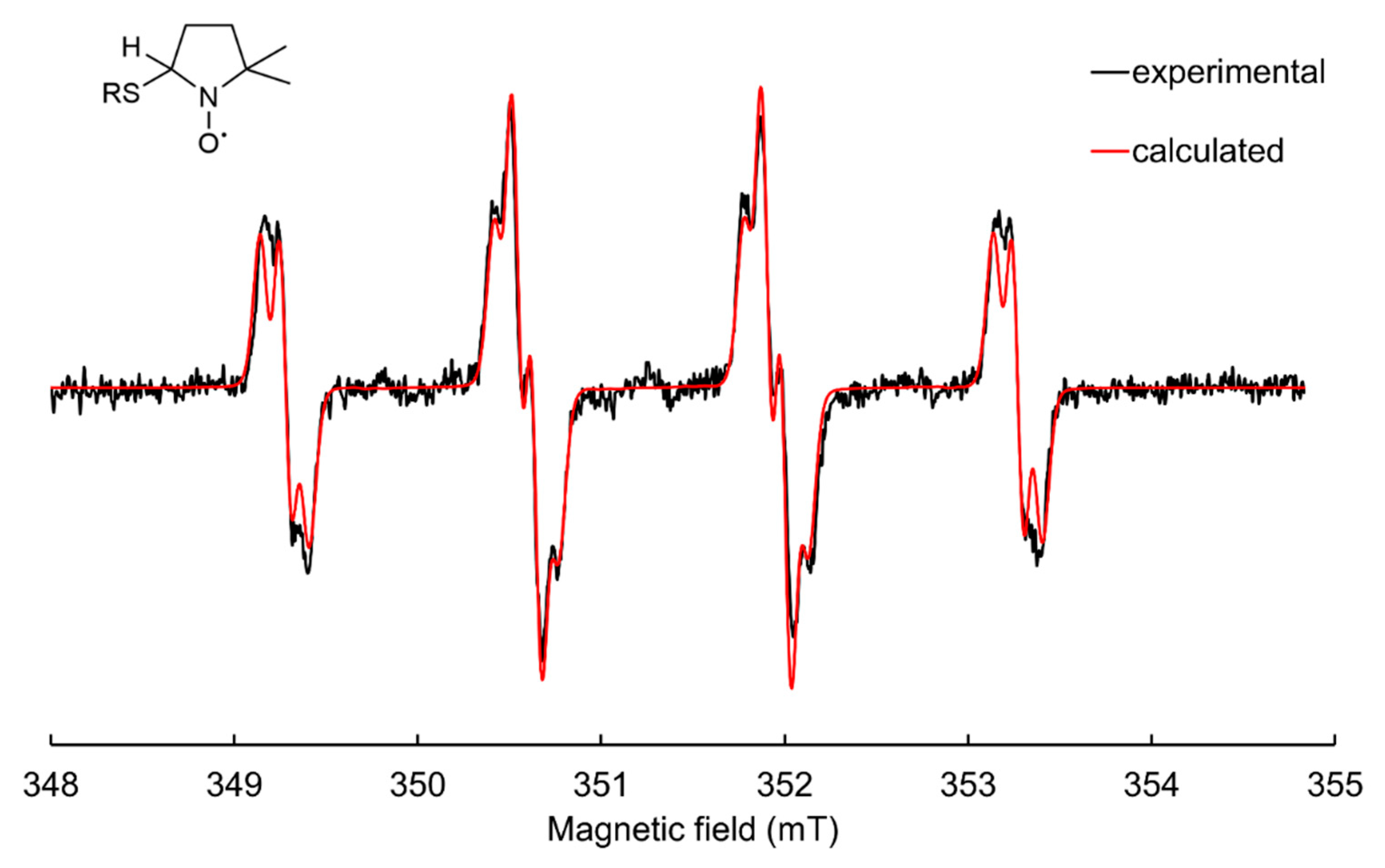
3.3.2. Thiyl Radical Spin Adducts of DMPO
3.3.3. Phosphorus-Centred Radical Spin Adducts of DMPO
3.3.4. (4-Methyl)phenyl Radical Spin Adduct of DMPO
4. Experimental Setup
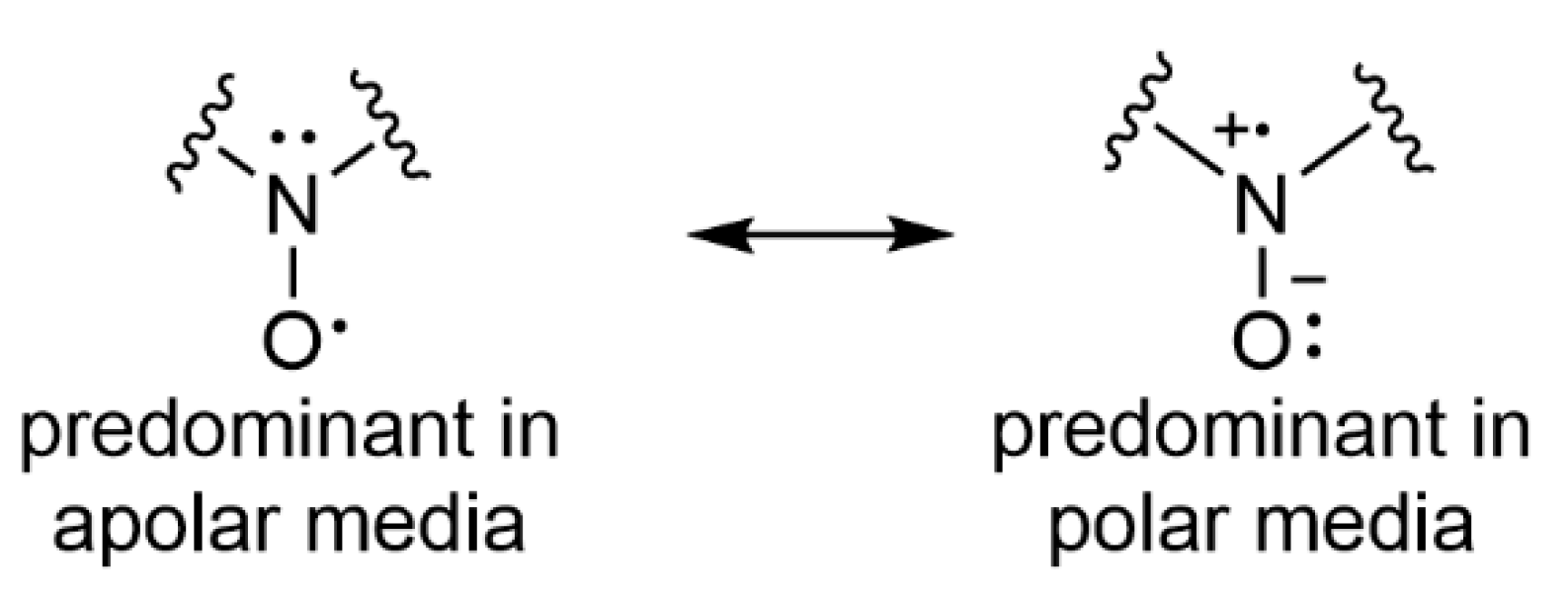
4.1. Solvent and Cell Choice
4.2. Spin Trap Concentration
4.3. Removal of Oxygen
4.4. Irradiation In Situ or Ex Situ
4.5. Acquisition Parameters
5. Interpretation of Results
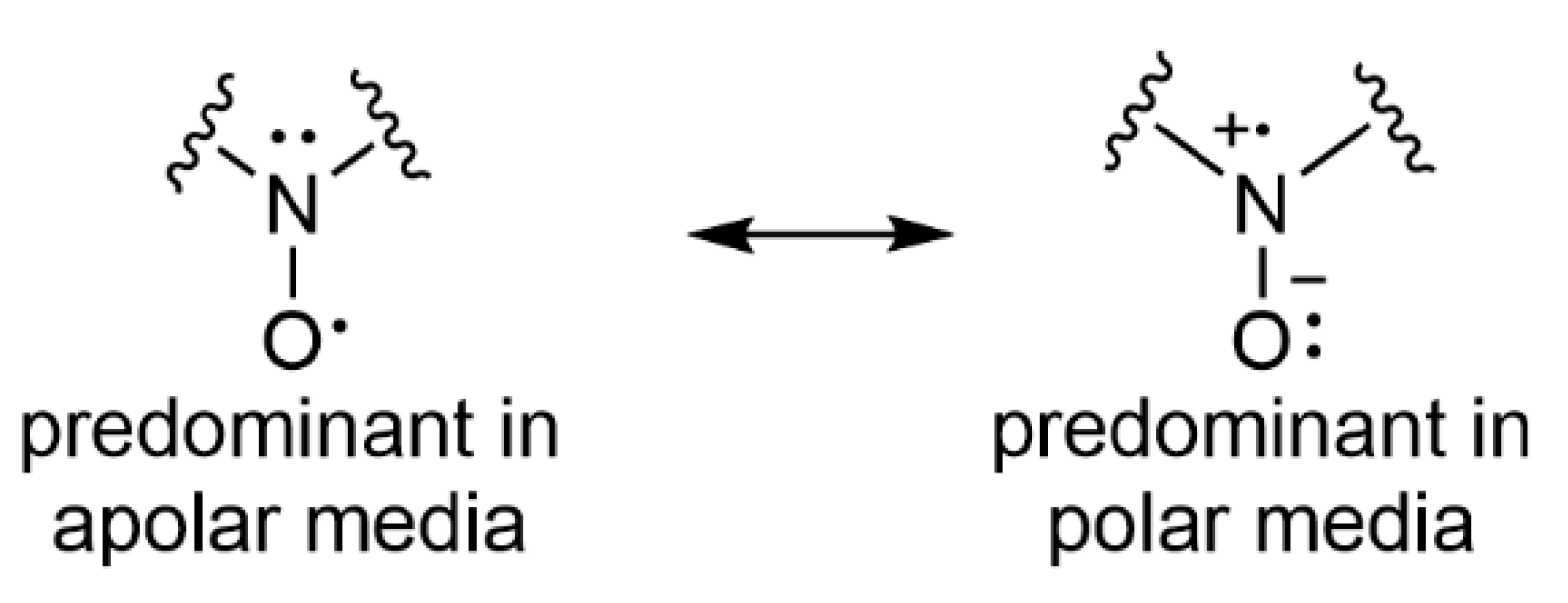
5.1. Signal Attribution
5.2. Kinetic Considerations
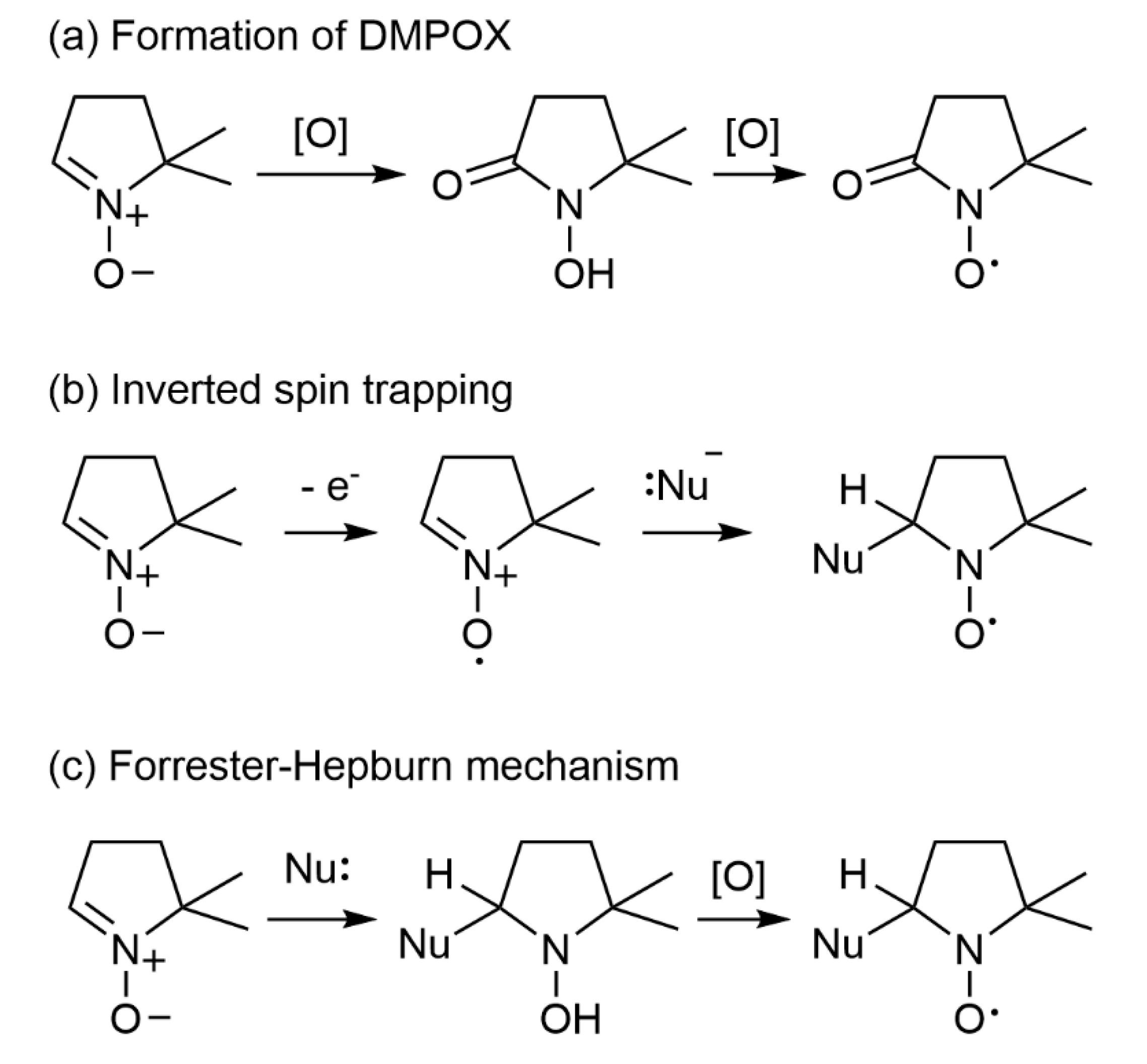
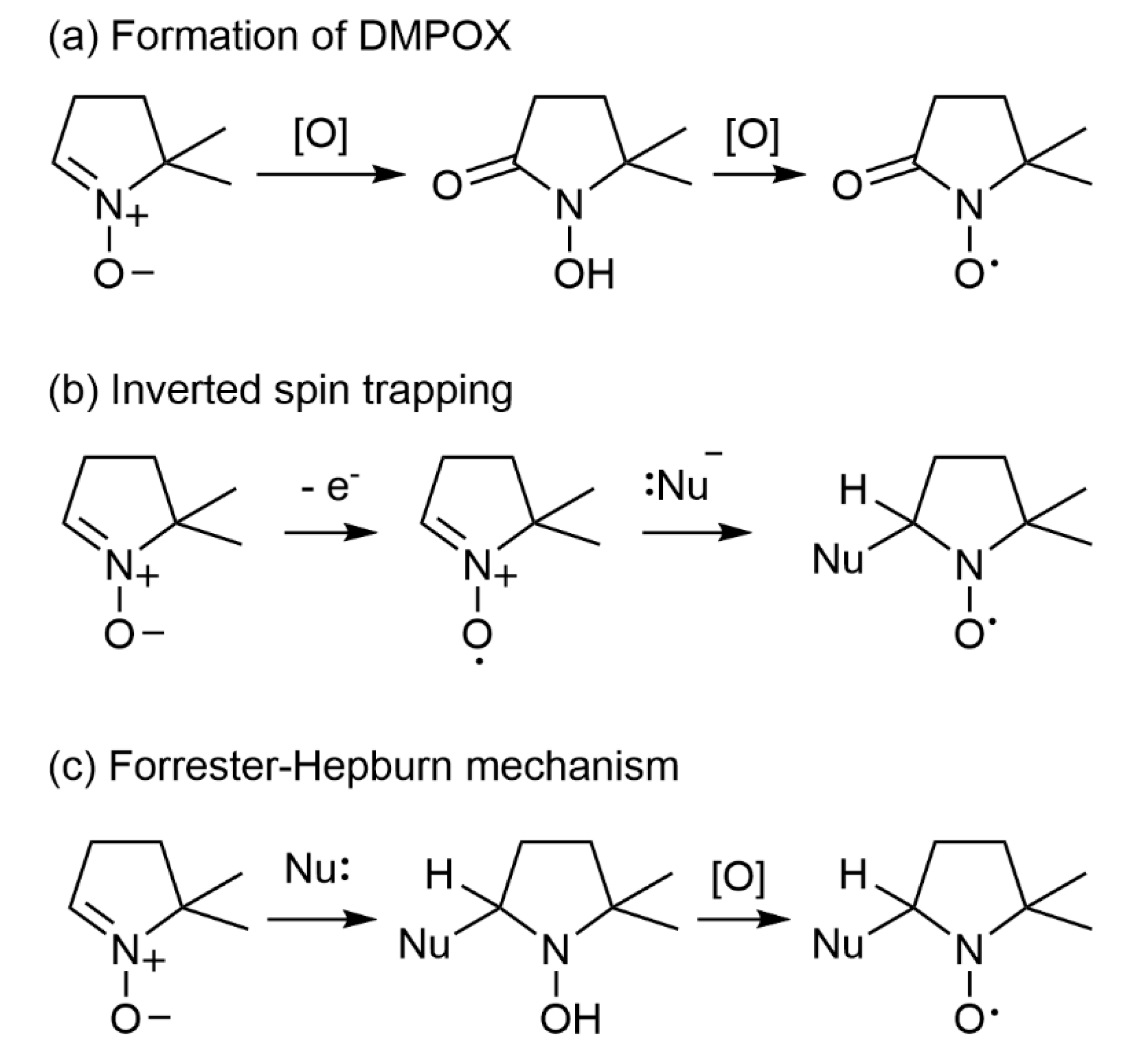
5.3. Potential Artefacts
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BDE | bond dissociation energy |
| BP | benzophenone |
| CP | cationic photopolymerization |
| CQ | camphorquinone |
| DEPMPO | 5-diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide |
| DMPA | 2,2′-dimethoxyphenyl acetophenone |
| DMPO | 5,5-dimethyl-1-pyrrolidine N-oxide |
| DMSO | dimethylsulfoxide |
| EPR | electron paramagnetic resonance |
| ESR | electron spin resonance |
| FRP | free radical photopolymerization |
| FRPCP | free radical promoted cationic photopolymerization |
| HFS | hyperfine splitting |
| Iod | bis(4-methylphenyl) iodonium hexafluorophosphate |
| Ir | iridium(III) complexes |
| LED | light-emitting diode |
| LFP | laser flash photolysis |
| MDEA | N-methyl diethanol amine |
| MNP | 1-methyl-1-nitrosopropane |
| ND | nitrosodurene |
| NIR | near infrared |
| NHC | N-heterocyclic carbene |
| NHC-BS | N-heterocyclic carbene-boryl sulfide |
| NP | nanoparticle |
| PBN | N-tert-α-phenyl-butylnitrone |
| PIS | photo-initiating system |
| PM | 4-allyloxy-3-methoxybenzoyl)diphenylphosphine oxide |
| PPN | diethyl 1-(N-benzylidene N-oxyamino) 1-methylethyl phosphonate |
| ST | spin trapping |
| SVD | singular value decomposition |
| TT | trimethylolpropane tris(3-mercaptopropionate) |
| TTMSS | tris(trimethylsilyl)silane |
References
- Pierau, L.; Elian, C.; Akimoto, J.; Ito, Y.; Caillol, S.; Versace, D.-L. Bio-sourced monomers and cationic photopolymerization–The green combination towards eco-friendly and non-toxic materials. Prog. Polym. Sci. 2022, 127, 101517. [Google Scholar] [CrossRef]
- Pierau, L.; Versace, D.-L. Light and Hydrogels: A New Generation of Antimicrobial Materials. Materials 2021, 14, 787. [Google Scholar] [CrossRef] [PubMed]
- Fouassier, J.-P.; Lalevée, J. Photoinitiators for Polymer Synthesis: Scope, Reactivity and Efficiency; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. [Google Scholar]
- Bao, Y.; Paunović, N.; Leroux, J. Challenges and Opportunities in 3D Printing of Biodegradable Medical Devices by Emerging Photopolymerization Techniques. Adv. Funct. Mater. 2022, 32, 2109864. [Google Scholar] [CrossRef]
- Versace, D.-L.; Breloy, L.; Palierse, E.; Coradin, T. Contributions of photochemistry to bio-based antibacterial polymer materials. J. Mater. Chem. B 2021, 9, 9624–9641. [Google Scholar] [CrossRef] [PubMed]
- Versace, D.-L.; Ramier, J.; Grande, D.; Andaloussi, S.A.; Dubot, P.; Hobeika, N.; Malval, J.-P.; Lalevee, J.; Renard, E.; Langlois, V. Versatile Photochemical Surface Modification of Biopolyester Microfibrous Scaffolds with Photogenerated Silver Nanoparticles for Antibacterial Activity. Adv. Health Mater. 2013, 2, 1008–1018. [Google Scholar] [CrossRef]
- Breloy, L.; Ouarabi, C.A.; Brosseau, A.; Dubot, P.; Brezova, V.; Andaloussi, S.A.; Malval, J.-P.; Versace, D.-L. β-Carotene/Limonene Derivatives/Eugenol: Green Synthesis of Antibacterial Coatings under Visible-Light Exposure. ACS Sustain. Chem. Eng. 2019, 7, 19591–19604. [Google Scholar] [CrossRef]
- Versace, D.-L.; Moran, G.; Belqat, M.; Spangenberg, A.; Méallet-Renault, R.; Abbad-Andaloussi, S.; Brezová, V.; Malval, J.-P. Highly Virulent Bactericidal Effects of Curcumin-Based μ-Cages Fabricated by Two-Photon Polymerization. ACS Appl. Mater. Interfaces 2020, 12, 5050–5057. [Google Scholar] [CrossRef]
- Jin, M.; Hong, H.; Xie, J.; Malval, J.-P.; Spangenberg, A.; Soppera, O.; Wan, D.; Pu, H.; Versace, D.-L.; Leclerc, T.; et al. π-conjugated sulfonium-based photoacid generators: An integrated molecular approach for efficient one and two-photon polymerization. Polym. Chem. 2014, 5, 4747–4755. [Google Scholar] [CrossRef]
- Jung, K.; Xu, J.; Zetterlund, P.B.; Boyer, C. Visible-Light-Regulated Controlled/Living Radical Polymerization in Miniemulsion. ACS Macro Lett. 2015, 4, 1139–1143. [Google Scholar] [CrossRef]
- Fu, C.; Xu, J.; Boyer, C. Photoacid-mediated ring opening polymerization driven by visible light. Chem. Commun. 2016, 52, 7126–7129. [Google Scholar] [CrossRef]
- Lorenzini, C.; Haider, A.; Kang, I.-K.; Sangermano, M.; Abbad-Andalloussi, S.; Mazeran, P.-E.; Lalevée, J.; Renard, E.; Langlois, V.; Versace, D.-L. Photoinduced Development of Antibacterial Materials Derived from Isosorbide Moiety. Biomacromolecules 2015, 16, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Scheiger, J.M.; Levkin, P.A. Design and Applications of Photoresponsive Hydrogels. Adv. Mater. 2019, 31, e1807333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malval, J.-P.; Morlet-Savary, F.; Chaumeil, H.; Balan, L.; Versace, D.-L.; Jin, M.; Defoin, A. Photophysical Properties and Two-Photon Polymerization Ability of a Nitroalkoxystilbene Derivative. J. Phys. Chem. C 2009, 113, 20812–20821. [Google Scholar] [CrossRef]
- Yagci, Y.; Jockusch, S.; Turro, N.J. Photoinitiated Polymerization: Advances, Challenges, and Opportunities. Macromolecules 2010, 43, 6245–6260. [Google Scholar] [CrossRef]
- Lalevee, J.; Roz, M.E.I.; Tehfe, M.A.; Alaaeddine, M.; Allonas, X.; Fouassier, J.-P. New Opportunities in Free Radical Photopolymerization and Free Radical Promoted Cationic Polymerization Based on the Silyl Radical Chemistry. J. Photopolym. Sci. Technol. 2009, 22, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Andrzejewska, E.; Grajek, K. Recent Advances in Photo-Induced Free-Radical Polymerization. MOJ Polym. Sci. 2017, 1, 58–60. [Google Scholar] [CrossRef] [Green Version]
- Breloy, L.; Brezová, V.; Barbieriková, Z.; Ito, Y.; Akimoto, J.; Chiappone, A.; Abbad-Andaloussi, S.; Malval, J.-P.; Versace, D.-L. Methacrylated Quinizarin Derivatives for Visible-Light Mediated Photopolymerization: Promising Applications in 3D-Printing Biosourced Materials under LED@405 nm. ACS Appl. Polym. Mater. 2022, 4, 210–228. [Google Scholar] [CrossRef]
- Breloy, L.; Brezová, V.; Richeter, S.; Clément, S.; Malval, J.-P.; Andalloussi, S.A.; Versace, D.-L. Bio-based porphyrins pyropheophorbide a and its Zn-complex as visible-light photosensitizers for free-radical photopolymerization. Polym. Chem. 2022, 13, 1658–1671. [Google Scholar] [CrossRef]
- Elian, C.; Brezová, V.; Sautrot-Ba, P.; Breza, M.; Versace, D.L. Lawsone Derivatives as Efficient Photopolymerizable Initiators for Free-Radical, Cationic Photopolymerizations, and Thiol—Ene Reactions. Polymers 2021, 13, 2015. [Google Scholar] [CrossRef]
- Peng, X.; Yao, M.; Xiao, P. Newly Synthesized Chromophore-linked Iodonium Salts as Photoinitiators of Free Radical Photopolymerization. Macromol. Chem. Phys. 2021, 222, 2100035. [Google Scholar] [CrossRef]
- Versace, D.-L.; Breloy, L.; Yagci, Y.; Yilmaz, I.; Yavuz, O. Design, Synthesis and Use of Phthalocyanines as a New Class of Visible-Light Photoinitiators for Free-Radical and Cationic Polymerizations. Polym. Chem. 2021, 12, 4291–4316. [Google Scholar] [CrossRef]
- Sautrot-Ba, P.; Jockusch, S.; Malval, J.-P.; Brezová, V.; Rivard, M.; Abbad-Andaloussi, S.; Blacha-Grzechnik, A.; Versace, D.-L. Quinizarin Derivatives as Photoinitiators for Free-Radical and Cationic Photopolymerizations in the Visible Spectral Range. Macromolecules 2020, 53, 1129–1141. [Google Scholar] [CrossRef]
- Breloy, L.; Mhanna, R.; Malval, J.-P.; Brezová, V.; Jacquemin, D.; Pascal, S.; Siri, O.; Versace, D.-L. Azacalixphyrins as an innovative alternative for the free-radical photopolymerization under visible and NIR irradiation without the need of co-initiators. Chem. Commun. 2021, 57, 8973–8976. [Google Scholar] [CrossRef] [PubMed]
- Iwamura, M.; Inamoto, N. Novel Formation of Nitroxide Radicals by Radical Addition to Nitrones. Bull. Chem. Soc. Jpn. 1967, 40, 703. [Google Scholar] [CrossRef] [Green Version]
- Janzen, E.G.; Blackburn, B.J. Detection and identification of short-lived free radicals by an electron spin resonance trapping technique. J. Am. Chem. Soc. 1968, 90, 5909–5910. [Google Scholar] [CrossRef]
- Chignell, C.F. Spin trapping studies of photochemical reactions. Pure Appl. Chem. 1990, 62, 301–305. [Google Scholar] [CrossRef]
- Ouari, O.; Hardy, M.; Karoui, H.; Tordo, P. Recent developments and applications of the coupled EPR/Spin trapping technique (EPR/ST). In Electron Paramagnetic Resonance; The Royal Society of Chemistry: London, UK, 2010; Volume 22, pp. 1–40. [Google Scholar] [CrossRef]
- Lauricella, R.; Allouch, A.; Roubaud, V.; Bouteiller, J.C.; Tuccio, B. A New Kinetic Approach to the Evaluation of Rate Constants for the Spin Trapping of Superoxide/Hydroperoxyl Radical by Nitrones in Aqueous Media. Org. Biomol. Chem. 2004, 2, 1304–1309. [Google Scholar] [CrossRef]
- Criqui, A.; Lalevée, J.; Allonas, X.; Fouassier, J.-P. Electron Spin Resonance Spin Trapping Technique: Application to the Cleavage Process of Photoinitiators. Macromol. Chem. Phys. 2008, 209, 2223–2231. [Google Scholar] [CrossRef]
- Janzen, E.G.; Stronks, H.J.; DuBose, C.M.; Poyer, J.L.; McCay, P.B. Chemistry and Biology of Spin-Trapping Radicals Associated with Halocarbon Metabolism in Vitro and in Vivo. Environ. Health Perspect. 1985, 64, 151. [Google Scholar] [CrossRef]
- Lauricella, R.; Tuccio, B. Detection and Characterisation of Free Radicals After Spin Trapping. In Electron Paramagnetic Resonance Spectroscopy: Applications; Bertrand, P., Ed.; Springer International Publishing: Cham, Switzerland, 2020; pp. 51–82. [Google Scholar] [CrossRef]
- Turro, N.J.; Khudyakov, I.V. Single-phase primary electron spin polarization transfer in spin-trapping reactions. Chem. Phys. Lett. 1992, 193, 546–552. [Google Scholar] [CrossRef]
- Terabe, S.; Kuruma, K.; Konaka, R. ChemInform Abstract: Spin Trapping by Use of Nitroso-Compounds Part 6, Nitrosodurene And Other Nitrosobenzene Derivatives. Chem. Inf. 1973, 4, 1252–1258. [Google Scholar] [CrossRef]
- Sautrot-Ba, P.; Brezová, V.; Malval, J.-P.; Chiappone, A.; Breloy, L.; Abbad-Andaloussi, S.; Versace, D.-L. Purpurin derivatives as visible-light photosensitizers for 3D printing and valuable biological applications. Polym. Chem. 2021, 12, 2627–2642. [Google Scholar] [CrossRef]
- Sueishi, Y.; Nishihara, Y. Spin Trapping Chemistry of the Diphenylphosphinyl Radical. J. Chem. Res. 2001, 2001, 84–86. [Google Scholar] [CrossRef]
- Chandra, H.; Davidson, I.M.T.; Symons, M.C.R. Unstable intermediates. Part 202. The use of spin traps to study trialkylsilyl and related radicals. J. Chem. Soc. Perkin Trans. 1982, 2, 1353–1356. [Google Scholar] [CrossRef]
- Tuccio, B.; Zeghdaoui, A.; Finet, J.-P.; Cerri, V.; Tordo, P. Use of new β-phosphorylated nitrones for the spin trapping of free radicals. Res. Chem. Intermed. 1996, 22, 393–404. [Google Scholar] [CrossRef]
- Lalevée, J.; Blanchard, N.; El-Roz, M.; Graff, B.; Allonas, X.; Fouassier, J.P. New Photoinitiators Based on the Silyl Radical Chemistry: Polymerization Ability, Esr Spin Trapping, and Laser Flash Photolysis Investigation. Macromolecules 2008, 41, 4180–4186. [Google Scholar] [CrossRef]
- Versace, D.L.; Tehfe, M.A.; Lalevée, J.; Casarotto, V.; Blanchard, N.; Morlet-Savary, F.; Fouassier, J.P. Silyloxyamines as Sources of Silyl Radicals: Esr Spin-Trapping, Laser Flash Photolysis Investigation, and Photopolymerization Ability. J. Phys. Org. Chem. 2011, 24, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Telitel, S.; Vallet, A.-L.; Schweizer, S.; Delpech, B.; Blanchard, N.; Morlet-Savary, F.; Graff, B.; Curran, D.P.; Robert, M.; Lacôte, E.; et al. Formation of N-Heterocyclic Carbene–Boryl Radicals through Electrochemical and Photochemical Cleavage of the B–S bond in N-Heterocyclic Carbene–Boryl Sulfides. J. Am. Chem. Soc. 2013, 135, 16938–16947. [Google Scholar] [CrossRef]
- Bouzrati-Zerelli, M.; Maier, M.; Dietlin, C.; Fabrice, M.-S.; Fouassier, J.P.; Klee, J.E.; Lalevée, J. A novel photoinitiating system producing germyl radicals for the polymerization of representative methacrylate resins: Camphorquinone/R 3 GeH/iodonium salt. Dent. Mater. 2016, 32, 1226–1234. [Google Scholar] [CrossRef]
- Baumann, H.; Timpe, H.J.; Zubarev, V.E.; Fok, N.V.; Mel’nikov, M.Y. Lichtinitiierte Polymer- Und Polymerisations-Reaktionen Xxii: Untersuchungen Zur Photolyse Von Photo-Initiatoren Mit Benzyliden-Tert.-Butylamin-N-Oxid Als Spin-Trap. J. Photochem. 1985, 30, 487–500. [Google Scholar] [CrossRef]
- Kura, H.; Oka, H.; Ohwa, M.; Matsumura, T.; Kimura, A.; Iwasaki, Y.; Ohno, T.; Matsumura, M.; Murai, H. Photochemistry and photocuring properties of thiol-substituted α-aminoalkylphenone as radical photoinitiator. J. Polym. Sci. Part B Polym. Phys. 2005, 43, 1684–1695. [Google Scholar] [CrossRef]
- Teshima, W.; Nomura, Y.; Tanaka, N.; Urabe, H.; Okazaki, M.; Nahara, Y. ESR study of camphorquinone/amine photoinitiator systems using blue light-emitting diodes. Biomaterials 2003, 24, 2097–2103. [Google Scholar] [CrossRef]
- Jockusch, S.; Landis, M.S.; Freiermuth, B.; Turro, N.J. Photochemistry and Photophysics of α-Hydroxy Ketones. Macromolecules 2001, 34, 1619–1626. [Google Scholar] [CrossRef]
- Versace, D.-L.; Dubot, P.; Cenedese, P.; Lalevée, J.; Soppera, O.; Malval, J.-P.; Renard, E.; Langlois, V. Natural biopolymer surface of poly(3-hydroxybutyrate-co-3-hydroxyvalerate)-photoinduced modification with triarylsulfonium salts. Green Chem. 2012, 14, 788–798. [Google Scholar] [CrossRef]
- Lalevée, J.; Poupart, R.; Bourgon, J.; Fouassier, J.-P.; Versace, D.-L. In situ production of visible light absorbing Ti-based nanoparticles in solution and in a photopolymerizable cationic matrix. Chem. Commun. 2015, 51, 5762–5765. [Google Scholar] [CrossRef]
- Garra, P.; Brunel, D.; Noirbent, G.; Graff, B.; Morlet-Savary, F.; Dietlin, C.; Sidorkin, V.F.; Dumur, F.; Duché, D.; Gigmes, D.; et al. Ferrocene-based (photo)redox polymerization under long wavelengths. Polym. Chem. 2019, 10, 1431–1441. [Google Scholar] [CrossRef]
- Lalevée, J.; Allonas, X.; Fouassier, J.; Tachi, H.; Izumitani, A.; Shirai, M.; Tsunooka, M. Investigation of the photochemical properties of an important class of photobase generators: The O-acyloximes. J. Photochem. Photobiol. A Chem. 2002, 151, 27–37. [Google Scholar] [CrossRef]
- Chandra, H.; Davidson, I.M.T.; Symons, M.C.R. Use of spin traps in the study of silyl radicals in the gas phase. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1983, 79, 2705–2711. [Google Scholar] [CrossRef]
- Detrembleur, C.; Versace, D.-L.; Piette, Y.; Hurtgen, M.; Jérôme, C.; Lalevée, J.; Debuigne, A. Synthetic and mechanistic inputs of photochemistry into the bis-acetylacetonatocobalt-mediated radical polymerization of n-butyl acrylate and vinyl acetate. Polym. Chem. 2011, 3, 1856–1866. [Google Scholar] [CrossRef]
- El-Roz, M.; Lalevée, J.; Allonas, X.; Fouassier, J.P. Mechanistic Investigation of the Silane, Germane, and Stannane Behavior When Incorporated in Type I and Type II Photoinitiators of Polymerization in Aerated Media. Macromolecules 2009, 42, 8725–8732. [Google Scholar] [CrossRef]
- Lalevée, J.; Peter, M.; Dumur, F.; Gigmes, D.; Blanchard, N.; Tehfe, M.-A.; Morlet-Savary, F.; Fouassier, J.P. Subtle Ligand Effects in Oxidative Photocatalysis with Iridium Complexes: Application to Photopolymerization. Chem.—A Eur. J. 2011, 17, 15027–15031. [Google Scholar] [CrossRef] [PubMed]
- Tehfe, M.A.; Lalevée, J.; Allonas, X.; Fouassier, J.P. Long Wavelength Cationic Photopolymerization in Aerated Media: A Remarkable Titanocene/Tris(trimethylsilyl)silane/Onium Salt Photoinitiating System. Macromolecules 2009, 42, 8669–8674. [Google Scholar] [CrossRef]
- Lalevée, J.; Blanchard, N.; Tehfe, M.-A.; Chany, A.-C.; Fouassier, J.-P. New Boryl Radicals Derived from N-Heteroaryl Boranes: Generation and Reactivity. Chem.—A Eur. J. 2010, 16, 12920–12927. [Google Scholar] [CrossRef]
- Baban, J.A.; Marti, V.P.; Roberts, B.P. Ligated Boryl Radicals. Part 2. Electron Spin Resonance Studies of Trialkylamin–Boryl Radicals. J. Chem. Soc. Perkin Trans. 1985, 2, 1723–1733. [Google Scholar] [CrossRef]
- Lacôte, E.; Curran, D.P.; Lalevée, J. NHC-Boranes: Air- and Water-tolerant Co-initiators for Type II Photopolymerizations. CHIMIA 2012, 66, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Jockusch, S.; Koptyug, I.V.; McGarry, P.F.; Sluggett, G.W.; Turro, N.J.; Watkins, D.M. A Steady-State and Picosecond Pump-Probe Investigation of the Photophysics of an Acyl and a Bis(acyl)phosphine Oxide. J. Am. Chem. Soc. 1997, 119, 11495–11501. [Google Scholar] [CrossRef]
- Jockusch, S.; Turro, N.J. Phosphinoyl Radicals: Structure and Reactivity. A Laser Flash Photolysis and Time-Resolved ESR Investigation. J. Am. Chem. Soc. 1998, 120, 11773–11777. [Google Scholar] [CrossRef]
- Sueishi, Y.; Miyake, Y. Spin Trapping of Phosphorus-Centered Radicals Produced by the Reactions of Dibenzoyl Peroxide with Organophosphorus Compounds. Bull. Chem. Soc. Jpn. 1997, 70, 397–403. [Google Scholar] [CrossRef]
- Lalevée, J.; Morlet-Savary, F.; Tehfe, M.A.; Graff, B.; Fouassier, J.P. Photosensitized Formation of Phosphorus-Centered Radicals: Application to the Design of Photoinitiating Systems. Macromolecules 2012, 45, 5032–5039. [Google Scholar] [CrossRef]
- Breloy, L.; Negrell, C.; Mora, A.-S.; Li, W.S.J.; Brezová, V.; Caillol, S.; Versace, D.-L. Vanillin derivative as performing type I photoinitiator. Eur. Polym. J. 2020, 132, 109727. [Google Scholar] [CrossRef]
- Graceffa, P. Spin trapping the cysteine thiyl radical with phenyl-N-t-butylnitrone. Biochim. Biophys. Acta (BBA)—Protein Struct. Mol. Enzym. 1988, 954, 227–230. [Google Scholar] [CrossRef]
- Ito, O.; Matsuda, M. Flash Photolysis Study for Substituent and Solvent Effects on Spin-Trapping Rates of Phenylthiyl Radicals with Nitrones. Bull. Chem. Soc. Jpn. 1984, 57, 1745–1749. [Google Scholar] [CrossRef] [Green Version]
- Frejaville, C.; Karoui, H.; Culcasi, M.; Pietri, S.; Lauricella, R.; Tordo, P. 5-Diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide (DEPMPO): A new phosphorylated nitrone for the efficient In Vitro and In Vivo spin trapping of oxygen-centred radicals. J. Chem. Soc. Chem. Commun. 1994, 15, 1793–1794. [Google Scholar] [CrossRef]
- Frejaville, C.; Karoui, H.; Tuccio, B.; Le Moigne, F.; Culcasi, M.; Pietri, S.; Lauricella, R.; Tordo, P. 5-(Diethoxyphosphoryl)-5-methyl-1-pyrroline N-oxide: A New Efficient Phosphorylated Nitrone for the in Vitro and in Vivo Spin Trapping of Oxygen-Centered Radicals. J. Med. Chem. 1995, 38, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Breloy, L.; Brezová, V.; Blacha-Grzechnik, A.; Presset, M.; Yildirim, M.S.; Yilmaz, I.; Yagci, Y.; Versace, D.-L. Visible Light Anthraquinone Functional Phthalocyanine Photoinitiator for Free-Radical and Cationic Polymerizations. Macromolecules 2020, 53, 112–124. [Google Scholar] [CrossRef]
- Breloy, L.; Losantos, R.; Sampedro, D.; Marazzi, M.; Malval, J.-P.; Heo, Y.; Akimoto, J.; Ito, Y.; Brezová, V.; Versace, D.-L. Allyl amino-thioxanthone derivatives as highly efficient visible light H-donors and co-polymerizable photoinitiators. Polym. Chem. 2020, 11, 4297–4312. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Bowman, C.N. Thiol–Ene Click Chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Lee, T.Y.; Roper, T. Thiol-enes: Chemistry of the past with promise for the future. J. Polym. Sci. Part A Polym. Chem. 2004, 42, 5301–5338. [Google Scholar] [CrossRef]
- Karlsson, H.; Lagercrantz, C.; Lemmich, J.; Torssell, K.; Shimizu, A. Spin Trapping of Some Phosphorus-centered Radical Species. Acta Chem. Scand. 1970, 24, 3411–3413. [Google Scholar] [CrossRef]
- Abe, Y.; Seno, S.-Y.; Sakakibara, K.; Hirota, M. Spin trapping of oxygen-centred radicals by substituted N-benzylidene-tert-butylamine N-oxides. J. Chem. Soc. Perkin Trans. 2 1991, 12, 897–903. [Google Scholar] [CrossRef]
- Murofushi, K.; Abe, K.; Hirota, M. Substituent effect on the spin-trapping reactions of substituted N-benzylidene-t-butylamine N-oxides. J. Chem. Soc. Perkin Trans. 2 1987, 12, 1829–1833. [Google Scholar] [CrossRef]
- Dalal, D.P.; Eaton, S.S.; Eaton, G.R. The effects of lossy solvents on quantitative EPR studies. J. Magn. Reson. 1981, 44, 415–428. [Google Scholar] [CrossRef]
- Buettner, G.R. Spin Trapping: ESR parameters of spin adducts 1474 1528V. Free Radic. Biol. Med. 1987, 3, 259–303. [Google Scholar] [CrossRef]
- Leinisch, F.; Jiang, J.; DeRose, E.F.; Khramtsov, V.V.; Mason, R.P. Investigation of spin-trapping artifacts formed by the Forrester-Hepburn mechanism. Free Radic. Biol. Med. 2013, 65, 1497–1505. [Google Scholar] [CrossRef] [Green Version]
- Buettner, G.R.; Kiminyo, K.P. Optimal EPR detection of weak nitroxide spin adduct and ascorbyl free radical signals. J. Biochem. Biophys. Methods 1992, 24, 147–151. [Google Scholar] [CrossRef]
- Duling, D.R. Simulation of Multiple Isotropic Spin-Trap EPR Spectra. J. Magn. Reson. Ser. B 1994, 104, 105–110. [Google Scholar] [CrossRef]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef]
- Kotake, Y.; Kuwata, K. Electron Spin Resonance Study on the Difference of Structure in Diastereomeric Nitroxyl Radicals. Bull. Chem. Soc. Jpn. 1981, 54, 394–398. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, E.; Rosen, G.M.; Rauckman, E.J. Spin Trapping of Superoxide and Hydroxyl Radical: Practical Aspects. Arch. Biochem. Biophys. 1980, 200, 1–16. [Google Scholar] [CrossRef]
- Ranguelova, K.; Mason, R.P. The fidelity of spin trapping with DMPO in biological systems. Org. Magn. Reson. 2011, 49, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Rosen, G.M.; Rauckman, E.J. Spin trapping of the primary radical involved in the activation of the carcinogen N-hydroxy-2-acetylaminofluorene by cumene hydroperoxide-hematin. Mol. Pharmacol. 1980, 17, 233–238. [Google Scholar] [PubMed]
- Chandra, H.; Symons, M.C.R. Hydration of spin-trap cations as a source of hydroxyl adducts. J. Chem. Soc. Chem. Commun. 1986, 16, 1301–1302. [Google Scholar] [CrossRef]
- Eberson, L. Inverted Spin Trapping’. Reactions between the Radical Cation of A-Phenyl-N-Tert-Butylnitrone and Ionic and Neutral Nucleophiles. J. Chem. Soc. Perkin Trans. 1992, 2, 1807–1813. [Google Scholar] [CrossRef]
- Zubarev, V.; Brede, O. Direct detection of the cation radical of the spin trap α-phenyl-N-tert-butylnitrone. J. Chem. Soc. Perkin Trans. 1994, 2, 1821–1828. [Google Scholar] [CrossRef]
- Forrester, A.R.; Hepburn, S.P. Spin Traps. A Cautionary Note. J. Chem. Soc. C 1971, 701–703. [Google Scholar] [CrossRef]
- Eberson, L. Inverted Spin Trapping. Part Iii. Further Studies on the Chemical and Photochemical Oxidation of Spin Traps in the Presence of Nucleophiles. J. Chem. Soc. Perkin Trans. 1994, 2, 171–176. [Google Scholar] [CrossRef]
- Tuccio, B.; Lauricella, R.; Fréjaville, C.; Bouteiller, J.-C.; Tordo, P. Decay of the hydroperoxyl spin adduct of 5-diethoxyphosphoryl-5-methyl-1-pyrroline N-oxide: An EPR kinetic study. J. Chem. Soc. Perkin Trans. 1995, 2, 295–298. [Google Scholar] [CrossRef]
- Potapenko, D.I.; Bagryanskaya, E.G.; Tsentalovich, Y.P.; Reznikov, V.A.; Clanton, T.L.; Khramtsov, V.V. Reversible Reactions of Thiols and Thiyl Radicals with Nitrone Spin Traps. J. Phys. Chem. B 2004, 108, 9315–9324. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peyrot, F.; Lajnef, S.; Versace, D.-L. Electron Paramagnetic Resonance Spin Trapping (EPR–ST) Technique in Photopolymerization Processes. Catalysts 2022, 12, 772. https://doi.org/10.3390/catal12070772
Peyrot F, Lajnef S, Versace D-L. Electron Paramagnetic Resonance Spin Trapping (EPR–ST) Technique in Photopolymerization Processes. Catalysts. 2022; 12(7):772. https://doi.org/10.3390/catal12070772
Chicago/Turabian StylePeyrot, Fabienne, Sonia Lajnef, and Davy-Louis Versace. 2022. "Electron Paramagnetic Resonance Spin Trapping (EPR–ST) Technique in Photopolymerization Processes" Catalysts 12, no. 7: 772. https://doi.org/10.3390/catal12070772