
Insights into the Capture of CO2 by Nickel Hydride Complexes




Abstract
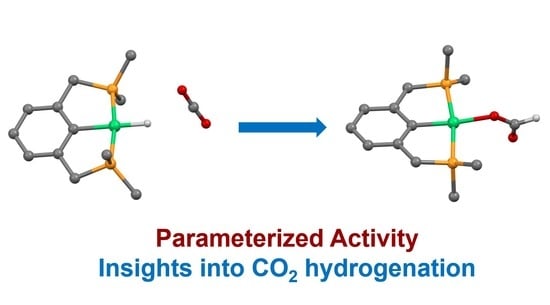
:
1. Introduction
2. Computational Methods
3. Results and Discussion
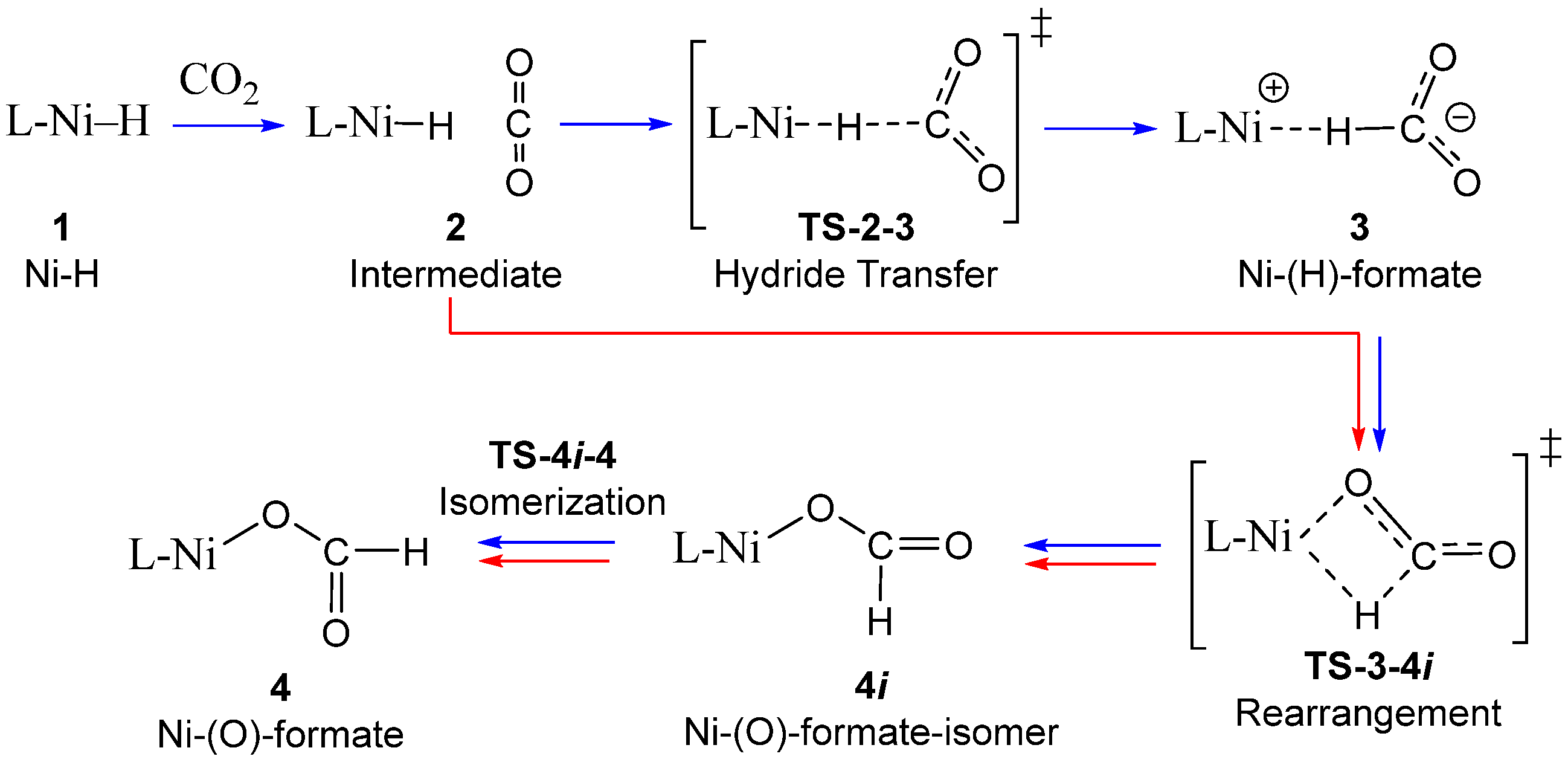
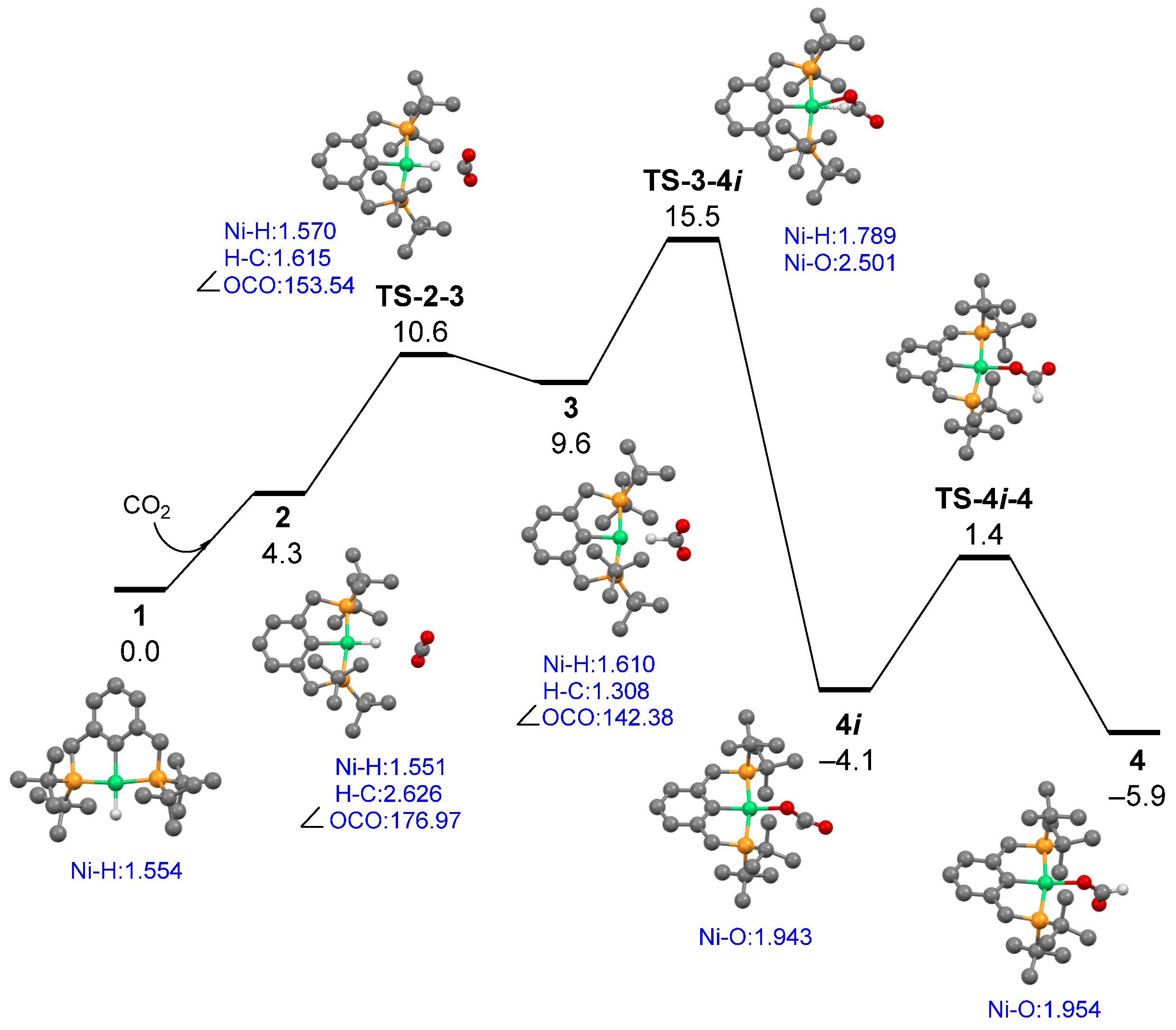
3.1. Mechanism of CO2 Insertion into Ni–H Bond of tBu2(PCP)Ni–H
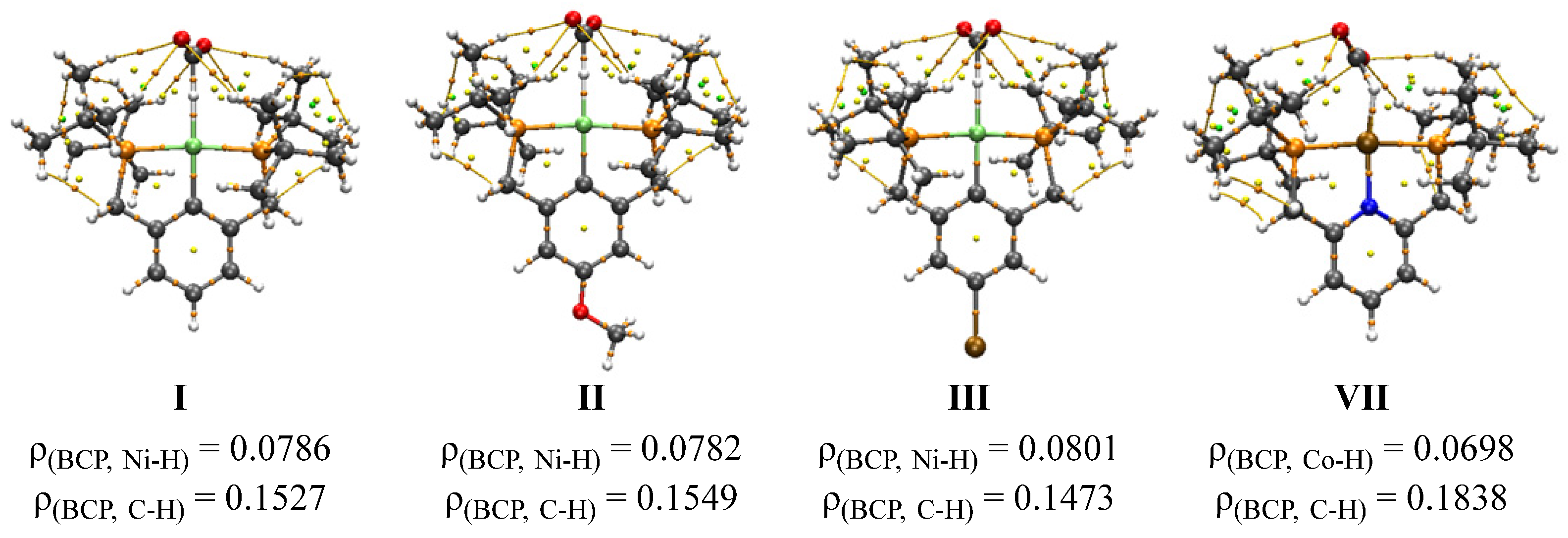
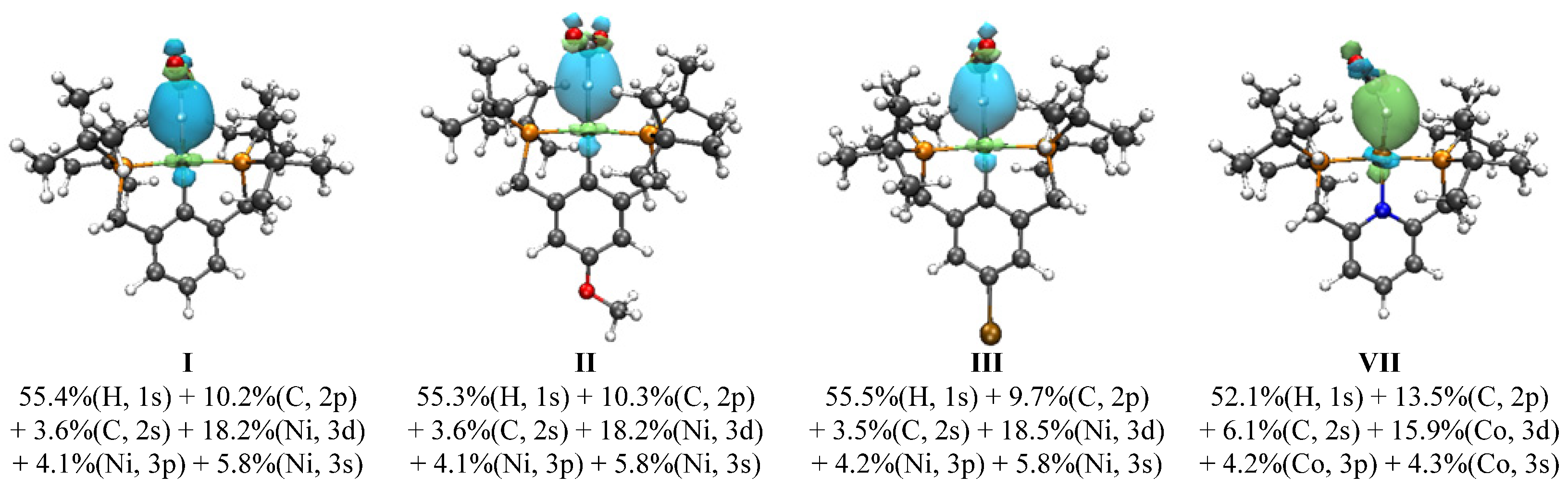
3.2. Analysis of Ni-(H)-Formate Intermidiate 3
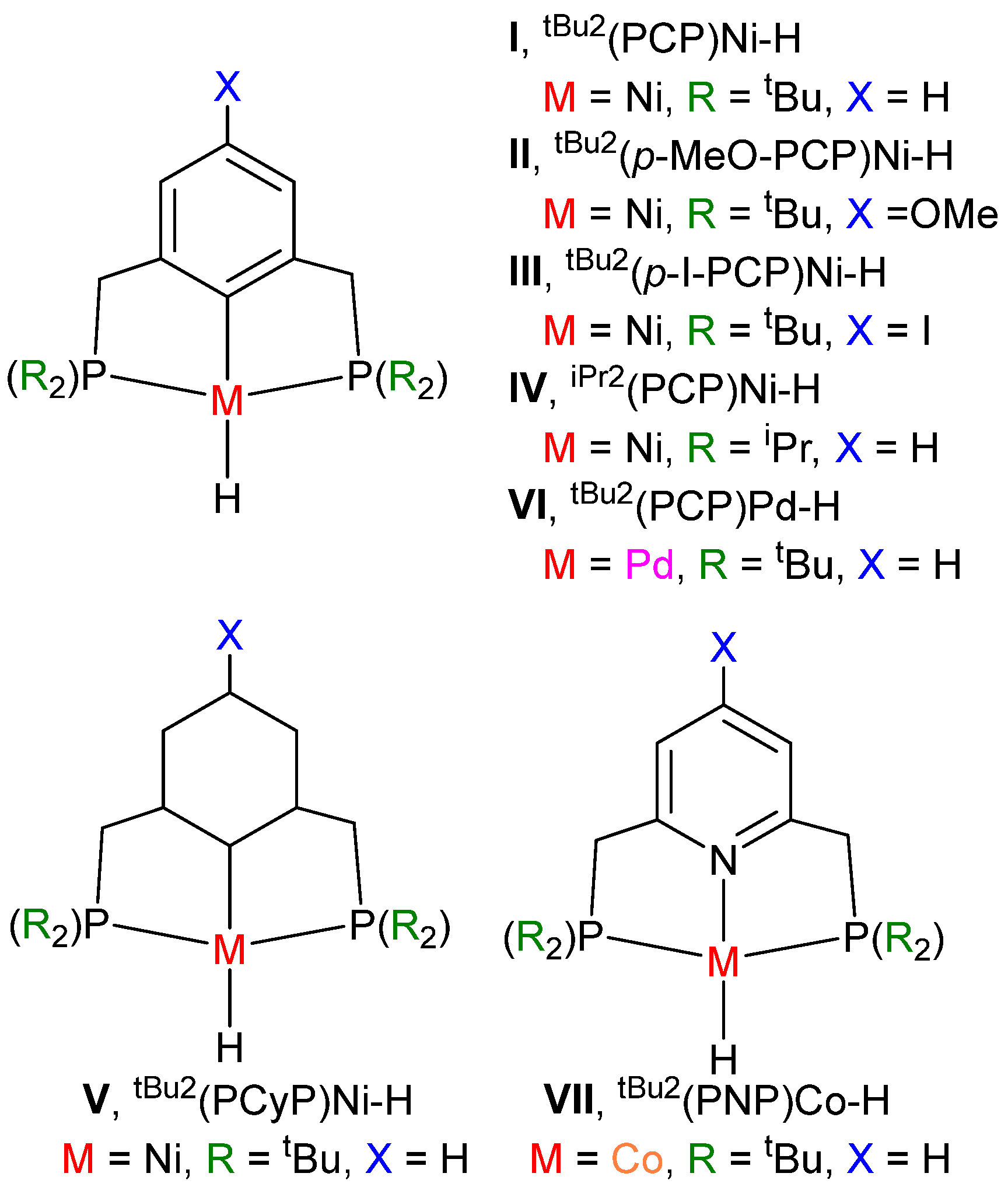
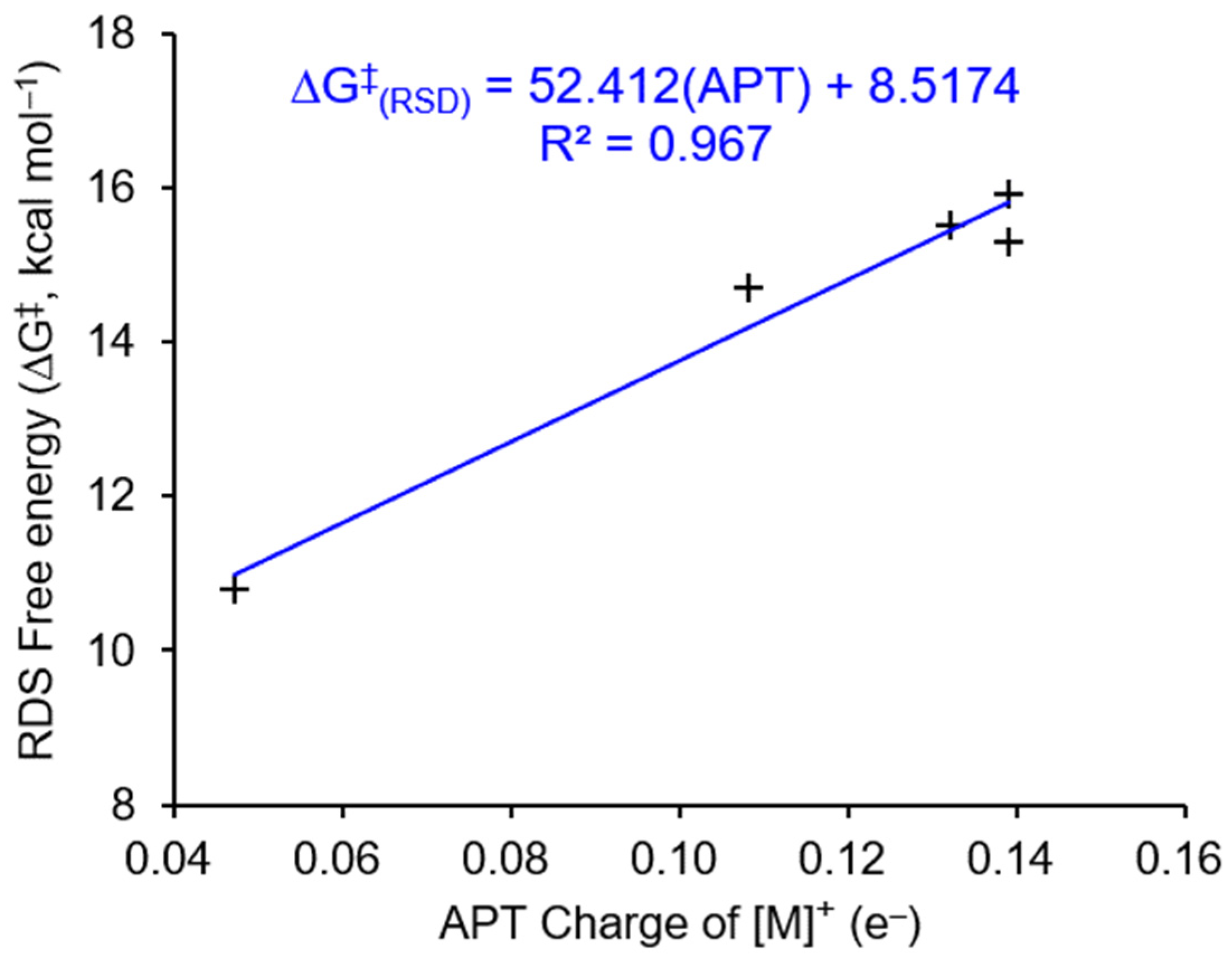
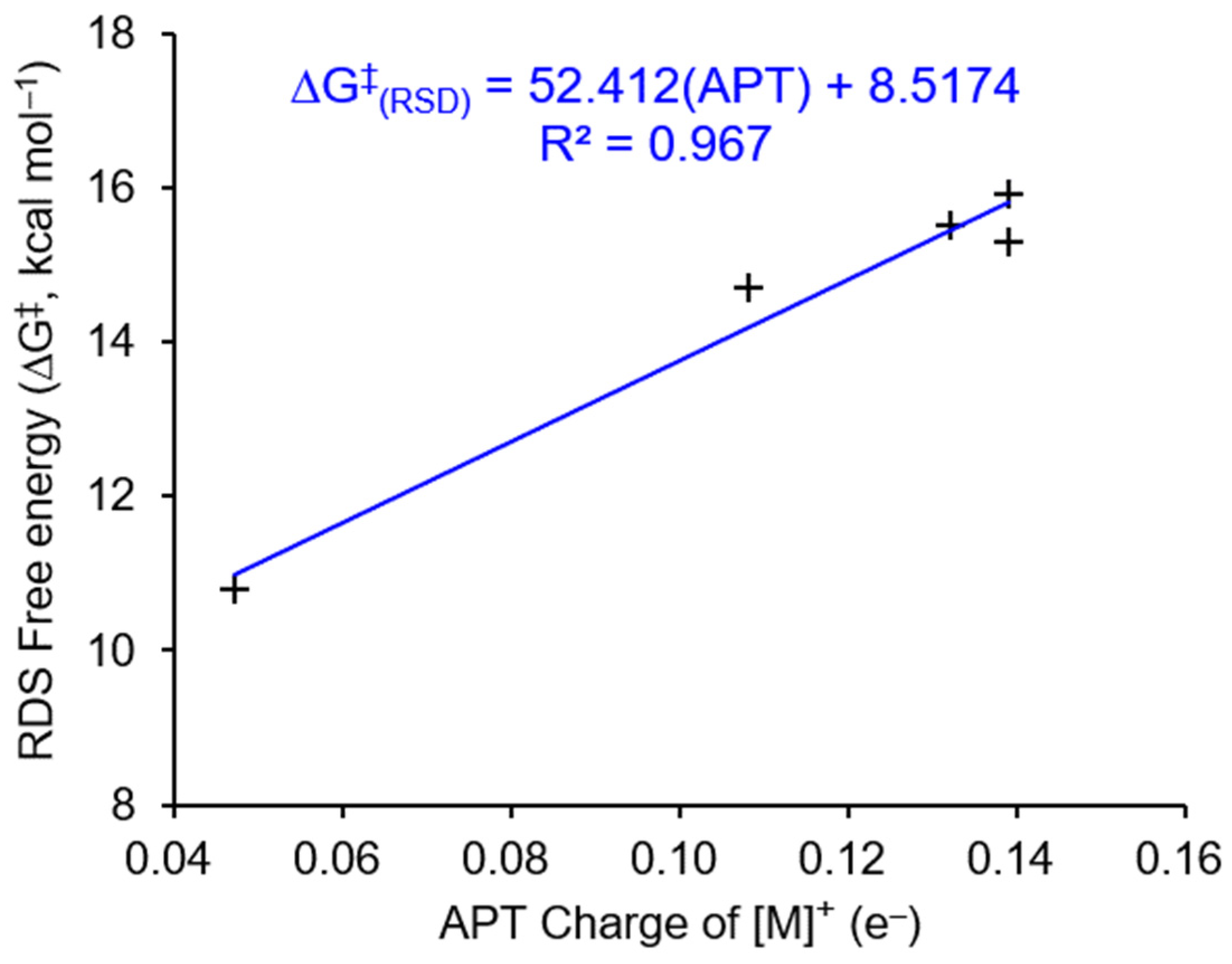
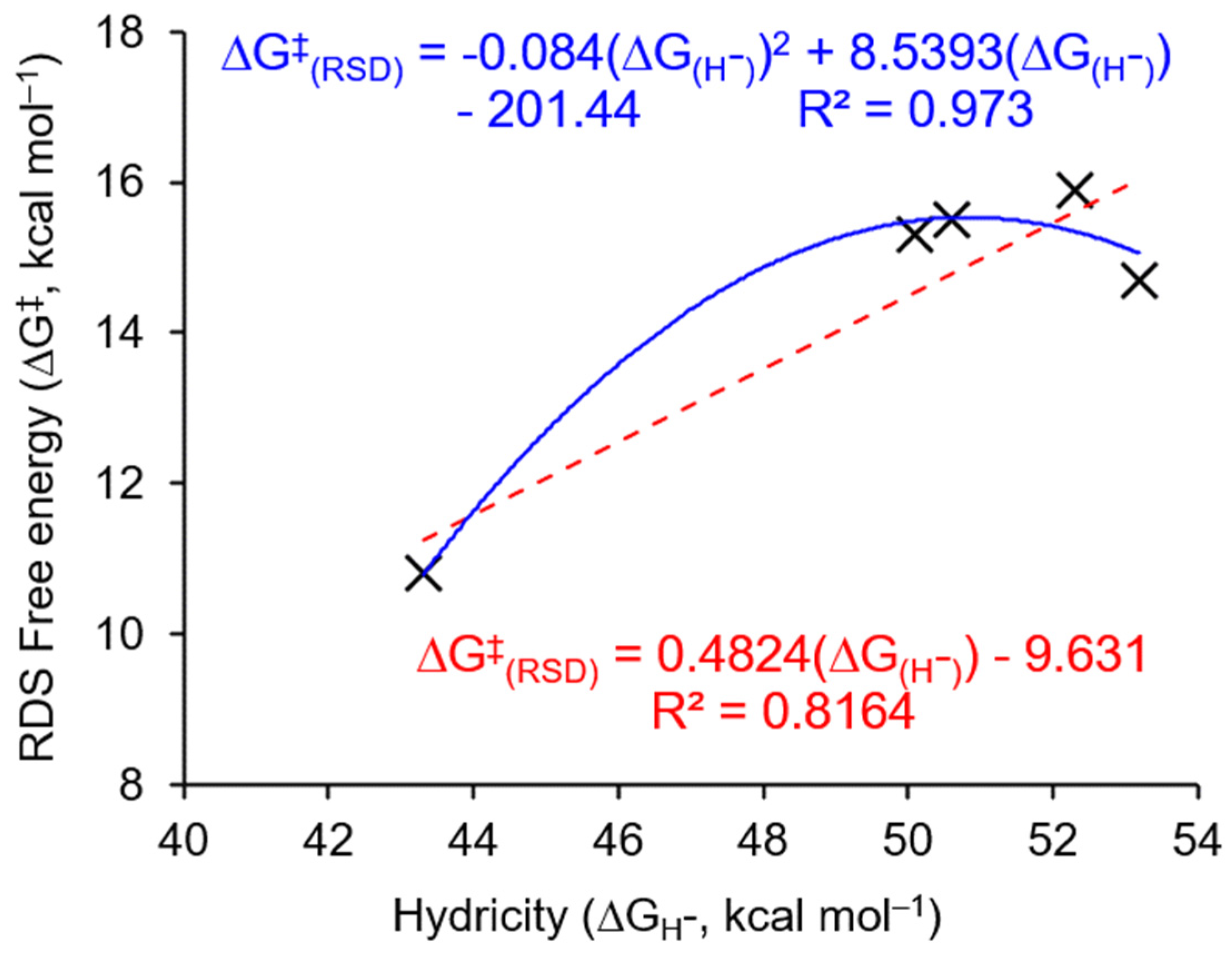
3.3. Parameterized Activity and Modeling of Ni–H Complexes for CO2 Insertion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hasan, M.M.F.; Rossi, L.M.; Debecker, D.P.; Leonard, K.C.; Li, Z.; Makhubela, B.C.E.; Zhao, C.; Kleij, A. Can CO2 and Renewable Carbon Be Primary Resources for Sustainable Fuels and Chemicals? ACS Sustain. Chem. Eng. 2021, 9, 12427–12430. [Google Scholar] [CrossRef]
- Saravanan, A.; Kumar, P.S.; Vo, D.-V.N.; Jeevanantham, S.; Bhuvaneswari, V.; Narayanan, V.A.; Yaashikaa, P.R.; Swetha, S.; Reshma, B. A comprehensive review on different approaches for CO2 utilization and conversion pathways. Chem. Eng. Sci. 2021, 236, 116515. [Google Scholar] [CrossRef]
- Valluri, S.; Claremboux, V.; Kawatra, S. Opportunities and challenges in CO2 utilization. J. Environ. Sci. 2022, 113, 322–344. [Google Scholar] [CrossRef]
- Kinzel, N.W.; Werlé, C.; Leitner, W. Transition Metal Complexes as Catalysts for the Electroconversion of CO2: An Organometallic Perspective. Angew. Chem. Int. Ed. 2021, 60, 11628–11686. [Google Scholar] [CrossRef]
- Navarro-Jaén, S.; Virginie, M.; Bonin, J.; Robert, M.; Wojcieszak, R.; Khodakov, A.Y. Highlights and challenges in the selective reduction of carbon dioxide to methanol. Nat. Rev. Chem. 2021, 5, 564–579. [Google Scholar] [CrossRef]
- Zhang, S.; Fan, Q.; Xia, R.; Meyer, T.J. CO2 Reduction: From Homogeneous to Heterogeneous Electrocatalysis. Acc. Chem. Res. 2020, 53, 255–264. [Google Scholar] [CrossRef]
- Kostera, S.; Peruzzini, M.; Gonsalvi, L. Recent Advances in Metal Catalyst Design for CO2 Hydroboration to C1 Derivatives. Catalysts 2021, 11, 58. [Google Scholar] [CrossRef]
- Bai, S.-T.; De Smet, G.; Liao, Y.; Sun, R.; Zhou, C.; Beller, M.; Maes, B.U.W.; Sels, B.F. Homogeneous and heterogeneous catalysts for hydrogenation of CO2 to methanol under mild conditions. Chem. Soc. Rev. 2021, 50, 4259–4298. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-H.; Himeda, Y.; Muckerman, J.T.; Manbeck, G.F.; Fujita, E. CO2 Hydrogenation to Formate and Methanol as an Alternative to Photo- and Electrochemical CO2 Reduction. Chem. Rev. 2015, 115, 12936–12973. [Google Scholar] [CrossRef]
- Jessop, P.G.; Joó, F.; Tai, C.-C. Recent advances in the homogeneous hydrogenation of carbon dioxide. Coord. Chem. Rev. 2004, 248, 2425–2442. [Google Scholar] [CrossRef]
- Schmeier, T.J.; Hazari, N.; Incarvito, C.D.; Raskatov, J.A. Exploring the reactions of CO2 with PCP supported nickel complexes. Chem. Commun. 2011, 47, 1824–1826. [Google Scholar] [CrossRef] [PubMed]
- Heimann, J.E.; Bernskoetter, W.H.; Hazari, N.; Mayer, J.M. Acceleration of CO2 insertion into metal hydrides: Ligand, Lewis acid, and solvent effects on reaction kinetics. Chem. Sci. 2018, 9, 6629–6638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heimann, J.E.; Bernskoetter, W.H.; Hazari, N. Understanding the Individual and Combined Effects of Solvent and Lewis Acid on CO2 Insertion into a Metal Hydride. J. Am. Chem. Soc. 2019, 141, 10520–10529. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Camaioni, D.M.; Dupuis, M.; Raugei, S.; Appel, A.M. Mechanistic insights into hydride transfer for catalytic hydrogenation of CO2 with cobalt complexes. Dalton Trans. 2014, 43, 11803–11806. [Google Scholar] [CrossRef]
- Siek, S.; Burks, D.B.; Gerlach, D.L.; Liang, G.; Tesh, J.M.; Thompson, C.R.; Qu, F.; Shankwitz, J.E.; Vasquez, R.M.; Chambers, N.; et al. Iridium and Ruthenium Complexes of N-Heterocyclic Carbene- and Pyridinol-Derived Chelates as Catalysts for Aqueous Carbon Dioxide Hydrogenation and Formic Acid Dehydrogenation: The Role of the Alkali Metal. Organometallics 2017, 36, 1091–1106. [Google Scholar] [CrossRef]
- Whittlesey, M.K.; Perutz, R.N.; Moore, M.H. Facile insertion of CO2 into the Ru-H bonds of Ru(dmpe)2H2 (dmpe = Me2PCH2CH2PMe2): Identification of three ruthenium formate complexes. Organometallics 1996, 15, 5166–5169. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Waldie, K.M.; Warnke, I.; De Crisci, A.G.; Batista, V.S.; Waymouth, R.M.; Chidsey, C.E.D. Experimental and Theoretical Study of CO2 Insertion into Ruthenium Hydride Complexes. Inorg. Chem. 2016, 55, 1623–1632. [Google Scholar] [CrossRef]
- Suh, H.-W.; Schmeier, T.J.; Hazari, N.; Kemp, R.A.; Takase, M.K. Experimental and Computational Studies of the Reaction of Carbon Dioxide with Pincer-Supported Nickel and Palladium Hydrides. Organometallics 2012, 31, 8225–8236. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results Obtained With The Correlation Energy Density Functionals Of Becke And Lee, Yang And Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Couty, M.; Hall, M.B. Basis sets for transition metals: Optimized outer p functions. J. Comput. Chem. 1996, 17, 1359–1370. [Google Scholar] [CrossRef]
- Check, C.E.; Faust, T.O.; Bailey, J.M.; Wright, B.J.; Gilbert, T.M.; Sunderlin, L.S. Addition of polarization and diffuse functions to the LANL2DZ basis set for p-block elements. J. Phys. Chem. A 2001, 105, 8111–8116. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular-Orbital Studies of Organic-Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The Influence of Polarization Functions on Molecular-Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Foresman, J.B.; Frisch, A. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian, Inc.: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Andrae, D.; Häusermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Wiedner, E.S.; Chambers, M.B.; Pitman, C.L.; Bullock, R.M.; Miller, A.J.M.; Appel, A.M. Thermodynamic hydricity of transition metal hydrides. Chem. Rev. 2016, 116, 8655–8692. [Google Scholar] [CrossRef] [PubMed]
- Ilic, S.; Alherz, A.; Musgrave, C.B.; Glusac, K.D. Thermodynamic and kinetic hydricities of metal-free hydrides. Chem. Soc. Rev. 2018, 47, 2809–2836. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Zhang, M.; Webster, C.E. Mechanistic Studies of Oxygen-Atom Transfer (OAT) in the Homogeneous Conversion of N2O by Ru Pincer Complexes. Inorganics 2022, 10, 69. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Dunlap, B.I. Fitting the Coulomb potential variationally in Xα molecular calculations. J. Chem. Phys. 1983, 78, 3140–3142. [Google Scholar] [CrossRef]
- Dunlap, B.I. Robust and variational fitting: Removing the four-center integrals from center stage in quantum chemistry. J. Mol. Struct. THEOCHEM 2000, 529, 37–40. [Google Scholar] [CrossRef]
- Zhang, M.; Liang, G. Understanding the Sigmatropic Shifts of Cyclopenta-2,4-dien-1-yltrimethylsilane in its Diels—Alder Addition. Org. Biomol. Chem. 2021, 19, 1732–1737. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Casals-Sainz, J.L.; Fernández-Alarcón, A.; Francisco, E.; Costales, A.; Pendás, Á.M. Bond Order Densities in Real Space. J. Phys. Chem. A 2020, 124, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Multiwfn, Version 3.8; 2021. Available online: http://sobereva.com/multiwfn/ (accessed on 2 December 2021).
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- VMD, Version 1.9.3; 2016. Available online: http://www.ks.uiuc.edu/Research/vmd/ (accessed on 4 December 2021).
- Falivene, L.; Credendino, R.; Poater, A.; Petta, A.; Serra, L.; Oliva, R.; Scarano, V.; Cavallo, L. SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps. Organometallics 2016, 35, 2286–2293. [Google Scholar] [CrossRef] [Green Version]
- SambVca, Version 2.1; 2019. Available online: https://www.molnac.unisa.it/OMtools/sambvca2.1/index.html (accessed on 12 November 2021).
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef] [Green Version]
- Poater, A.; Ragone, F.; Giudice, S.; Costabile, C.; Dorta, R.; Nolan, S.P.; Cavallo, L. Thermodynamics of N-Heterocyclic Carbene Dimerization: The Balance of Sterics and Electronics. Organometallics 2008, 27, 2679–2681. [Google Scholar] [CrossRef]
- Poater, A.; Ragone, F.; Mariz, R.; Dorta, R.; Cavallo, L. Comparing the Enantioselective Power of Steric and Electrostatic Effects in Transition-Metal-Catalyzed Asymmetric Synthesis. Chem. Eur. J. 2010, 16, 14348–14353. [Google Scholar] [CrossRef]
- Zhang, M.; Liang, G.; Xing, M. Theoretical Investigation of Hydrogen Bonds Assisted Tetradentate N4 Copper(I) Chloride and trans-1,2-Peroxodicopper Complexes. Eur. J. Inorg. Chem. 2021, 2021, 2194–2200. [Google Scholar] [CrossRef]
- Witte, J.; Mardirossian, N.; Neaton, J.B.; Head-Gordon, M. Assessing DFT-D3 Damping Functions Across Widely Used Density Functionals: Can We Do Better? J. Chem. Theory Comput. 2017, 13, 2043–2052. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; An, K.; Wang, Y.; Wu, Y.-D.; Zhang, X.; Yu, Z.-X.; He, W. A Combined Computational and Experimental Study of Rh-Catalyzed C–H Silylation with Silacyclobutanes: Insights Leading to a More Efficient Catalyst System. J. Am. Chem. Soc. 2021, 143, 3571–3582. [Google Scholar] [CrossRef] [PubMed]
- Zheng, D.; Wang, F. Performing Molecular Dynamics Simulations and Computing Hydration Free Energies on the B3LYP-D3(BJ) Potential Energy Surface with Adaptive Force Matching: A Benchmark Study with Seven Alcohols and One Amine. ACS Phys. Chem. Au 2021, 1, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Hazari, N.; Heimann, J.E. Carbon Dioxide Insertion into Group 9 and 10 Metal–Element σ Bonds. Inorg. Chem. 2017, 56, 13655–13678. [Google Scholar] [CrossRef] [PubMed]
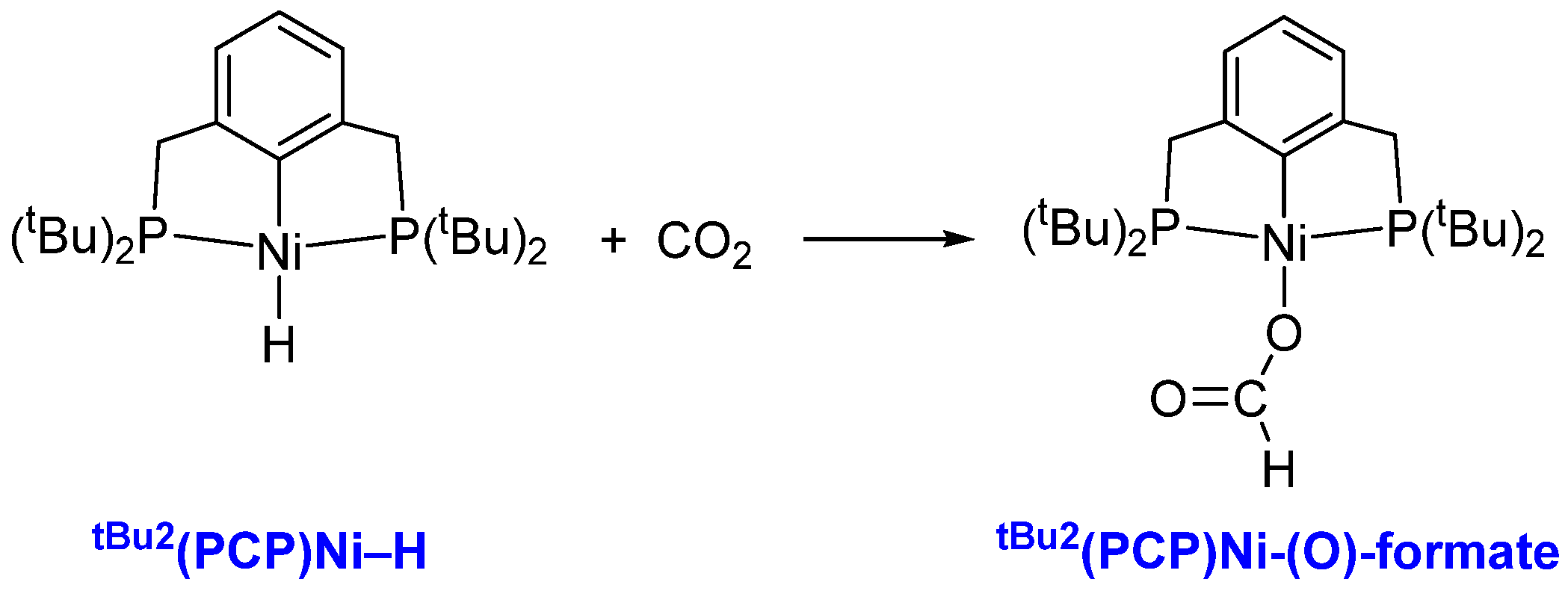

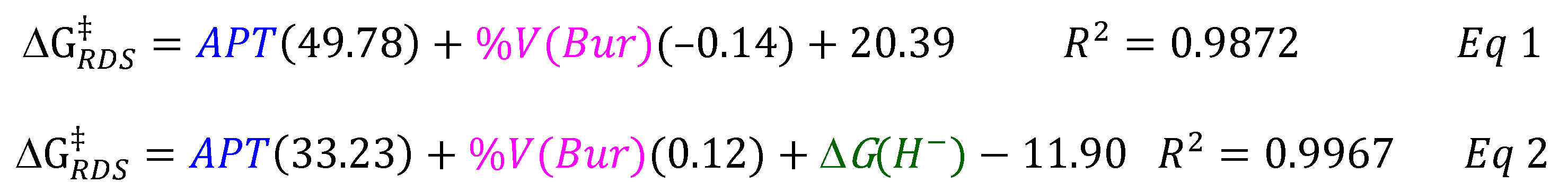

{kind=link}
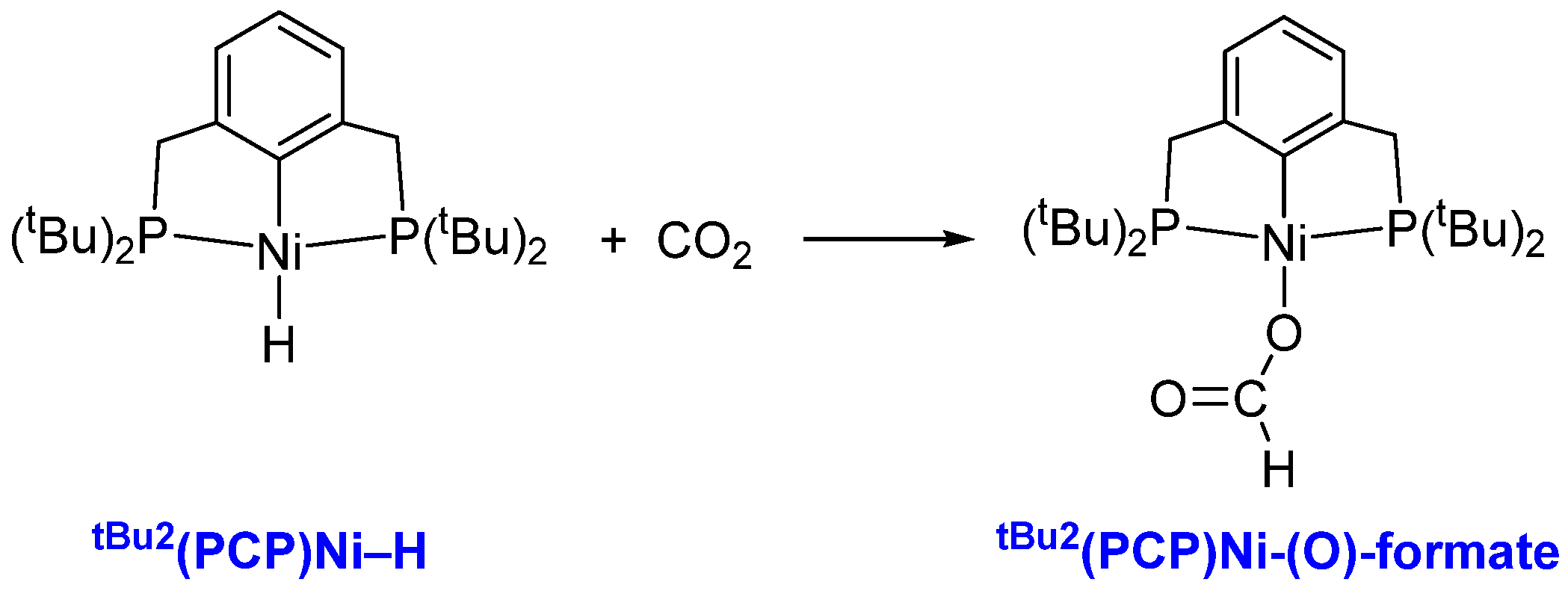{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Hydricity | TS-2-3 | TS-3-4i | APT | %VBur | k (M−1 s−1) |
|---|---|---|---|---|---|---|
| I, tBu2(PCP)Ni–H | 50.6 | 10.6 | 15.5 | 0.132 | 81.4 | 6.8 ± 0.7 |
| II, tBu2(p-MeO-PCP)Ni–H | 50.1 | 10.7 | 15.3 | 0.139 | 81.4 | 11.7 ± 1 |
| III, tBu2(p-I-PCP)Ni–H | 52.3 | 11.1 | 15.9 | 0.139 | 81.4 | 1.6 ± 0.2 |
| IV, iPr2(PCP)Ni–H | 53.2 | 11.5 | 14.7 | 0.108 | 77.4 | 4400 |
| V, tBu2(PCyP)Ni–H | 43.3 | 10.4 | 10.8 | 0.047 | 83.4 | - |
| VI, tBu2(PCP)Pd–H | 48.7 | 10.5 | 15.1 | 0.052 | 81.3 | - |
| VII, tBu2(PNP)Co–H | 41.2 | 8.4 | 8.9 | −0.116 | 77.7 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Liang, X.; Wang, Y.; Yang, H.; Liang, G. Insights into the Capture of CO2 by Nickel Hydride Complexes. Catalysts 2022, 12, 790. https://doi.org/10.3390/catal12070790
Zhang M, Liang X, Wang Y, Yang H, Liang G. Insights into the Capture of CO2 by Nickel Hydride Complexes. Catalysts. 2022; 12(7):790. https://doi.org/10.3390/catal12070790
Chicago/Turabian StyleZhang, Min, Xiaoqing Liang, Yaozheng Wang, Hongyu Yang, and Guangchao Liang. 2022. "Insights into the Capture of CO2 by Nickel Hydride Complexes" Catalysts 12, no. 7: 790. https://doi.org/10.3390/catal12070790
APA StyleZhang, M., Liang, X., Wang, Y., Yang, H., & Liang, G. (2022). Insights into the Capture of CO2 by Nickel Hydride Complexes. Catalysts, 12(7), 790. https://doi.org/10.3390/catal12070790