Abstract
The geometric and electronic structures of different octahedron RuRh clusters are studied using density functional theory calculations. The binding energy, electronic structure, and energy gap of the clusters have been obtained to determine the possible stable structures. The results show that the Ru4Rh2 cluster is the most stable structure which has D4h symmetry with the largest ionization potential, smallest affinity energy and larger energy gap. Furthermore, the information on adsorption and dissociation of multiple nitrogen molecules and the density of state for the octahedral Ru4Rh2 cluster is analyzed. The dissociation barrier of three nitrogen molecules further decreases to 1.18 eV with an increase in the number of N2 molecules. The co-adsorption of multiple N2 molecules facilitates the dissociation of N2 on the Ru4Rh2 cluster. The strong interaction between the antibonding orbital of N2 and the d orbital of the Ru4Rh2 cluster is illustrated by calculating and analyzing the results of PDOS, which stretches the N−N bond length and reduces the activation energy to dissociation. The antibonding orbital of the nitrogen molecule shows distinct and unique catalytic activity for the dissociation of the adsorbed nitrogen molecule on the octahedral Ru4Rh2 cluster.
1. Introduction
Ammonia is an important inorganic chemical product that sustains the global food supply chain. It is widely used in the manufacture of nitrogen-containing compounds, such as fertilizer and ammoniated feed, and plays an important role in the global economy [1,2,3]. However, the process of reducing N2 to ammonia is slow and needs to be carried out under high temperatures (400–600 °C), high pressure (20–40 MPa) and catalytic conditions, resulting in high energy consumption and serious environmental pollution [4,5]. Accordingly, it is of great significance to devise an environmentally benign and low energy consumption synthetic method of ammonia for the sustainable development of the global economy. The dissociation of N2 on the catalyst surface is the rate-limiting step for ammonia synthesis [6,7]. Therefore, it is necessary to further optimize the performance of the catalyst toward N2 dissociation and understand the reaction mechanism. In past decades, great progress has been made in the development of new catalysts. The direct dissociation of N2 is facilitated by the strong interaction of N2 molecules on the surfaces of Ru(0001) [8], Ru() [9] and Ru() [10], indicating that the ruthenium catalyst has recently become an industrial catalyst after Pd(110) [11], Fe(111) [12] and W(110) [13]. Although some studies have shown that the catalytic activity of the Ru step surface is better than that of the terrace surface, the stability of the material has not been considered in the literature. Stability is an important parameter that determines the long-term performance of the material during the reaction process.
Recently, it has been shown that sub-nanoscale, atomically dispersed, and fully exposed metal cluster catalysts can not only provide adjacent metal atoms as catalytic sites, but also maintain full atomic utilization efficiency, providing a variety of structural possibilities and catalytic feasibility [14,15,16]. Studying the formation, structure, and properties of clusters not only plays a bridging role in the transition between atomic and molecular physics and condensed matter physics, but also plays an important role in developing the theory of interaction between atoms and molecules, material science and environmental science. Furthermore, various sub-nano to nano-scale clusters with different structures have been experimentally and theoretically studied [17,18]. Theoretical studies on the dissociation of N2 on clusters of ruthenium, rhodium, aluminum, and tantalum have been reported [19,20,21,22,23]. Then, ruthenium-rhodium binary alloys are often used as catalysts in the industry. Early theoretical studies showed that Ru6 and Rh6 clusters with octahedral geometry had high catalytic activity in several reactions [24,25,26,27]. RuRh clusters further enhance the catalytic activity and also reduces the disadvantages that are present in pure Rh catalysts [25,28]. It is important to study the geometric structure and stability of ruthenium-rhodium clusters for their material reaction and catalytic mechanism.
In this paper, the geometric structure and stability of octahedral RumRhn (m + n = 6) clusters are systematically studied by density functional theory (DFT). It is predicted that the properties of RumRhn clusters are different from those of pure clusters, and provide some theoretical references for the study of larger RuRh clusters and their related chemical reactions. Overall Ru4Rh2 has the best energy and electronic stability and better chemical stability. The structure of nitrogen adsorbed on the Ru4Rh2 cluster was further systematically studied, and the potential energy surface of nitrogen bond breaking on the Ru4Rh2 cluster was calculated. Our work shows that the Ru4Rh2 cluster is an N2 dissociation active catalyst, which revises the previous practice of Ru2Rh4 [25] for catalytic reactions.
2. Results and Discussion
2.1. Geometric Structure of Clusters
The original pure Ru6 and Rh6 structures are the globally lowest energy structures obtained by the Birmingham cluster genetic algorithm [29]. The resulting lowest global energy structure is an octahedron. Bimetallic RuRh clusters were obtained using Rh atoms instead of Ru atoms in Ru6 clusters. The global optimization of the octahedron structure of six-atom bimetallic RumRhn clusters was carried out using the BFGS structure optimization algorithm [30] of the DFT method. The ground state structures of these RumRhn clusters are obtained according to the principle of minimum energy. Figure 1 shows the geometric structure of RumRhn clusters. Table 1 lists the symmetry, binding energy, bond order and average bond length of neutral RumRhn clusters. As shown in Figure 1 and Table 1, the binding energy Eb of RumRhn clusters increases with an increasing Ru atom number and tends to converge to a certain point. This is because the cohesive energy of Ru is 6.74 eV/atom, which is greater than that of Rh is 5.75 eV/atom [31], suggesting that the constructed structure model is reasonable. The symmetry distribution of RumRhn clusters is symmetrical. The symmetry of Ru6 is the same as that of Rh6, which are both Oh. The symmetry of Ru5Rh1 is the same as that of Ru1Rh5, both of which belong to C4v. When the Ru/Rh atomic ratio of clusters is 1:2 or 2:1, each ratio has two different structures. In the first structure, two Rh or two Ru atoms are located in the axial direction, and the symmetry of Ru4Rh2 and Ru2Rh4 clusters is D4h. In the second structure, two Rh or two Ru atoms are adjacent, and the symmetry of Ru4Rh2-I and Ru2Rh4-I clusters is C2v. Since the binding energy of Ru4Rh2 and Ru2Rh4 clusters is greater than that of Ru4Rh2-I and Ru2Rh4-I clusters, Ru4Rh2 and Ru2Rh4 clusters with higher stability will be studied below. The Ru3Rh3 has two possible geometries with the symmetries C2v and C3v.
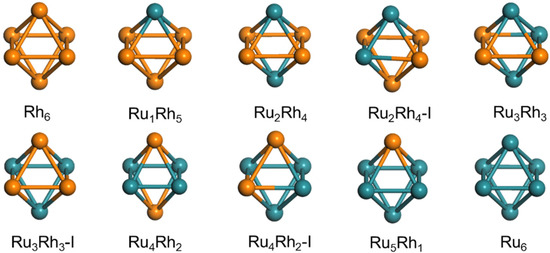
Figure 1.
Optimized geometric structure of octahedral RumRhn clusters, green balls represent Ru atoms and orange balls represent Rh atoms. The geometric and energetic parameters for these clusters are reported in Table 1. Ru4Rh2 is a magic cluster.

Table 1.
Optimized symmetry PG, binding energy Eb (eV), bond order σ, and average bond length d (Å) of octahedral RumRhn clusters, including the bond length d (Å) of the Ru-Ru, Ru-Rh, and Rh-Rh. When the atomic number ratio of Ru and Rh is 2:1, 1:1 and 1:2, each proportion corresponds to two structures, represented as Ru4Rh2 and Ru4Rh2-I, Ru3Rh3 and Ru3Rh3-I, and Ru2Rh4 and Ru2Rh4-I.
The bond order parameter σ of octahedral RumRhn clusters varies with composition as shown in Table 1 and Figure 2. In Ru6, Ru5Rh1, Ru1Rh5, and Ru6 clusters, the bond order σ > 0. In particular, the positive σ values of Ru5Rh1 and Ru1Rh5 indicate different degrees of separation between Ru and Rh atoms. The bond order σ = 0 of Ru4Rh2-I, Ru3Rh3-I and Ru2Rh4-I indicate a distorted mixing of clusters. The bond order σ < 0 of Ru4Rh2, Ru3Rh3, and Ru2Rh4 indicate that the clusters are mixed. Therefore, we prove that ruthenium and rhodium can be mixed at the nanoscale. The bond order σ = 0 and σ < 0 correspond to Figure 2c–e. For all the optimized structures, the bond length of Ru-Ru is shorter than the theoretical value of 2.70 [32]. All cluster model bond lengths show small contraction, as with some metal cluster models, which can be ascribed to surface stress [33,34]. The bond length between metal and metal in the cluster model is shorter than that between bulk metals [35]. The binding energies Eb per atom for RumRhn clusters reflect the degree of deviation from bulk metals in cluster stability. It can be seen from Table 1 that Eb decreases with the increase in the number of Rh atoms, indicating that the stability of metal clusters increases with an increase in the number of Ru atoms. For clusters with the same number of Ru and Rh atoms, the number of Ru-Ru and Ru-Rh bonds is positively correlated with the binding energy of clusters. Therefore, the stability of octahedral RumRhn clusters is mainly determined by the number of Ru−Ru bonds and Ru−Rh bonds.
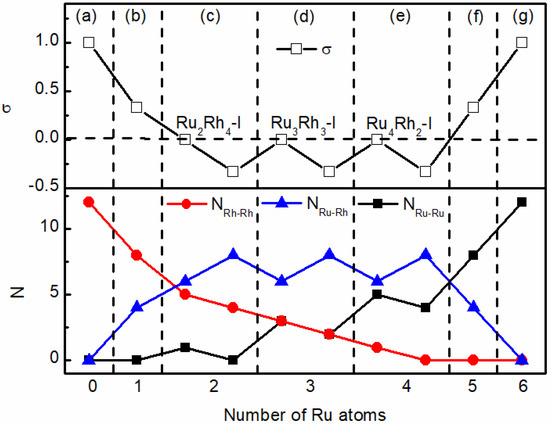
Figure 2.
Bond order parameters σ and number N of Ru−Ru bonds, Rh−Rh bonds and Ru−Rh bonds of octahedral RumRhn clusters varying with the number of Ru atoms. (a) Ru6, (b) Ru1Rh5, (c) Ru2Rh4, Ru2Rh4-I, (d) Ru3Rh3, Ru3Rh3-I, (e) Ru4Rh2, Ru4Rh2-I, (f) Ru5Rh1 and (g) Rh6. Ru2Rh4, Ru3Rh3, and Ru4Rh2 are mixed or alloyed clusters.
2.2. Stability of Clusters
The electronic states of small metal clusters possess the characteristics of discrete energy levels instead of bands. The reciprocal of the highest occupied molecular orbital (HOMO)-lowest unoccupied molecular orbital (LUMO) gap can reflect the activity of electrons and the degree of deformation of their distribution [36,37,38]. That is, the HOMO−LUMO gap (Eg) is inversely correlated with the ability to participate in chemical reactions, suggesting that Eg determines the chemical stability of the cluster. Therefore, it is necessary to study the energy level distribution of clusters. Figure 3 shows the HOMO, LUMO and corresponding orbital energy levels of the RuRh cluster. Figure 4 shows the calculated Eg, i.e., the difference between the calculated HOMO and LUMO energy levels. The Eg of RumRhn clusters becomes smaller than that of the pure Ru or Rh clusters, which means that the chemical activity of RumRhn clusters is higher. According to Figure 4, Ru6 has the highest chemical stability among the studied RumRhn clusters, with the largest energy gap of 0.47 eV. Such a large energy gap indicates that electrons find it difficult to transition from an occupied orbital to a vacant orbital. This means that clusters of large energy gaps have high chemical stability [39]. We find that the Eg value of the Ru3Rh3 and Ru4Rh2 cluster is slightly lower than that of the Ru6 cluster, indicating that their ability to participate in chemical reactions is higher than Ru6, that is, the stability of the Ru3Rh3 and Ru4Rh2 cluster is relatively higher than that of other RumRhn clusters.
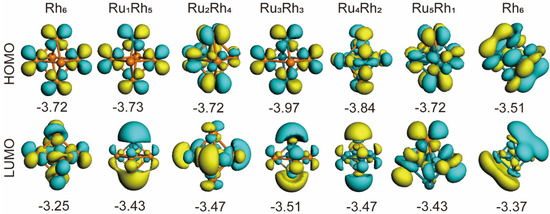
Figure 3.
HOMO and LUMO for the octahedral RumRhn clusters system (the contour value is 0.03 electron/Å3).
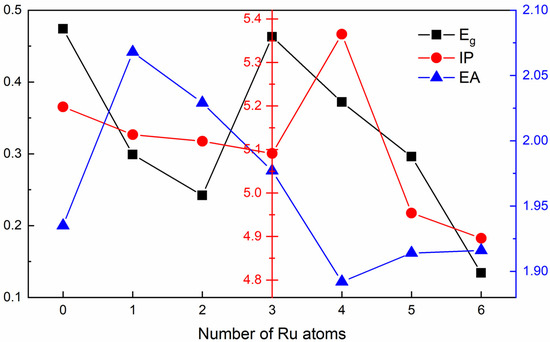
Figure 4.
The Eg (eV), IP (eV) and EA (eV) for the octahedral RumRhn clusters system varying with the number of Ru atoms. Compared with pure Rh6 clusters, alloying RumRhn clusters have smaller energy gap and stronger chemical activation. Ru4Rh2 has highest IP and lowest EA in all clusters.
The IP and EA can also be used to determine the electronic stability of the clusters. The IP of a cluster is defined as the energy needed for ionizing an electron away from the cluster. The energy required for electron ionization increases with the increase in the IP value, resulting in a more difficult expression of adiabatic electron ionization affinity, indicating that clusters tend to generate negative ions [40,41]. The stability of clusters decreases with the increase in the tendency of a cluster to generate negative ions. As shown in Figure 4, with the increase in the number of Ru atoms, the IP of RumRhn clusters first decreases, then increases, and finally tends to decrease. Among them, the IP of the Ru4Rh2 cluster is the largest. The low IP indicates that cations can easily form clusters by losing electrons. The ability of other RumRhn clusters to lose electrons increases with the increase in the number of Ru atoms, and the stability of RumRhn clusters are decreased, except Ru4Rh2. Moreover, the electron affinity EA of the clusters increases first to Ru1Rh5, then decreases to Ru4Rh2, and finally it tends to increase with the increase in the number of Ru atoms in the clusters. This phenomenon indicates that the cluster has a small tendency to acquire electrons to form anions, which corresponds to high stability. From the above-mentioned observations, we can conclude that Ru4Rh2 clusters have the largest IP (5.37 eV) and smallest EA (1.89 eV), which is more stable in terms of reactivity.
In order to better characterize the structural stability of clusters, two parameters, namely the mixing energy and quadratic difference energy, are used for comparison. The mixing energy Emix is used to characterize the degree of mixing, or the segregation stability. A negative value of Emix means that mixing is favorable. Emix is the analogue of the formation energy of bulk alloys, used in the case of nanoclusters. Another index of energetic stability is the quadratic difference in energy Δ2, whers the Δ2 describes the relative stability of the cluster as compared to the neighboring compositions. The value of Δ2 is positively correlated with the closeness of bonding between atoms, and the high closeness of bonding represents the high stability of clusters. In addition, the magic number structure is often the maximum value in the Δ2 image. Figure 5 compares the Emix and Δ2 values of all RumRhn clusters. It is found that when the number of Ru atoms is four, the Emix value becomes the smallest and the value of Δ2 become the largest. Therefore, the Ru4Rh2 cluster has the highest energy stability among all RumRhn clusters. Hence we will take Ru4Rh2 as the main research object in the following study to analyze its catalytic activity for N2 decomposition.
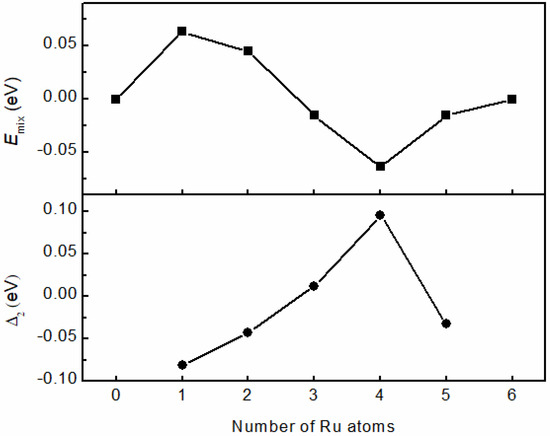
Figure 5.
The plot of mixing energy Emix and quadratic difference energy Δ2 of octahedral RumRhn clusters varying with the number of Ru atoms. Ru4Rh2 is a magic cluster.
2.3. Adsorption and Dissociation of N2 on Ru4Rh2
Figure 6 gives the configurations of N2 adsorbed on the octahedral Ru4Rh2 cluster. The effective adsorption sites of Ru4Rh2 metal clusters vary with atoms as shown in Figure 6. T1 and T2 are the top sites, at the top position of the Ru and Rh atoms in the cluster, respectively. B1 and B2 are two bridge sites, which refer to the bridges between the Ru and Ru, and Ru and Rh atoms, respectively. Two Ru atoms and one Rh atom make up the hollow adsorption site (H). As shown in Figure 6, the adsorption configurations of N2 molecules on clusters can be divided into two types: End-on (standing) and side-on (lying) conformations. The optimal adsorption site was found by placing a single N2 molecule at each location of each configuration and minimizing the local DFT. By placing a single N2 molecule at every position of each configuration, we can find the best adsorption site based on adsorption energy.
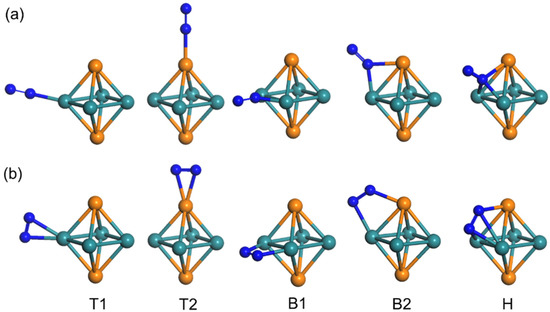
Figure 6.
(a) End-on and (b) side-on configurations of N2 adsorbed sites (T1, T2, B1, B2, H) on octahedral Ru4Rh2 clusters. Green balls represent Ru atoms, orange balls represent Rh atoms, and blue balls represent N atoms. B2 site of side-on configuration has the longest N−N bond length.
For the end-on conformations, as shown in Figure 6a and Table 2, the energy of N2 adsorption on T1 and T2 sites is −1.07 eV and −1.02 eV, and the bond length of N−N is 1.135 Å and 1.131 Å, respectively. Besides, the energy of N2 adsorption on the B2 site (−0.69 eV) is clearly smaller than that on the T1 and T2 sites. The adsorption of N2 at the B1 site is unstable and shifts to the T1 site. Similarly, the adsorption of N2 at the H site is unstable and shifts to the T2 site. For side-on conformations, as shown in Figure 6b and Table 2, B2 has adsorption energy of −0.77 eV, and the bond length of N−N is 1.169 Å, which is in accordance with the values from previous results ~1.130 Å. [42]. The N–Ru bond distance of 2.135 Å is also in line with the available investigations [43]. The adsorption of N2 at B1 and H sites is unstable and shifts to the end-on T1 and B2 sites, respectively. Finally, the adsorption energies on the T1 and T2 sites of the end-on configuration are −0.65 eV and −0.47 eV, respectively, with the same bond length of 1.142 Å. In contrast to B2 sites, the bond lengths of N−N on T1 and T2 sites are shorter, so the dissociation at these two sites is not studied for the time being.
Table 2.
Adsorption energy Ea (eV), bond length d (Å), vibrational wavenumbers ν (cm−1) and total charge number Δq (|e|) of N2 adsorbed on Ru4Rh2 clusters.
For these adsorption configurations, only the molecules with side-on configurations can dissociate. Therefore, we have considered the dissociation path at the B2 adsorption site and draw the calculated dissociation potential energy surface as shown in Figure 7. The adsorption energy of the nitrogen molecule at the B2 site on the Ru4Rh2 cluster is −0.77 eV. The N2 molecule dissociates from the B2 site to two H sites with an exothermicity of −1.42 eV; the dissociation barrier is 1.38 eV, which is less than 1.59 eV of the pure Ru6 cluster in this work and 1.80 eV of the terrace Ru(0001) of the previous study [44]. This indicates that the catalytic performance of the alloyed clusters is better than that of the pure Ru. We explain this phenomenon by comparing the d-band centers of the two clusters and the Mulliken charge transfer on the alloyed Ru4Rh2 cluster. In comparison with the pure Ru cluster, the charge transfer from the Ru atom (0.037) to the Rh atom (−0.073) in the Ru4Rh2 cluster results in the up-shift of the d-band center from −1.76 eV to −1.36 eV. In addition, the atomic size caused by the length of the Ru−Rh bond (2.607 Å) in Ru4Rh2 cluster is larger than that of the Ru−Ru bond (2.508 Å) in the Ru6 cluster (Table 1), which also causes the center of the d-band to move up, thus enhancing the catalytic activity.
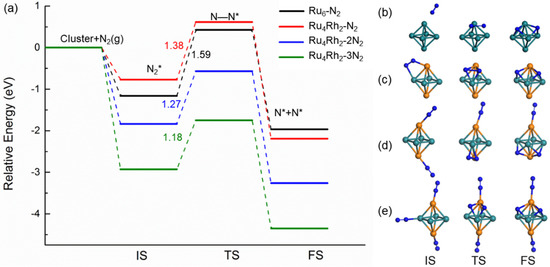
Figure 7.
(a) Potential energy surface from density functional calculations comparing N2 dissociation on Ru6 and Ru4Rh2 clusters, where * denotes adsorption. Geometric structure of the initial state (IS), transition state (TS), and final state (FS) of a N2 molecule in elementary reactions on (b) Ru6 and (c) Ru4Rh2 clusters, including (d) two and (e) three N2 molecules in elementary reactions on Ru4Rh2 cluster. The plot shows that alloying Ru4Rh2 cluster relative to Ru6 cluster leads to enhancement in activity, and N2 coadsorption on Ru4Rh2 cluster is beneficial to N2 dissociation.
In order to further verify the reliability of the results, the bond lengths, electronic configurations, and vibrational wavenumbers of N2 on the Ru4Rh2 clusters were studied. Table 2 shows that the bond length of the N2 molecule at B2 is elongated to 1.169 Å compared with 1.11 Å of the gas−phase nitrogen molecule [20]. It will undergo more deformation when nitrogen molecules attach to the cluster, which makes the dissociation barrier smaller. We also calculated the Mulliken charge of the adsorbed N2 molecule. A positive value represents the transfer of charge from the metal to the nitrogen molecule. As shown in Table 2, the number of electronic configurations of a nitrogen molecule in B2 is 0.711 |e|, which is larger than that of other sites, indicating a strong correlation with the breakage of the nitrogen bond in this adsorption configuration. We calculated the vibrational wave number of the nitrogen molecule as listed in Table 2. Compared with the vibration wave number of the gas−phase nitrogen molecule (2359 cm−1) [20] and that of others listed in Table 2, the calculated vibration wave number of the nitrogen molecule at B2 is 1556 cm−1 indicating the adsorption potential of the nitrogen molecule at B2 is the weakest among all adsorption sites, and the dissociation barrier is smallest when the nitrogen molecule dissociates.
2.4. Adsorption and Dissociation of Multiple N2 on Ru4Rh2
Because of the highly symmetrical structure of the Ru4Rh2 cluster, it is possible for several nitrogen molecules to adsorb together on the Ru4Rh2 cluster. According to the above calculations, nitrogen molecules have the largest adsorption energy at the end-on configuration of T2, which makes it unlikely to decompose into nitrogen atoms. When two nitrogen molecules co-adsorb on Ru4Rh2, one at T2 and the other at T1, the adsorption energy obtained is −1.84 eV. The dissociation path structure and relative potential energy are shown in Figure 7. Compared with the dissociation barrier of one nitrogen molecule of 1.38 eV, the dissociation barrier of two N2 molecules at the most active B2 site is slightly reduced to 1.27 eV. When three nitrogen molecules are adsorbed to Ru4Rh2, one nitrogen molecule at T1, one at top T2, and the other at bottom T2, the adsorption energy obtained is −2.93 eV and the energy barrier of nitrogen bond breaking is further reduced to 1.18 eV. This result implies that for multiple nitrogen molecules adsorbed on Ru4Rh2 clusters together, the dissociation of adsorbed nitrogen molecules will be accelerated according to the real conditions that may occur. To explain this situation, we compare the N−N bond lengths in the transition state (TS) structures for three cases. In a single N2 adsorption system, the N−N bond length for the reactant and TS is 1.169 and 1.680 Å, respectively, indicating the bond length elongation of 0.511 Å. For the two N2 adsorption systems the N−N bond length elongation of 0.630 Å between the reactant state (1.130 Å) and the TS (1.760 Å) is obtained. On the other hand, for the three N2 adsorption systems, the N−N bond length elongation between the reactants (1.132 Å) and TS (1.895 Å) is 0.763 Å. To further illustrate that multiple nitrogen molecules can accelerate the dissociation of adsorbed nitrogen molecules, we added four nitrogen molecule dissociation path structures and relative potential energy as shown in Figure S1. When the four nitrogen molecules were adsorbed on the T1, T1, T2, and T2 positions of Ru4Rh2, the adsorption energy was −3.28 eV and the nitrogen dissociation energy barrier was further reduced to 1.05 eV. The elongation of the N bond length between the reactants N−N bond length (1.135 Å) and TS (1.994 Å) is 0.859 Å, which is larger than the tensile length of the three nitrogen atoms adsorbed. From the observed results, we can conclude that deformation occurs in N2 after the formation of TS indicating the adsorption of multiple N2 can increase the degree of N2 structural deformation, which is conducive to accelerating the reaction process.
The partial density of states (PDOS) of several nitrogen molecules co-adsorbed on the Ru4Rh2 cluster is shown in Figure 8, including the p orbital of a nitrogen molecule and the d orbital of the metal atom, and it is compared with that of the Ru4Rh2 cluster without adsorbed N2 molecule as shown in Figure 8a. Figure 8b shows a single nitrogen molecule that has a maximum peak from −5.9 to −8.1 eV in the 1π and 5σ orbital at the B2 site, which is hybridized with the d orbital of the Ru4Rh2 cluster. In Figure 8c,d and Figure we find that the maximum value of the 5σ orbital decreases for the co-adsorption of two N2 and three N2 molecules, especially for three N2 molecules (Figure 8d). This observation indicates that the strong interaction between the 5σ orbital of the nitrogen molecule and the d orbital of Ru4Rh2 with several nitrogen molecules co-adsorption can reduce the strength of the N−N bond. In Figure 8d, the antibonding orbital (2π*) of the nitrogen molecule has obvious peaks. In the region of +2.5 eV above the Fermi level, it shows that the unoccupied antibonding orbital 2π* from the d electron of Ru to the nitrogen molecule adsorbs more of the nitrogen molecule. This suggests that Ru, which has abundant d-orbital electron properties, can receive electrons from the σ bond of N2 and give them to the antibonding orbital of N2, thus effectively weakening the bond strength of N2. This is consistent with previous studies [45]. As shown in Figure 8b-d the calculated p-band centers of a nitrogen molecule are −5.42, −5.16, and −5.81 eV; and that of the d-band centers of Ru atoms are −1.89, −1.68 and −1.56 eV. This result also supports the co-adsorption of N2 on Ru4Rh2 and the stronger interaction between the nitrogen molecule and the Ru atom. This is consistent with the prediction of the d-band center theory [46]. The calculated wave numbers of single, two and three nitrogen molecules are 1540, 1468, and 1382 cm−1, respectively. The weakest N−N bond of the three molecules provides clear evidence. With the larger co-adsorption of N2, many more electrons are transferred to the antibonding orbital of N2, which is testified by our calculations.Further, we cansuggest that the mechanism of ammonia formation is facilitated, after multiple N2 co-adsorptions on the Ru4Rh2 cluster. The reaction mechanism of ammonia synthesis in the clusters is a complex and multi-step process. The dissociation of the N2 molecule is only the first step toward the formation of NH3. This study provides a catalyst that can facilitate the N2 dissociation reaction, without high temperatures and pressures.
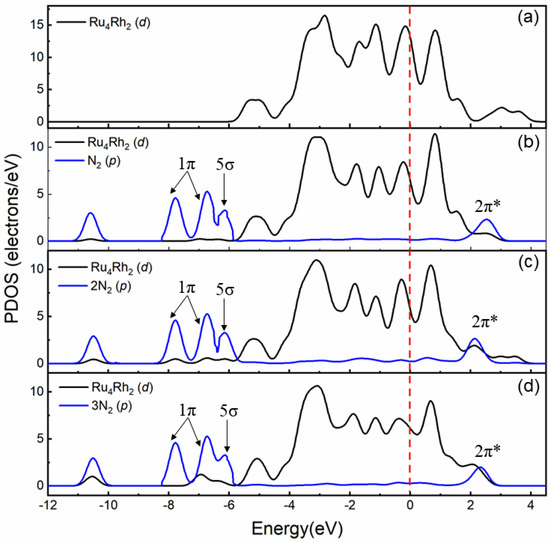
Figure 8.
Partial density of states (PDOS) on the N2 molecule (p orbital) (blue line) adsorbed on the Ru4Rh2 cluster (d orbital) (black line) under conditions: (a) Without adsorbed, (b) N2 adsorbed, (c) two N2 and (d) three N2 co-adsorbed, respectively, where 1π and 5σ represent the bonding orbitals, and 2π* represents the antibonding orbitals of N2 molecules. The Fermi level was shifted to 0 eV. The plot shows that the hybridization between p orbital of N2 and d orbital of Ru4Rh2 cluster becomes increasingly strong with the increase in the number of N2 adsorption.
3. Materials and Methods
DFT calculations were carried out using the atomic orbital-based DMol3 package [47]. It was performed using the revised Perdew−Burke−Ernzerhof (RPBE) exchange and correlation functional within generalized gradient approximation (GGA) [48]. This function aptly describes the electronic structure of the transition metal and is widely used [49]. The DFT semi-core pseudo potentials (DSPPs) core treatment was implemented for relativistic effects [50]. Moreover, double numerical plus polarization (DNP) was chosen as the basis set. The spin-polarization method was used for all calculations. The convergence tolerance of energy is 1×10−6 Ha, the maximum force is 0.002 Ha Å−1, and the maximum displacement is 0.005 Å. The range for the orbital cutoff was set to 5.0 Å. The transition states (TS) were obtained by LST/QST tools in Dmol3 code [51].
In order to study the electronic properties of metal clusters, and quantitatively study the separation of clusters, the ionization potential (IP), electron affinity (EA) and bond order parameter (σ) [52] are defined as follows:
where, is the total energy of the cluster, is the total energy of the cations with the same configuration of the cluster, and is the total energy of the anions with the same configuration of the cluster. , , and are the numbers of Ru-Ru, Ru-Rh, and Rh-Rh bonds, respectively. The ionization potential reflects how easily a cluster can lose an electron, while the electron affinity potential measures the ability of a cluster to gain an electron [40,41]. Positive values of σ indicate segregation of the atoms in the cluster while σ would be zero for disorderly mixed clusters. Mixed and onion-like phases of clusters result in negative values of σ [52].
To compare the relative stability of clusters, the binding energies (Eb) per atom for RumRhn clusters, mixing energy (Emix) and quadratic difference energies (Δ2) of RumRhn clusters are defined for six-atom nanoscale systems as follows: [53].
where (m + n = 6) is the total energy of each cluster. and are the energies of a single Ru atom and a single Rh atom, while and are the total energies of pure Ru and pure Rh clusters, respectively. Stable structures are identified by more negative and values. Maxima of indicates structures of special relative stability which is compared to those of the same size and nearby compositions.
The formula for calculating the adsorption energy (Ea) is as follows:
where is the energy of substances adsorbed on the surface, is the energy of a clean surface, and is the energy of free molecules.
4. Conclusions
The geometric structure of different octahedron RuRh clusters was optimized by the DFT method. The possible structures were determined and their stability and electronic properties were studied. It is found that the average binding energy of clusters increases with an increase in the number of Ru atoms. The stability of six-atom RumRhn clusters is mainly determined by the number of Ru−Ru bonds and Ru−Rh bonds. The ability of RumRhn clusters to lose electrons increases with the increase in the number of Ru atoms. Overall, Ru4Rh2 has the best energy and electronic stability and better chemical activation stability. The adsorption and dissociation of the octahedral Ru4Rh2 cluster for several nitrogen molecules were studied. The dissociation barrier of nitrogen molecules further decreases to 1.18 eV with an increase in the number of N2 molecules (for three nitrogen molecules). The strong interaction between the p orbital of N2 and the d orbital of the Ru4Rh2 cluster in illustrated by calculating and analyzing the results of the PDOS. The antibonding orbital of the nitrogen molecule shows distinct and unique catalytic activity for the dissociation of adsorbed nitrogen molecules on the octahedral Ru4Rh2 cluster.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal12080881/s1. Figure S1. Potential energy surface comparing N2 dissociation on Ru4Rh2 cluster. Geometric structure of the IS, TS, and FS of four N2 molecule in elementary reactions on Ru4Rh2 cluster.
Author Contributions
Conceptualization, N.Z. and R.J.; data curation, N.Z.; investigation, N.Z. and L.H.; methodology, N.Z. and L.M.; software, H.Z.; writing—original draft preparation, N.Z.; writing—review and editing, N.Z. and R.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the China Postdoctoral Science Foundation (2020M673335), Fundamental Research Funds for Central Universities (GK201902001, GK202103055, GK202201003), and Funded Projects for the Academic Leaders and Academic Backbones of Shaanxi Normal University (18QNGG008).
Acknowledgments
The authors acknowledge the support of this study by the Free Exploring Research Project for PhD students of Shaanxi Normal University (2021TS004), and the State Key Laboratory of New Ceramic and Fine Processing Tsinghua University (KF202006).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fang, H.; Liu, D.; Luo, Y.; Zhou, Y.; Liang, S.; Wang, X.; Lin, B.; Jiang, L. Challenges and opportunities of Ru-based catalysts toward the synthesis and utilization of ammonia. ACS Catal. 2022, 12, 3938–3954. [Google Scholar] [CrossRef]
- Chang, F.; Guan, Y.; Chang, X.; Guo, J.; Wang, P.; Gao, W.; Wu, G.; Zheng, J.; Li, X.; Chen, P. Alkali and alkaline earth hydrides-driven N2 activation and transformation over Mn nitride catalyst. J. Am. Chem. Soc. 2018, 140, 14799–14806. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Meng, H.; Li, F.; Zhao, J. Fe3 cluster anchored on the c2n monolayer for efficient electrochemical nitrogen fixation. Catalysts 2020, 10, 974. [Google Scholar] [CrossRef]
- Honkala, K.; Hellman, A.; Remediakis, I.N.; Logadottir, A.; Carlsson, A.; Dahl, S.; Christensen, C.H.; Nørskov, J.K. Ammonia synthesis from first-principles calculations. Science 2005, 307, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Nattino, F.; Costanzo, F.; Kroes, G.-J. N2 dissociation on W(110): An ab initio molecular dynamics study on the effect of phonons. J. Chem. Phys. 2015, 142, 104702. [Google Scholar] [CrossRef] [PubMed]
- Martirez, J.M.P.; Carter, E.A. Thermodynamic constraints in using Aum (m = Fe, Co, Ni, and Mo) alloys as N2 dissociation catalysts: Functionalizing a plasmon-active metal. ACS Nano 2016, 10, 2940–2949. [Google Scholar] [CrossRef]
- Martirez, J.M.P.; Carter, E.A. Excited-state N2 dissociation pathway on Fe-functionalized Au. J. Am. Chem. Soc. 2017, 139, 4390–4398. [Google Scholar] [CrossRef]
- Diekhöner, L.; Mortensen, H.; Baurichter, A.; Luntz, A.C.; Hammer, B. Dynamics of high-barrier surface reactions: Laser-assisted associative desorption of N2 from Ru(0001). Phys. Rev. Lett. 2000, 84, 4906–4909. [Google Scholar] [CrossRef]
- Shetty, S.; Jansen, A.P.J.; van Santen, R.A. Active sites for N2 dissociation on ruthenium. J. Phys. Chem. C 2008, 112, 17768–17771. [Google Scholar] [CrossRef]
- Kim, Y.K.; Morgan, G.A.; Yates, J.T. Site-specific dissociation of n2 on the stepped Ru(109) surface. Surf. Sci. 2005, 598, 14–21. [Google Scholar] [CrossRef]
- Brüning, K.; Granow, J.; Reiniger, M.; Heiland, W.; Vicanek, M.; Schlathölter, T. Dissociation of fast N2 molecules at a Pd(110) surface. Surf. Sci. 1998, 402–404, 215–218. [Google Scholar] [CrossRef]
- Logadottir, A.; Nørskov, J.K. The effect of strain for N2 dissociation on fe surfaces. Surf. Sci. 2001, 489, 135–143. [Google Scholar] [CrossRef]
- Alducin, M.; Díez Muiño, R.; Busnengo, H.F.; Salin, A. Dissociative adsorption of N2 on W(110): Theoretical study of the dependence on the incidence angle. Surf. Sci. 2007, 601, 3726–3730. [Google Scholar] [CrossRef][Green Version]
- Wang, X.; Qiu, S.; Feng, J.; Tong, Y.; Zhou, F.; Li, Q.; Song, L.; Chen, S.; Wu, K.-H.; Su, P.; et al. Confined Fe–Cu clusters as sub-nanometer reactors for efficiently regulating the electrochemical nitrogen reduction reaction. Adv. Mater. 2020, 32, 2004382. [Google Scholar] [CrossRef]
- Sun, Q.; Chen, B.W.J.; Wang, N.; He, Q.; Chang, A.; Yang, C.-M.; Asakura, H.; Tanaka, T.; Hülsey, M.J.; Wang, C.-H.; et al. Zeolite-encaged pd–mn nanocatalysts for co2 hydrogenation and formic acid dehydrogenation. Angew. Chem. Int. Ed. 2020, 59, 20183–20191. [Google Scholar] [CrossRef]
- Meng, F.; Peng, M.; Chen, Y.; Cai, X.; Huang, F.; Yang, L.; Liu, X.; Li, T.; Wen, X.; Wang, N.; et al. Defect-rich graphene stabilized atomically dispersed Cu3 clusters with enhanced oxidase-like activity for antibacterial applications. Appl. Catal., B 2022, 301, 120826. [Google Scholar] [CrossRef]
- Vojvodic, A.; Nørskov, J.K. New design paradigm for heterogeneous catalysts. Nati. Sci. Rev. 2015, 2, 140–143. [Google Scholar] [CrossRef]
- Zhu, C.; Du, D.; Eychmüller, A.; Lin, Y. Engineering ordered and nonordered porous noble metal nanostructures: Synthesis, assembly, and their applications in electrochemistry. Chem. Rev. 2015, 115, 8896–8943. [Google Scholar] [CrossRef]
- Li, H.-J.; Yeh, C.-H.; Ho, J.-J. The highly effective catalytic behavior of a metal nanocluster Ru79 on the dissociation of a N2 molecule—A quantum-chemical calculation. Catal. Commun. 2014, 52, 5–9. [Google Scholar] [CrossRef]
- Yeh, C.-H.; Lin, Y.-C.; Ho, J.-J. Highly effective catalysis of the double-icosahedral Ru19 cluster for dinitrogen dissociation – a first-principles investigation. Phys. Chem. Chem. Phys. 2014, 16, 7394–7400. [Google Scholar] [CrossRef]
- Wu, G.; Yang, M.; Guo, X.; Wang, J. Comparative DFT study of N2 and NO adsorption on vanadium clusters Vn (n = 2–13). J. Comput. Chem. 2012, 33, 1854–1861. [Google Scholar] [CrossRef] [PubMed]
- Kumar Yadav, M.; Mookerjee, A. Nitrogen absorption and dissociation on small tantalum clusters. Phys. B 2010, 405, 3940–3942. [Google Scholar] [CrossRef]
- Kulkarni, B.S.; Krishnamurty, S.; Pal, S. Size- and shape-sensitive reactivity behavior of Aln (n = 2–5, 13, 30, and 100) clusters toward the n2 molecule: A first-principles investigation. J. Phys. Chem. C 2011, 115, 14615–14623. [Google Scholar] [CrossRef]
- Kerpal, C.; Harding, D.J.; Lyon, J.T.; Meijer, G.; Fielicke, A. N2 activation by neutral ruthenium clusters. J. Phys. Chem. C 2013, 117, 12153–12158. [Google Scholar] [CrossRef]
- Ghatak, K.; Sengupta, T.; Krishnamurty, S.; Pal, S. Computational investigation on the catalytic activity of Rh6 and Rh4Ru2 clusters towards methanol activation. Theor. Chem. Acc. 2015, 134, 1597. [Google Scholar] [CrossRef]
- Villaseñor-González, P.; Dorantes-Dávila, J.; Dreyssé, H.; Pastor, G.M. Size and structural dependence of the magnetic properties of rhodium clusters. Phys. Rev. B 1997, 55, 15084–15091. [Google Scholar] [CrossRef]
- Ruzankin, S.F.; Avdeev, V.I.; Dobrynkin, N.M.; Zhidomirov, G.M.; Noskov, A.S. Modeling active centers in ammonia synthesis. DFT study of dissociative adsorption of N2 on Ru clusters. J. Struct. Chem. 2003, 44, 341–350. [Google Scholar] [CrossRef]
- Chang, C.-C.; Ho, J.-J. Catalytic enhancement in dissociation of nitric oxide over rhodium and nickel small-size clusters: A dft study. Phys. Chem. Chem. Phys. 2014, 16, 5393–5398. [Google Scholar] [CrossRef]
- Roberts, C.; Johnston, R.L.; Wilson, N.T. A genetic algorithm for the structural optimization of morse clusters. Theor. Chem. Acc. 2000, 104, 123–130. [Google Scholar] [CrossRef]
- Pfrommer, B.G.; Côté, M.; Louie, S.G.; Cohen, M.L. Relaxation of crystals with the quasi-newton method. J. Comput. Phys. 1997, 131, 233–240. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 8th ed.; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2005; p. 704. [Google Scholar]
- Yang, J.; Deng, K.; Xiao, C.; Wang, K. Low-spin and high-spin states of ru13 clusters. Phys. Lett. A 1996, 212, 253–257. [Google Scholar] [CrossRef]
- Mays, C.W.; Vermaak, J.S.; Kuhlmann-Wilsdorf, D. On surface stress and surface tension: II. Determination of the surface stress of gold. Surf. Sci. 1968, 12, 134–140. [Google Scholar] [CrossRef]
- Lamber, R.; Wetjen, S.; Jaeger, N.I. Size dependence of the lattice parameter of small palladium particles. Phys. Rev. B 1995, 51, 10968–10971. [Google Scholar] [CrossRef]
- Montano, P.A.; Shenoy, G.K.; Alp, E.E.; Schulze, W.; Urban, J. Structure of copper microclusters isolated in solid argon. Phys. Rev. Lett. 1986, 56, 2076–2079. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Parr, R.G. Hardness, softness, and the fukui function in the electronic theory of metals and catalysis. Proc. Natl. Acad. Sci. USA 1985, 82, 6723–6726. [Google Scholar] [CrossRef]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1874. [Google Scholar] [CrossRef]
- Wu, Q.; Jia, X.; Wong, M. Effects of number, type and length of the alkyl-chain on the structure and property of indazole derivatives used as corrosion inhibitors. Mater. Today Chem 2022, 23, 100636. [Google Scholar] [CrossRef]
- Jalbout, A.F.; Fernandez, S. Part ii. Gaussian, complete basis set and density functional theory stability evaluation of the singlet states of Cn (n=1–6): Energy differences, HOMO–LUMO band gaps, and aromaticity. J. Mol. Struct. 2002, 584, 169–182. [Google Scholar] [CrossRef]
- Kang, Y.; Jeon, S.H.; Cho, Y.; Han, S. Ab initio calculation of ionization potential and electron affinity in solid-state organic semiconductors. Phys. Rev. B 2016, 93, 035131. [Google Scholar] [CrossRef]
- Pašteka, L.F.; Eliav, E.; Borschevsky, A.; Kaldor, U.; Schwerdtfeger, P. Relativistic coupled cluster calculations with variational quantum electrodynamics resolve the discrepancy between experiment and theory concerning the electron affinity and ionization potential of gold. Phys. Rev. Lett. 2017, 118, 023002. [Google Scholar] [CrossRef]
- Mortensen, J.J.; Morikawa, Y.; Hammer, B.; Nørskov, J.K. Density functional calculations of n2 adsorption and dissociation on a Ru(0001) surface. J. Catal. 1997, 169, 85–92. [Google Scholar] [CrossRef]
- Dooling, D.J.; Nielsen, R.J.; Broadbelt, L.J. A density-functional study of the interaction of nitrogen with ruthenium clusters. Chem. Eng. Sci. 1999, 54, 3399–3409. [Google Scholar] [CrossRef]
- Dahl, S.; Logadottir, A.; Egeberg, R.C.; Larsen, J.H.; Chorkendorff, I.; Törnqvist, E.; Nørskov, J.K. Role of steps in N2 activation on Ru(0001). Phys. Rev. Lett. 1999, 83, 1814–1817. [Google Scholar] [CrossRef]
- Hirakawa, H.; Hashimoto, M.; Shiraishi, Y.; Hirai, T. Photocatalytic conversion of nitrogen to ammonia with water on surface oxygen vacancies of titanium dioxide. J. Am. Chem. Soc. 2017, 139, 10929–10936. [Google Scholar] [CrossRef] [PubMed]
- Lima, F.; Zhang, J.; Shao, M.; Sasaki, K.; Vukmirovic, M.; Ticianelli, E.; Adzic, R. Catalytic activity-d-band center correlation for the O2 reduction reaction on platinum in alkaline solutions. J. Phys. Chem. C 2007, 111, 404–410. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Norskov, J.K. Improved adsorption energetics within density-functional theory using revised perdew-burke-ernzerhof functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, F.Y.; Wu, X.Q. Global optimization and oxygen dissociation on polyicosahedral Ag32Cu6 core-shell cluster for alkaline fuel cells. Sci. Rep. 2015, 5, 11984. [Google Scholar] [CrossRef]
- Delley, B. Hardness conserving semilocal pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Halgren, T.A.; Lipscomb, W.N. The synchronous-transit method for determining reaction pathways and locating molecular transition states. Chem. Phys. Lett. 1977, 49, 225–232. [Google Scholar] [CrossRef]
- Molayem, M.; Grigoryan, V.G.; Springborg, M. Global minimum structures and magic clusters of cumagn nanoalloys. J. Phys. Chem. C 2011, 115, 22148–22162. [Google Scholar] [CrossRef]
- Rossi, G.; Rapallo, A.; Mottet, C.; Fortunelli, A.; Baletto, F.; Ferrando, R. Magic polyicosahedral core-shell clusters. Phys. Rev. Lett. 2004, 93, 105503. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).