Abstract
The in situ formation and activation of Cr(III) catalysts based on unsymmetrical PNP ligands yield efficient catalytic systems for selective ethylene tri-/tetramerization. The electronic nature (electron-withdrawing or electron-donating) and position (para or meta) of the substituents over the phenyl rings of the PNP, the nature of cocatalyst (DMAO/AlEt3 and MMAO-3A), and reaction conditions have been observed to have a marked impact on catalytic performance, particularly catalytic activity. Ligand L2, bearing 4-(trifluoromethyl)phenyl substituents, yielded 33.6 kg(product).g(Cr)−1·h−1 catalytic activity with 57.7% C8 selectivity under optimal conditions. Ligand L4, having para-tolyl substituents, yielded 43.3 kg(product).g(Cr)−1·h−1 with 59.0% C8 selectivity under optimum conditions. Changing the positions of both the electron-withdrawing and electron-donating substituents from para to meta over the phenyls of the PNP may lead to both catalytic systems exhibiting poor performance.
1. Introduction
Short-chain linear α-olefins (LAOs), particularly 1-hexene and 1-octene, are widely used as comonomers in the production of linear low-density polyethylene (LLDPE); therefore, they are in high demand [1,2]. In contrast to conventional technologies, which improve the product slate of oligomers, selective ethylene trimerization and tetramerization produce highly active and selective 1-hexene and 1-octene fractions, respectively; therefore, over the last two decades, they have interested the industrial and academic communities [3,4,5].
Ligand architecture is probably the most important factor that governs the catalytic performance of selective ethylene oligomerization. A ligand scaffold may, therefore, be modified to effectively tune catalytic activity and product selectivity [6,7,8]. In 2004, a subtle modification to the versatile PNP ligand transformed the selective ethylene trimerization system to a selective ethylene tetramerization system, provided 70% C8 selectivity and a high catalytic activity [9]. Since then, the PNP scaffold has been extensively investigated for selective ethylene oligomerization, and consequently, many efficient catalyst systems have been explored [10,11,12].
Herein, we report a series of new unsymmetrical PNP L1–L6 ligands bearing electron-withdrawing/donating moieties with phenyls of the PNP scaffold. The in situ mixing of L1–L6 ligands with CrCl3(THF)3 and activation with an aluminum-based activator (DMAO/AlEt3, MMAO-3A) successfully yielded efficient catalytic systems for selective ethylene tri-/tetramerization. We found that the electronic nature of the substituents attached to the phenyls, the position of these moieties over phenyl rings, the nature of the cocatalyst, and the reaction conditions strongly influenced the catalytic performance of these systems. Ligand L4, bearing electron-donating para-tolyl groups on the phenyls of the PNP, afforded the highest catalytic activity of 43.3 kg(product).g(Cr)−1·h−1 with 59.0% C8 selectivity with 1.0 MPa ethylene, at a temperature of 50 °C and with 2.4 μmol catalyst loading, in the presence of a 700 equivalent MMAO cocatalyst.
2. Results and Discussion
In the presence of the DMAO/AlEt3 or MMAO-3A cocatalyst, the stoichiometric mixing of L1–L6 ligands with CrCl3(THF)3 yielded Cr(III)-based active catalysts. The Cr(III) catalysts formed in situ and based on L1–L6 ligands were then subjected to ethylene oligomerization testing; the screening results of these complexes are presented in Table 1.

Table 1.
Evaluation of L1–L3 ligands for selective ethylene oligomerization a.
The results of the ethylene oligomerization of L1–L3 ligands, bearing electron-withdrawing groups on the phenyls of the PNP, demonstrate the role of electron-withdrawing substituents in controlling the catalytic performance of unsymmetrical PNP-based Cr(III) complexes (Table 1). The L1 ligand, bearing 4-fluorophenyl groups, produced a 63.7% high selective C8 fraction; however, it exhibited a very low catalytic activity of 9.3 kg(product).g(Cr)−1·h−1 (Table 1, entry 1). The L2 ligand, having 4-(trifluoromethyl)phenyl substituents, remarkably improved the catalytic activity, but slightly reduced C8 selectivity and could enhance the formation of the C6 fraction (Table 1, entry 2). These results suggest that the attachment of the trifluoromethyl groups at the para position of the phenyls of the PNP stabilized the catalytic system, consequently promoting the catalytic activity of the L2-based system. The presence of 3,5-bis(trifluoromethyl)phenyl moieties on the PNP scaffold of ligand L3 again reduced the catalytic activity and enhanced the C8 selectivity (Table 1, entry 3). The screening results of the L3-based system suggest that changing the position of the electron-withdrawing trifluoromethyl groups from the para (as in L2) to meta (as in L3) position of the phenyl rings destabilized the catalytic system, consequently producing a lower catalytic activity than the L2-based catalyst (Table 1; compare entry 2 with entry 3) [8].
Ligands L4–L6, bearing electron-donating groups on the phenyls of the PNP, demonstrated the role of electron-donating substituents in controlling the catalytic performance of unsymmetrical PNP-based Cr(III) complexes (Table 2). The L4 ligand, which had para-tolyl groups, exhibited a catalytic activity of 19.2 kg(product).g(Cr)−1·h−1 and C8 selectivity of 65.2% (Table 2, entry 1). The presence of electron-donating 3,5-dimethyl groups over the phenyls of the PNP in ligand L5 considerably reduced the catalytic activity and also C8 selectivity, but promoted the formation of the C6 fraction (Table 2, entry 2). These results again suggested that methyl substituents at the para position of the phenyl rings stabilized the catalytic system and enhanced the catalytic activity, whereas the presence of methyl groups at the meta position of the phenyls destabilized the catalytic system, consequently offering a low catalytic activity (Table 2, compare entry 1 with entry 2) [8]. The L6 ligand, having 4-methoxyphenyl substituents, restored the catalytic activity up to 18.5 kg(product).g(Cr)−1·h−1 with 55.3% C8 selectivity (Table 2, entry 3). A comparison of the screening results of the L4- and L6-based systems suggested that a moderate electron-donating substituent (methyl) led to a better catalytic performance than a strong electron-donating substituent (methoxy) when present at the para position of the phenyl rings (Table 2; compare entry 1 with entry 3) [13].
Table 2.
Evaluation of L4–L6 ligands for selective ethylene oligomerization a.
Along with the valuable C6 and C8 fractions, the ethylene oligomerization of unsymmetrical PNP L1–L6 ligands also produced C4, cyclic-C6 (methylcyclopentane and methylenecyclopentane), and solid PE in every run. Similarly to other catalytic systems, the unsymmetrical PNP L1–L6 ligands produced methylcyclopentane and methylenecyclopentane fractions at an almost 1:1 ratio [3,14,15]. The presence of a small amount of solid PE suggests that, along with the catalytic active species, an independent polymerization catalyst was also present in the catalytic system [16]. We did not observe the higher branched co-oligomer (C10+) fractions in the oligomerization product of the L1–L6-based catalysts. The formation of C10+ products could be attributed to the ligand selectivity effect; therefore, unsymmetrical PNP L1–L6 ligands may hinder the secondary co-trimerization and co-tetramerization reactions of 1-hexene and 1-octene with ethylene, and may consequently suppress the formation of C10+ fractions [8,17]. Alternatively, the catalytic activity of L1–L6 may not be high enough (due to the mild screening conditions) to promote the formation of C10+ fractions, because the formation of these side-products is proportional to the productivity of the reaction [3].
Preliminary evaluations of L1–L6-based catalysts suggested that L2- and L4-based catalysts did not show completely opposite catalytic performance under the same reaction conditions. On the contrary, both exhibited high activity and high selectivity of 1-octene. It was shown that both electron-withdrawing and electron-donating substituents, with a substituent at the para position on the phenyl group of PNP ligand, yielded high catalytic performance. On the other hand, both electron-withdrawing and electron-donating substituents exhibited poor catalytic performance when these substituents were in the meta position of phenyl (as in ligands L3 and L5). This suggests that the electronic effect is more than a steric effect on L1–L6-based catalysts with asymmetric structures. When the substituent with large steric hindrance is in the para position of the phenyl group of the PNP ligand, it can stabilize the active center and improve the catalytic performance. However, when the substituent with large steric hindrance is in the meta position of the phenyl group of the PNP ligand, it may affect the stability of the catalytic system, resulting in deactivation of the active center and a reduction in the catalytic activity. To further study the stability mechanism of Cr(III)-based catalysts with asymmetric structures, more substituents with different steric and electronic effects should be introduced into ligands in the future.
The efficient catalytic systems based on L2 and L4 ligands were further optimized under various reaction conditions (Al/Cr molar ratio, catalyst mass loading, and different nature of cocatalyst). To investigate the role of the Al/Cr molar ratio, the L2-based catalyst was evaluated under different Al/Cr molar ratios (Table 3). The catalytic activity of the L2-based system increased with the increasing Al/Cr molar ratio up to a certain limit (Table 3, entries 1–3), and then dramatically decreased (Table 3, entry 4). The highest catalytic activity for L2 was obtained at an Al/Cr ratio of 700 (Table 3, entry 3). These results suggested that a very low Al/Cr ratio fails to activate the catalyst system completely, whereas a very high Al/Cr molar ratio interferes with the catalytic active species, consequently reducing the activity. The Al/Cr ratio slightly affected the product selectivity of the L2-based catalyst, and a higher Al/Cr ratio marginally promoted the formation of the C8 product at the expense of C6 selectivity (Table 3, entries 3–4) [18,19].
Table 3.
Evaluation of the effect of Al/Cr molar ratios on the catalytic performance of the L2-based catalyst a.
Similarly to the L2-based system, the catalytic system based on the L4 ligand also displayed the highest catalytic activity with an Al/Cr molar ratio of 700 (Table 4, entry 3), and any increase or decrease in the Al/Cr molar ratio led to low catalytic activity in the L4-based system (Table 4, entries 1–2 and 4). In case of the L4-based system, a low Al/Cr ratio promoted the formation of the C6 fraction (Table 4, entry 1), whereas a higher Al/Cr ratio enhanced the C8 selectivity at the expense of the C6 product (Table 4, entries 2–4).
Table 4.
Evaluation of the effect of Al/Cr molar ratios on the catalytic performance of the L4-based catalyst a.
Table 5 displays the influence of mass loading on the catalytic performance of the L2-based catalyst.
Table 5.
The effect of catalyst loading on the catalytic performance of the L2-based catalyst a.
The L2 catalyst yielded the highest catalytic activity of 33.6 kg(product).g(Cr)−1·h−1 with 57.7% C8 selectivity at the optimal concentration of 2.4 μmol catalyst loading (Table 5, entry 2). Any increase or decrease in the catalyst mass loading from the optimal concentration led to a considerably low catalytic activity of the L2-based system (Table 5, entries 1 and 3). Variation in the catalyst mass loading failed to influence the product selectivity of the L2-based catalyst. The C6 and C8 selectivity values remained almost similar in all catalyst loading conditions (Table 5, entries 1–3) [19].
Similar to the L2-based system, the L4-based catalyst also offered the highest catalytic activity of 43.3 kg(product).g(Cr)−1·h−1 at 2.4 μmol of catalyst loading (Table 6, entry 2), and any increase or decrease in the catalyst loading remarkably reduced the catalytic activity of the L4-based system (Table 6, entries 1 and 3). Unlike the L2-based catalyst, the product selectivity of the L4-based system was affected by catalyst loading, and increasing the mass loading slightly promoted the formation of the C6 product at the expense of C8 selectivity (Table 6, entry 3).
Table 6.
Evaluation of the effect of mass loading on the catalytic performance of the L4-based catalyst a.
The cocatalyst/catalyst molar ratio considerably influences the catalytic performance of Cr(III)-based catalysts [20,21]. To investigate the effect of the nature of cocatalyst on the catalytic performance of unsymmetrical PNP-based Cr(III) complexes, the L2 ligand was assessed in the presence of DMAO/AlEt3 and MMAO-3A cocatalysts (Table 7).
Table 7.
Evaluation of the effect of cocatalyst on the catalytic performance of L2-based catalysts a.
Compared with the MMAO-3A cocatalyst, the L2-based system offered high catalytic activity and improved C8 selectivity when activated with the DMAO/AlEt3 cocatalyst (Table 7; compare entry 1 with entry 2). The activation of the L2-based system with MMAO-3A promoted the formation of a C6 fraction at the expense of C8 selectivity (Table 7, entry 2).
Compared with the L2-based system, which yielded high catalytic activity when activated only with DMAO/AlEt3, the L4-based catalytic system exhibited high catalytic activity when activated with the MMAO-3A or DMAO/AlEt3 cocatalyst (Table 8, entry 1,2). Regardless of the activation with different cocatalysts (DMAO/AlEt3 or MMAO-3A), the L4-based system offered almost similar product selectivity toward the valuable C6 and C8 fractions. Although both DMAO/AlEt3 and MMAO-3A are widely used as cocatalysts for ethylene oligomerization, the reduction ability of alkyl aluminum is higher than that of alkoxyaluminum. When AlEt3 with a strong reducing ability is introduced into DMAO, DMAO/AlEt3 can reduce the metal center to a low-valence state, thereby activating the catalytic center for ethylene tetramerization [22,23,24]. These results indicate that, relative to the steric hindrance effect of substituents, the electronic effect of substituents plays a major role in the activation process. The L2-based catalyst with an electron-withdrawing group needed to be activated by DMAO/AlEt3 with a stronger reducing ability to improve the catalyst activity, whereas the electron-donating group could facilitate the activation process. In the future, we could select more cocatalysts according to the different properties of the substituents on the phenyl group of PNP to study the activation mechanism of the catalyst.
Table 8.
The effect of cocatalyst on the catalytic performance of the L4-based catalyst a.
Compared with the reported chromium catalysts [12] with α-olefin (1-C6 + 1-C8) selectivity (>90 wt.%) at a reaction rate of 2000 kg·gCr−1·h−1 (ethylene, 45bar), it showed a much lower activity, 43.3 kg·gCr−1·h−1 (ethylene, 1 MPa), but higher α-olefin (1-C6 + 1-C8) selectivity (91.0%). As we know [25], with the increase in ethylene pressure, the activity will increase sharply and the selectivity of 1-octene will also increase. A series of new Cr (III) catalysts based on asymmetric PNP L1–L6 ligands has high α-olefin selectivity and competitive activity, showing good application prospects.
3. Experimental
3.1. General Comments
All experimental procedures were carried out in pre-dried flasks under an inert atmosphere using the standard Schlenk technique or a purified nitrogen-filled glove box. All anhydrous solvents were passed through a multi-column purification system and all other reagents were purchased from Aldrich and used as received. Dried Methylalumiumoxan (DMAO) and Modified Methylalumiumoxan type 3A (MMAO-3A) were purchased from Akzo Nobel and used as received. The NMR characterization of ligands was performed using a Bruker ACANCEⅢ400M nuclear magnetic resonance spectrometer (400 MHz for 1H, 162 MHz for 31P, and 101 MHz for 13C in CDCl3, Billerica, MA, USA) at 300 K.
3.2. Ligand Preparation; General Procedure
The unsymmetrical PNP L1–L6 ligands were prepared using the simple salt metathesis method. Treatment with excess triethylamine (Et3N) and the subsequent slow addition of chlorodiphenylphosphane (Ph2PCl) transformed the cyclopentanamine (cyclopentane-NH2) to an N-cyclopentyl-1,1-diphenylphosphanamine {Ph2P(cyclopentyl)NH} intermediate. The lithiation of Ph2P(cyclopentyl)NH by butyllithium yielded the lithium cyclopentyl(diphenylphosphaneyl)amide {Ph2P(cyclopentyl)NLi}. The corresponding L1–L6 {Ph2P(cyclopentyl)NP(R1)(R2)} ligands were obtained by treating the lithium cyclopentyl(diphenylphosphaneyl)amide with the corresponding chlorophosphane {(R1)(R2)PCl}reagents (Scheme 1). The detailed synthesis procedures of L1–L6 ligands are discussed in detail in Section 3.3. The details of experiments and characterizations were listed in Supplementary Materials (Figures S1–S18).
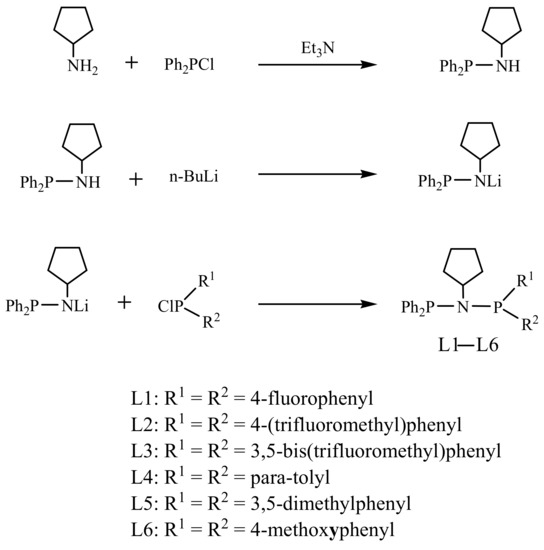
Scheme 1.
Synthesis of unsymmetrical PNP L1–L6 ligands.
3.3. Synthesis of L1–L6 Ligands; Detailed Procedure
3.3.1. N-(bis(4-fluorophenyl)phosphaneyl)-N-cyclopentyl-1,1-diphenylphosphanamine (L1)
Lithium cyclopentyl(diphenylphosphaneyl)amide was prepared through the lithiation of N-cyclopentyl-1,1-diphenylphosphanamine [26]. A hexane solution (10 mL) of chlorobis(4-fluorophenyl)phosphane (0.50 g, 1.9 mmol) was added to a solution of lithium cyclopentyl(diphenylphosphaneyl)amide (0.53 g, 1.9 mmol) in n-hexane at −35 °C, under stirring. Stirring was carried out constantly overnight at room temperature; then, the reaction mixture was filtered to remove lithium chloride salt. The solution was vacuum-dried, washed with n-hexane (3 × 10 mL), and dried again under vacuum conditions to obtain a yellow oily product. The colorless solid ligand L1 was isolated after recrystallization in hexane at −35 °C at a 41% yield. 1H NMR (400 MHz, CDCl3, 298 K): δ 1.39 (Br, 2H, N-CH-CH2), 1.52 (Br, 2H, N-CH-CH2), 1.73 (Br, 2H, N–CH–CH2–CH2), 1.87 (Br, 2H, N–CH–CH2–CH2), 3.79 (m, 1H, N–CH), 7.01–7.06 (m, 4H, Ph–H), 7.29–7.35 (br, 14H, Ph–H). 13C NMR (100 MHz, CDCl3, 298 K): δ 24.03, 34.07, 62.57 (Cyclopentyl, 5C); 162.09, 164.58 (–C–F, 2C); 115.24, 128.12, 128.75, 132.62, 132.84, 134.62, 135.60, 139.75(–Ph–C, 24C) 31P NMR (162 MHz, CDCl3, 298 K): δ 48.45 (Br), 50.07 (Br). Anal. Calcd for C29H27F2NP2 (found): C, 71.16 (71.34); H, 5.56 (5.72); N, 2.86 (2.69).
3.3.2. N-(bis(4-(trifluoromethyl)phenyl)phosphaneyl)-N-cyclopentyl-1,1-diphenylphosphanamine (L2)
The synthesis procedure for L2 was similar to that followed for L1, except that chlorobis(4-(trifluoromethyl)phenyl)phosphine (0.80 g, 2.2 mmol) was treated with lithium cyclopentyl(diphenylphosphaneyl)amide (0.61 g, 2.2 mmol). The product was obtained as a white solid in a 38% yield. 1H NMR (400 MHz, CDCl3, 298 K): δ 1.37 (Br, 2H, N–CH–CH2), 1.55 (Br, 2H, N–CH–CH2), 1.76 (Br, 2H, N–CH–CH2–CH2), 1.83 (Br, 2H, N–CH–CH2–CH2), 3.78 (m, 1H, N–CH), 7.32–7.43 (m, 14H, Ph–H), 7.54–7.56 (m, 4H, CF3–C–CH2). 13C NMR (100 MHz, CDCl3, 298 K): δ 24.04, 34.21, 62.97 (Cyclopentyl, 5C); 122.72 (–CF3, 2C); 124.94, 125.43, 128.26, 129.01, 130.73, 131.05, 132.87, 139.20, 144.33 (–Ph–C, 24C).31P NMR (162 MHz, CDCl3, 298 K): δ 48.90–51.56 (d). Anal. Calcd for C31H27F6NP2 (found): C, 63.16 (63.35); H, 4.62 (4.88); N, 2.38 (2.19).
3.3.3. N-(bis(3,5-bis(trifluoromethyl)phenyl)phosphaneyl)-N-cyclopentyl-1,1-diphenylphosphanamine (L3)
The synthesis procedure for L3 was similar to that followed for L1, except that bis(3,5-bis(trifluoromethyl)phenyl)chlorophosphane (0.50 g, 1.0 mmol) was treated with lithium cyclopentyl(diphenylphosphaneyl)amide (0.27 g, 1.0 mmol). The product was obtained as a white solid in a 32% yield. 1H NMR (400 MHz, CDCl3, 298 K): δ 1.39 (Br, 2H, N–CH–CH2), 1.53 (Br, 2H, N–CH–CH2), 1.73 (Br, 2H, N–CH–CH2–CH2), 1.85 (Br, 2H, N–CH–CH2–CH2), 3.69 (m, 1H, N–CH), 7.35 (m, 10H, Ph–H), 7.72 (m, 4H, CF3–C–CH–C–P),7.86 (m, 2H, CF3–C–CH–C–CF3). 13C NMR (100 MHz, CDCl3, 298 K):δ 23.84, 34.14, 63.11(Cyclopentyl, 5C); 124.47(–CF3, 4C); 123.07, 128.55, 129.46, 132.42, 132.64, 142.01, 142.25.(–Ph–C, 24C). 31P NMR (162 MHz, CDCl3, 298 K): δ 51.04 (s). Anal. Calcd for C33H25F12NP2 (found): C, 54.63 (54.77); H, 3.47 (3.62); N, 1.93 (1.82).
3.3.4. N-cyclopentyl-N-(di-p-tolylphosphaneyl)-1,1-diphenylphosphanamine (L4)
The synthesis procedure for L4 was similar to that followed for L1, except that chlorobis(p-tolyl)phosphine (0.50 g, 2.0 mmol) was treated with lithium cyclopentyl(diphenylphosphaneyl)amide (0.55 g, 2.0 mmol). The product was obtained as a white solid in a 35% yield. 1H NMR (400 MHz, CDCl3, 298 K): δ 1.33 (Br, 2H, N–CH–CH2), 1.47 (Br, 2H, N–CH–CH2), 1.67 (Br, 2H, N–CH–CH2–CH2), 1.84 (Br, 2H, N–CH–CH2–CH2), 2.35 (s, 6H, –CH3), 3.78 (m, 1H, N–CH), 7.09–7.11 (m, 8H, CH3–Ph–H), 7.25–7.36 (br, 10H, Ph–H). 13C NMR (100 MHz, CDCl3, 298 K):δ21.37 (2C, –CH3), 24.05, 34.02, 62.60 (Cyclopentyl, 5C); 127.98, 128.49, 128.80, 132.75, 132.97, 136.71, 136.83, 138.43, 140.16, 140.29 (–Ph–C, 24C) 31P NMR (162 MHz, CDCl3, 298 K): δ 48.89–49.28 (d). Anal. Calcd for C31H33NP2 (found): C, 77.32 (77.57); H, 6.91 (7.03); N, 2.91 (2.69).
3.3.5. N-(bis(3,5-dimethylphenyl)phosphaneyl)-N-cyclopentyl-1,1-diphenylphosphanamine (L5)
The synthesis procedure for L5 was similar to that followed for L1, except that chlorobis(3,5-dimethylphenyl)phosphine (0.50 g, 1.8 mmol) was treated with lithium cyclopentyl(diphenylphosphaneyl)amide (0.49 g, 1.8 mmol). The product was obtained as a white solid in a 39% yield. 1H NMR (400 MHz, CDCl3, 298 K): δ 1.33 (Br, 2H, N–CH–CH2), 1.50 (Br, 2H, N–CH–CH2), 1.67 (Br, 2H, N–CH–CH2–CH2), 1.84 (Br, 2H, N–CH–CH2–CH2), 2.23 (s, 12H, –CH3), 3.79 (m, 1H, N–CH), 6.94 (s, 6H, (CH3)2–Ph–H), 7.30–7.37(m, 10H, Ph–H). 13C NMR (100 MHz, CDCl3, 298 K): δ21.34 (4C, -CH3), 24.05, 34.05, 62.80 (Cyclopentyl, 5C); 162.09, 164.58 (–C–F, 2C); 127.92, 128.46, 130.26, 130.38, 130.60, 132.83, 133.05, 137.40, 139.66, 140.20. (–Ph–C, 24C) 31P NMR (162 MHz, CDCl3, 298 K): δ −22.75 (s). Anal. Calcd for C33H37NP2 (found): C, 77.78 (77.92); H, 7.32 (7.89); N, 2.75 (2.56).
3.3.6. N-(bis(4-methoxyphenyl)phosphaneyl)-N-cyclopentyl-1,1-diphenylphosphanamine (L6)
The synthesis procedure for L6 was similar to that followed for L1, except that chlorobis(4-methoxyphenyl)phosphine (0.50 g, 1.8 mmol) was treated with lithium cyclopentyl(diphenylphosphaneyl)amide (0.49 g, 1.8 mmol). The product was obtained as a colorless oil in a 41% yield. 1H NMR (400 MHz, CDCl3, 298 K): δ 1.32 (Br, 2H, N–CH–CH2), 1.49 (Br, 2H, N–CH–CH2), 1.68 (Br, 2H, N–CH–CH2–CH2), 1.83 (Br, 2H, N–CH–CH2–CH2), 3.79 (m, 1H, N–CH), 3.82 (s, 6H, O–CH3), 6.58–6.87 (Br, 4H, CH3O–C–CH–), 7.26–7.35 (m, 14H, Ph–H). 13C NMR (100 MHz, CDCl3, 298 K):δ 24.01, 33.91, 55.19 (Cyclopentyl, 5C); 55.19(–OCH3, 2C); 113.63, 128.01, 128.45, 132.71, 134.34, 140.11(–Ph–C, 24C); 160.05(–C–OCH3, 1C) 31P NMR (162 MHz, CDCl3, 298 K): δ 49.17 (s). Anal. Calcd for C31H33NO2P2 (found): C, 72.50 (72.47); H, 6.48 (6.49); N, 2.73 (2.66).
3.4. Ethylene Oligomerization General Procedure
A transparent glass autoclave reactor (145 mL) equipped with a magnetic stir bar and temperature-measuring device was used for ethylene oligomerization. After repeated evacuation and N2 purging, 20 mL methylcyclohexane was injected into the reactor; then, the catalyst solution of methylcyclohexane was injected into the reactor followed by 1.0 MPa pressurized ethylene, which was left to react for 30 min. After the completion of the reaction, the ethylene was released while the reactor was in an ice-water bath and the reaction was quenched with acidified MeOH (50 mL). After the separation of the organic phase, n-heptane was added as an internal standard and the product was analyzed using an Agilent 7890A gas chromatograph.
4. Conclusions
The Cr(III) catalysts formed and activated in situ and based on unsymmetrical PNP L1–L6 ligands yielded a selective ethylene tri-/tetramerization reaction with high catalytic activity and C8 selectivity. The electronic nature of the substituents (either electron-withdrawing or electron-donating) on the phenyls of the PNP, the position of these substituents over the phenyl ring, the nature of the cocatalyst, and the reaction conditions considerably influenced the catalytic performance, particularly the catalytic activity, of the unsymmetrical PNP L1–L6-based catalysts. We observed that both electron-withdrawing and electron-donating substituents, when present at the para position of the phenyl rings, stabilized the catalytic system (as in L2 and L4 ligands) and improved the catalytic performance, whereas the presence of these groups at the meta positions of the phenyls (as in L3 and L5 ligands) destabilized the catalytic systems and led to a poor catalytic performance. Among the L1–L6-based Cr(III) catalysts, the catalytic system based on L4, having para-tolyl groups, yielded the highest catalytic activity of 43.3 kg(product).g(Cr)−1·h−1 with 59% C8 selectivity with 1.0 MPa ethylene, at 50 °C temperature, with 2.4 μmol of catalyst loading, and in the presence of a 700 equivalent of MMAO-3A cocatalyst.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal12090944/s1, Experimental section, Figure S1: 1H NMR Spectrum of L1, Figure S2: 31P NMR Spectrum of L1, Figure S3: 13C NMR Spectrum of L1, Figure S4: 1H NMR Spectrum of L2, Figure S5: 31P NMR Spectrum of L2, Figure S6: 13C NMR Spectrum of L2, Figure S7: 1H NMR Spectrum of L3, Figure S8: 31P NMR Spectrum of L3, Figure S9: 13C NMR Spectrum of L3, Figure S10: 1H NMR Spectrum of L4, Figure S11: 31P NMR Spectrum of L4, Figure S12: 13C NMR Spectrum of L4, Figure S13: 1H NMR Spectrum of L5, Figure S14: 31P NMR Spectrum of L5, Figure S15: 13C NMR Spectrum of L5, Figure S16: 1H NMR Spectrum of L6, Figure S17: 31P NMR Spectrum of L6, Figure S18: 13C NMR Spectrum of L6.
Author Contributions
Conceptualization, T.J. and C.C.; oligomerization and characterization, Y.Z., H.F. and J.Z.; writing—original draft preparation, Y.Z., C.C. and H.F.; writing—review and editing, C.C. and F.A.; supervision, T.J.; project administration, C.C.; funding acquisition, T.J. and C.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China [22071178, 22050410271] and PetroChina Innovation Foundation [2019D-5007-0409].
Data Availability Statement
The data that support the findings of this study are available in the Supplementary Material of this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Agapie, T. Selective ethylene oligomerization: Recent advances in chromium catalysis and mechanistic investigations. Coord. Chem. Rev. 2011, 255, 861–880. [Google Scholar] [CrossRef]
- McGuinness, D.S. Olefin oligomerization via metallacycles: Dimerization, trimerization, tetramerization, and beyond. Chem. Rev. 2011, 111, 2321–2341. [Google Scholar] [CrossRef] [PubMed]
- Overett, M.J.; Blann, K.; Bollmann, A.; Dixon, J.T.; Haasbroek, D.; Killian, E.; Maumela, H.; McGuinness, D.S.; Morgan, D.H. Mechanistic investigations of the ethylene tetramerisation reaction. J. Am. Chem. Soc. 2005, 127, 10723–10730. [Google Scholar] [CrossRef] [PubMed]
- Belov, G. Tetramerization of ethylene to octene-1 (a review). Petrol. Chem. 2012, 52, 139–154. [Google Scholar] [CrossRef]
- Ma, X.; Liu, Y.; Wang, Z.; Zhao, X.; Mi, P. Ethylene tri-/tetramerization catalysts supported by diphosphinoindole ligands. J. Organomet. Chem. 2022, 958, 122175. [Google Scholar] [CrossRef]
- Jiang, T.; Zhang, S.; Jiang, X.; Yang, C.; Niu, B.; Ning, Y. The effect of N-aryl bisphosphineamine ligands on the selective ethylene tetramerization. J. Mol. Catal. A Chem. 2008, 279, 90–93. [Google Scholar] [CrossRef]
- Blann, K.; Bollmann, A.; Dixon, J.T.; Hess, F.M.; Killian, E.; Maumela, H.; Morgan, D.H.; Neveling, A.; Otto, S.; Overett, M.J. Highly selective chromium-based ethylene trimerisation catalysts with bulky diphosphinoamine ligands. Chem. Commun. 2005, 5, 620–621. [Google Scholar] [CrossRef]
- Overett, M.J.; Blann, K.; Bollmann, A.; Dixon, J.T.; Hess, F.; Killian, E.; Maumela, H.; Morgan, D.H.; Neveling, A.; Otto, S. Ethylene trimerisation and tetramerisation catalysts with polar-substituted diphosphinoamine ligands. Chem. Commun. 2005, 5, 622–624. [Google Scholar] [CrossRef]
- Bollmann, A.; Blann, K.; Dixon, J.T.; Hess, F.; Killian, E.; Maumela, H.; McGuinness, D.S.; Morgan, D.H.; Neveling, A.; Otto, S. Ethylene tetramerization: A new route to produce 1-octene in exceptionally high selectivities. J. Am. Chem. Soc. 2004, 126, 14712–14713. [Google Scholar] [CrossRef]
- Peitz, S.; Peulecke, N.; Aluri, B.R.; Hansen, S.; Müller, B.H.; Spannenberg, A.; Rosenthal, U.; Al-Hazmi, M.H.; Mosa, F.M.; Wöhl, A.; et al. A selective Chromium catalyst system for the trimerization of ethene and its coordination chemistry. Eur. J. Inorg. Chem. 2010, 2010, 1167–1171. [Google Scholar] [CrossRef]
- Lee, J.H.; Baek, J.W.; Lee, D.G.; Ko, J.H.; Lee, D.G.; Cho, K.S.; Lee, J.W.; Lee, B.Y. Preparation of extremely active ethylene tetramerization catalyst [iPrN(PAr2)2−CrCl2]+[B(C6F5)4]− (Ar = −C6H4-p-SiR3). Catalysts 2021, 11, 1122. [Google Scholar] [CrossRef]
- Jaseer, E.A.; Garcia, N.; Barman, S.; Khawaji, M.; Xu, W.; Alasiri, H.; Peedikakkal, A.M.P.; Akhtar, M.N.; Theravalappil, R. Highly efficient ethylene tetramerization using Cr catalysts constructed with trifluoromethyl-substituted N-Aryl PNP ligands. ACS Omega 2022, 7, 16333–16340. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Z.; Zhong, L.; Qiu, P.Y.; Dong, Q.; Cheng, R.H.; Vanderbilt, J.; Liu, B.P. Spin surface crossing between Chromium(I)/sextet and Chromium(III)/quartet without deprotonation in SNS-Cr mediated ethylene trimerization. Organometallics 2011, 30, 5297–5302. [Google Scholar] [CrossRef]
- Kuhlmann, S.; Blann, K.; Bollmann, A.; Dixon, J.T.; Killian, E.; Maumela, M.C.; Maumela, H.; Morgan, D.H.; Prétorius, M.; Taccardi, N.; et al. N-substituted diphosphinoamines: Toward rational ligand design for the efficient tetramerization of ethylene. J. Catal. 2007, 245, 279–284. [Google Scholar] [CrossRef]
- Britovsek, G.J.; McGuinness, D.S.; Wierenga, T.S.; Young, C.T. Single- and double-coordination mechanism in ethylene tri- and tetramerization with Cr/PNP catalysts. ACS Catal. 2015, 5, 4152–4166. [Google Scholar] [CrossRef]
- Lee, H.; Joe, Y.; Park, H. Chromium catalysts for ethylene trimerization/tetramerization functionalized with ortho-fluorinated arylphosphine ligand. Catal. Commun. 2019, 121, 15–18. [Google Scholar] [CrossRef]
- Lee, H.; Hong, S.H. Polyhedral oligomeric silsesquioxane-conjugated bis(diphenylphosphino)amine ligand for chromium(III) catalyzed ethylene trimerization and tetramerization. Appl. Catal. A Gen. 2018, 560, 21–27. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Zhang, X.; Wu, W.; Zhang, G.; Xu, S.; Shi, M. Switchable ethylene tri-/tetramerization with high activity: Subtle effect presented by backbone-substituent of carbon-bridged diphosphine ligands. ACS Catal. 2013, 3, 2311–2317. [Google Scholar] [CrossRef]
- Kim, S.K.; Kim, T.J.; Chung, J.H.; Hahn, T.K.; Chae, S.S.; Lee, H.S.; Cheong, M.; Kang, S.O. Bimetallic ethylene tetramerization catalysts derived from chiral DPPDME ligands: Syntheses, structural characterizations, and catalytic performance of [(DPPDME)CrCl3]2(DPPDME = S,S- and R,R-chiraphos and meso-achiraphos). Organometallics 2010, 29, 5805–5811. [Google Scholar] [CrossRef]
- McGuinness, D.S.; Overett, M.; Tooze, R.P.; Blann, K.; Dixon, J.T.; Slawin, A.M.Z. Ethylene Tri- and Tetramerization with Borate cocatalysts: effects on activity, selectivity, and catalyst degradation pathways. Organometallics 2007, 26, 1108–1111. [Google Scholar] [CrossRef]
- Lifschitz, A.M.; Hirscher, N.A.; Lee, H.B.; Buss, J.A.; Agapie, T. Ethylene tetramerization catalysis: Effects of Aluminum-induced isomerization of PNP to PPN ligands. Organometallics 2017, 36, 1640–1648. [Google Scholar] [CrossRef]
- Rucklidge, A.J.; McGuinness, D.S.; Tooze, R.P.; Slawin, A.M.Z.; Pelletier, J.D.A.; Hanton, M.J.; Webb, P.B. Ethylene tetramerization with cationic chromium(I) complexes. Organometallics 2007, 26, 2782–2787. [Google Scholar] [CrossRef]
- Jiang, T.; Liu, X.; Ning, Y.; Chen, H.; Luo, M. Performance of various aluminoxane activators in ethylene tetramerization based on PNP/Cr(III) catalyst system. Catal. Comm. 2007, 8, 1145–1148. [Google Scholar] [CrossRef]
- Kulangara, S.V.; Haveman, D.; Vidjayacoumar, B.; Korobkov, I.; Gambarotta, S.; Duchateau, R. Effect of cocatalysts and solvent on selective ethylene oligomerization. Organometallics 2015, 34, 1203–1210. [Google Scholar] [CrossRef]
- Alam, F. Selective Ethylene Oligomerization Catalysts Based on Cr(III) Complexes Stabilized by Silicon-Bridged Diphosphine Ligands. Ph.D. Thesis, Tianjin University of Science and Technology, Tianjin, China, 2019. [Google Scholar]
- Boulens, P.; Pellier, E.; Jeanneau, E.; Reek, J.N.; Olivier-Bourbigou, H.; Breuil, P.A.R. Self-assembled organometallic Nickel complexes as catalysts for selective dimerization of ethylene into 1-butene. Organometallics 2015, 34, 1139–1142. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).