
An Iron-Doped Calcium Titanate Cocatalyst for the Oxygen Reduction Reaction

Abstract
:1. Introduction
2. Results
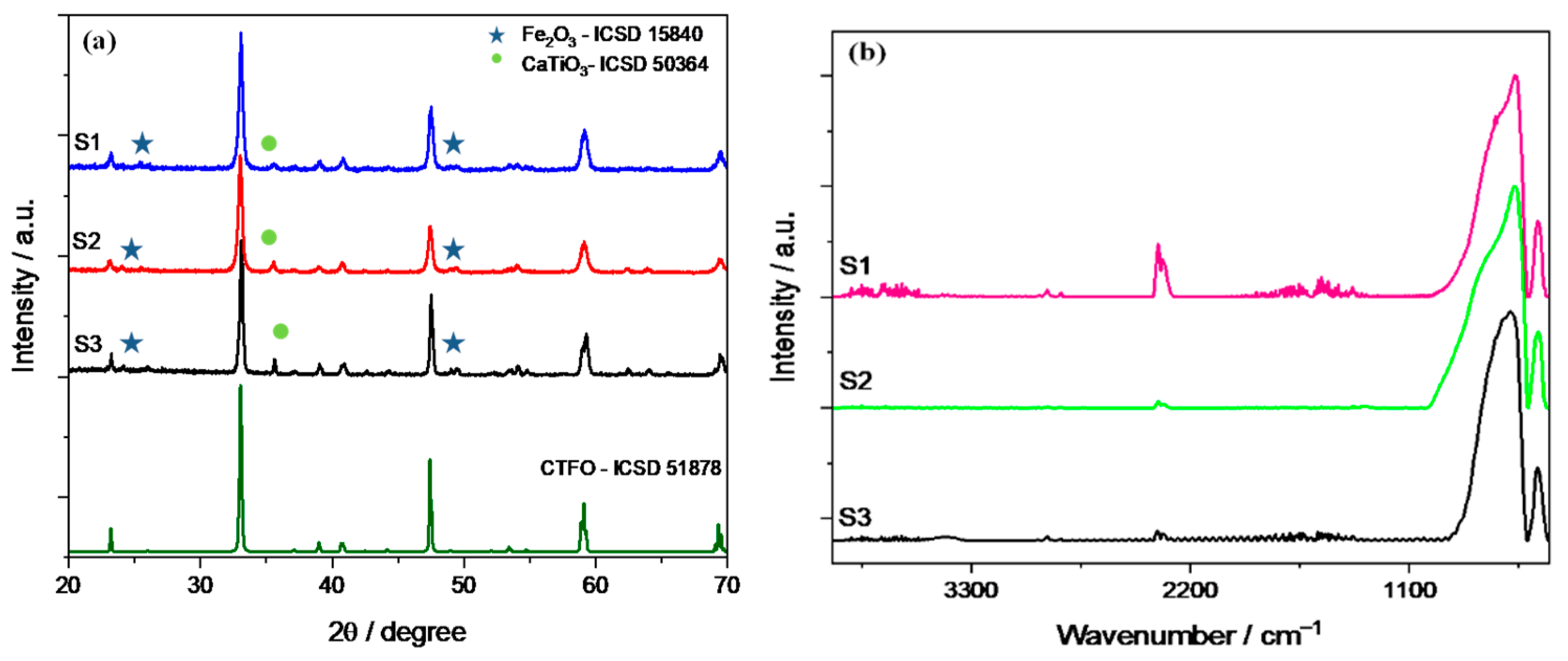
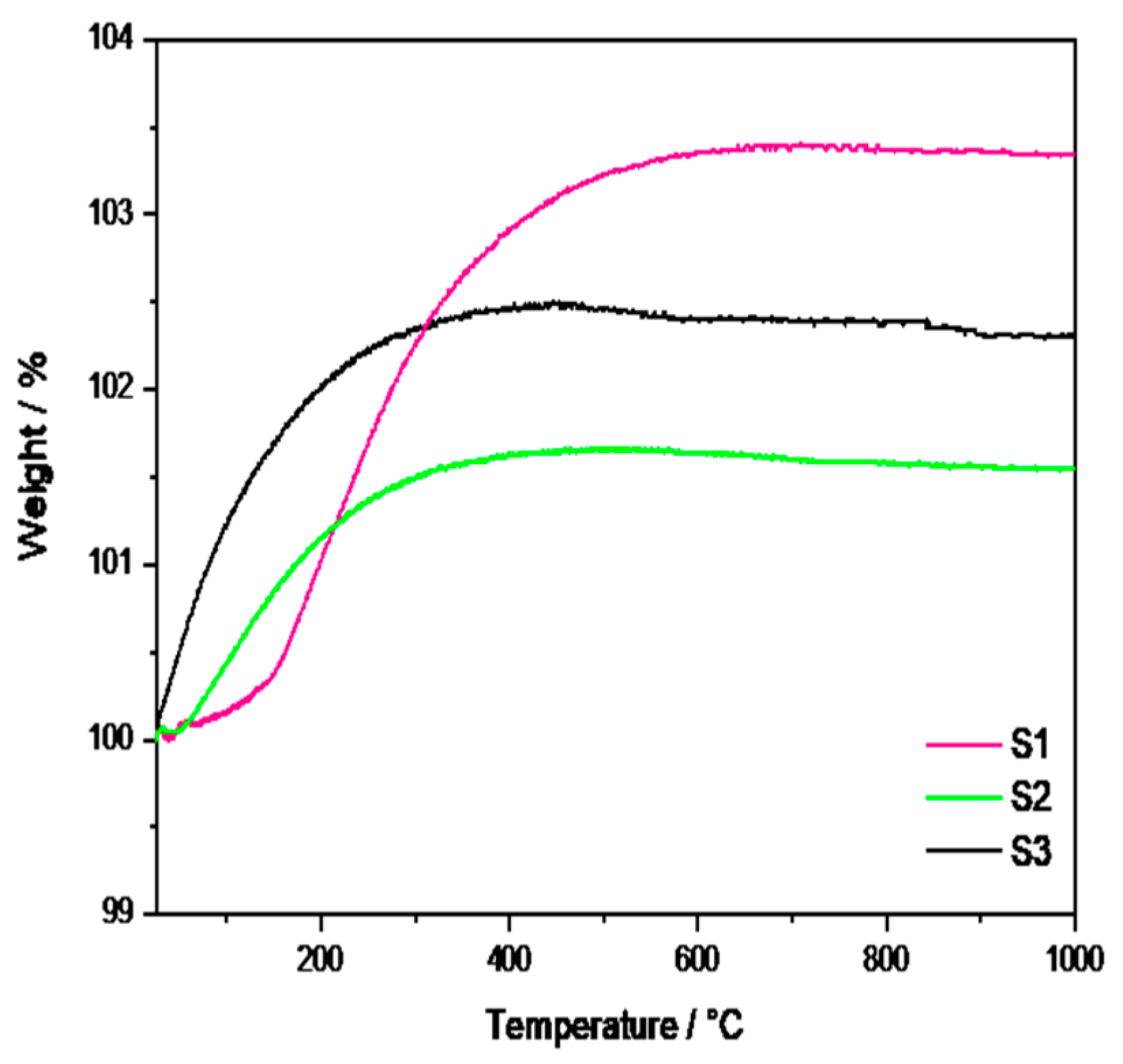
2.1. Physicochemical Characterization
2.2. Ex Situ Characterization of the Catalyst Inks
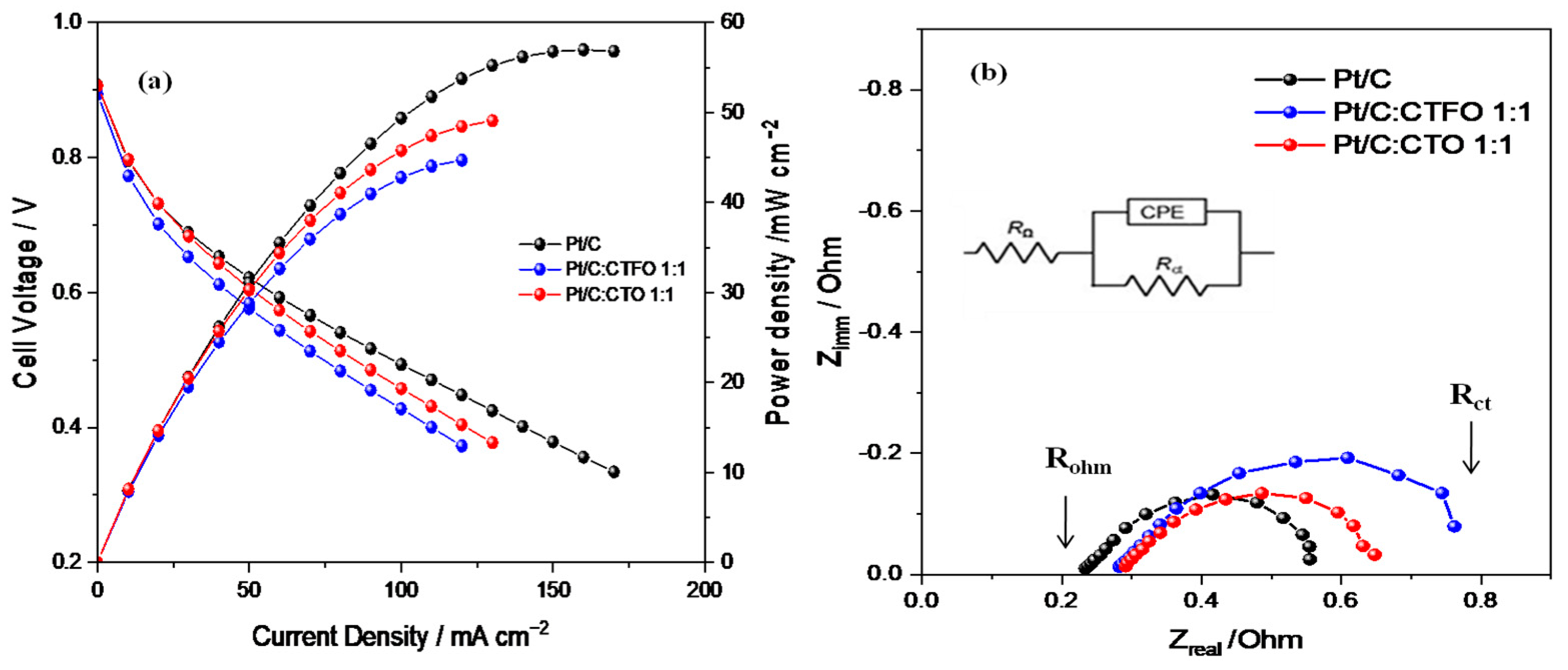
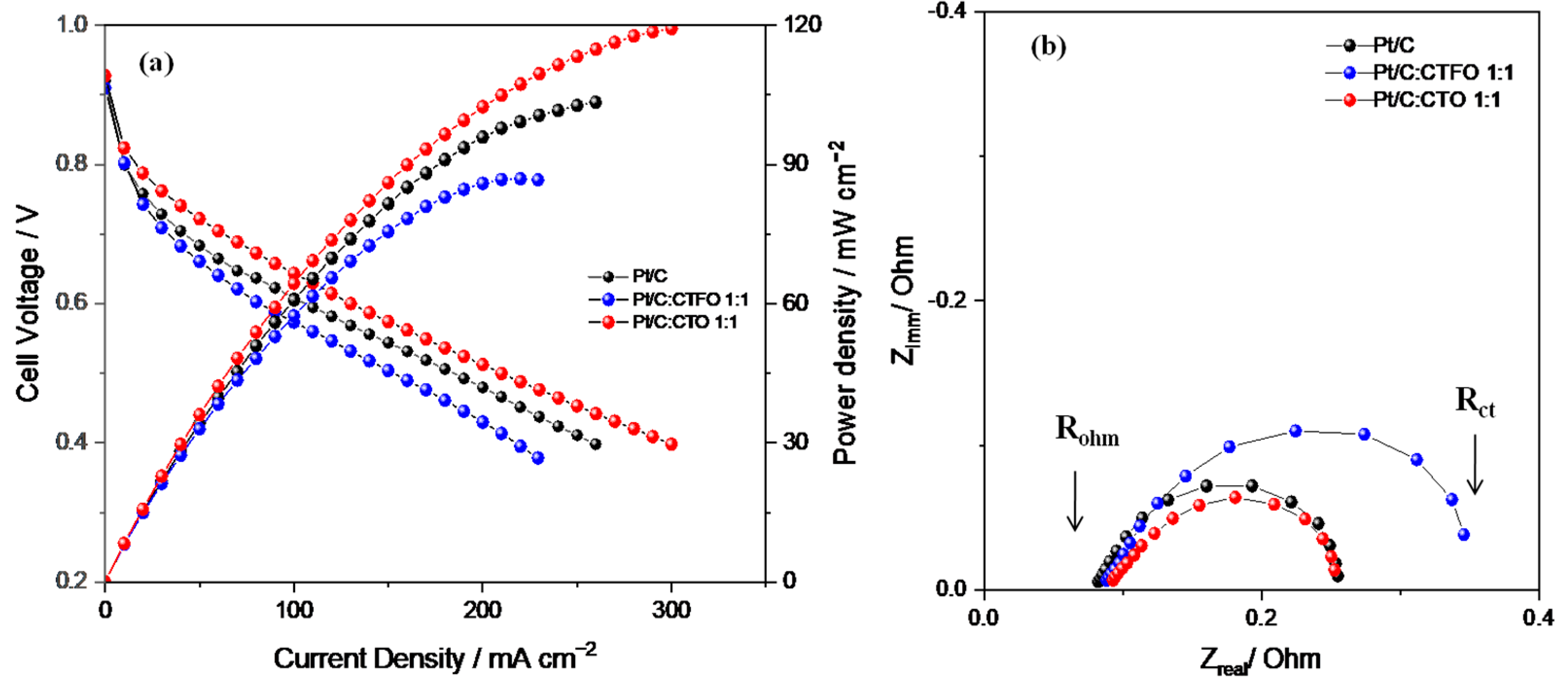
2.3. Fuel Cell Tests
3. Materials and Methods
3.1. Synthesis of CaTi1−xFexO3−δ
3.2. Physical and Chemical Characterizations
3.3. Electrochemical Investigations
3.3.1. Rotating Disk Electrode (RDE) Analysis
3.3.2. Proton Exchange Membrane Fuel Cell (PEMFC) Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Qu, E.; Hao, X.; Xiao, X.; Han, D.; Huang, S.; Huang, Z.; Wang, S.; Meng, Y. Proton exchange membranes for high temperature proton exchange membrane fuel cells: Challenges and perspectives. J. Power Sources 2022, 533, 231386–231406. [Google Scholar] [CrossRef]
- Hosseini, S.E.; Wahid, M.A. Hydrogen production from renewable and sustainable energy resources: Promising green energy carrier for clean development. Renew. Sustain. Energy Rev. 2016, 57, 850–866. [Google Scholar] [CrossRef]
- Siracusano, S.; Oldani, C.; Navarra, M.A.; Tonella, S.; Mazzapioda, L.; Briguglio, N.; Aricò, A.S. Chemically stabilized extrused and recast short side chain Aquivion® proton exchange membranes for high current density operation in water electrolysis. J. Membr. Sci. 2019, 578, 136–148. [Google Scholar] [CrossRef]
- Tellez-Cruz, M.M.; Escorihuela, J.; Solorza-Feria, O.; Compañ, V. Proton Exchange Membrane Fuel Cells (PEMFCs): Advances and Challenges. Polymers 2021, 13, 3064. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Chu, T.; Wang, T.; Wan, K.; Yang, D.; Li, B.; Ming, P.; Zhang, C. Preparation, Performance and Challenges of Catalyst Layer for Proton Exchange Membrane Fuel Cell. Membranes 2021, 11, 879. [Google Scholar] [CrossRef] [PubMed]
- Nanadegani, F.S.; Lay, E.N.; Sunden, B. Effects of an MPL on water and thermal management in a PEMFC. Int. J. Energy Res. 2019, 43, 274–296. [Google Scholar] [CrossRef] [Green Version]
- Owejan, J.P.; Trabold, T.A.; Mench, M.M. Oxygen transport resistance correlated to liquid water saturation in the gas diffusion layer of PEM fuel cells. Int. J. Heat Mass Transf. 2014, 71, 585–592. [Google Scholar] [CrossRef]
- Sasiwimonrit, K.; Chang, W.C. To improve the high temperature polymer electrolyte membrane fuel cells performance by altering the properties of catalyst layer. Int. J. Hydrog. Energy 2020, 45, 14491–14499. [Google Scholar] [CrossRef]
- He, Y.; Wu, G. PGM-Free Oxygen-Reduction Catalyst Development for Proton Exchange Membrane Fuel Cells: Challenges, Solutions, and Promises. Acc. Mater. Res. 2022, 3, 224–236. [Google Scholar] [CrossRef]
- Nie, Y.; Li, L.; We, Z. Recent advancements in Pt and Pt-free, catalysts for oxygen reduction reaction. Chem. Soc. Rev. 2015, 44, 2168–2201. [Google Scholar] [CrossRef]
- Hou, J.; Yang, M.; Ke, C.; Wei, G.; Priest, C.; Qiao, Z.; Wu, G.; Zhang, J. Platinum-Group-Metal Catalysts for Proton Exchange Membrane Fuel Cells: From Catalyst Design to Electrode Structure Optimization. Energy Chem. 2020, 2, 100023. [Google Scholar] [CrossRef]
- Lo Vecchio, C.; Aricò, A.S.; Monforte, G.; Baglio, V. EDTA-derived Co-N-C and Fe-N-C electrocatalysts for the oxygen reduction reaction in acid environment. Renew. Energy 2018, 120, 342–349. [Google Scholar] [CrossRef]
- Lo Vecchio, C.; Sebastiàn, D.; Làzaro, M.J.; Aricò, A.S.; Baglio, V. Methanol-Tolerant M–N–C Catalysts for Oxygen Reduction Reactions in Acidic Media and Their Application in Direct Methanol Fuel Cells. Catalysts 2018, 8, 650. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.Y.; Ganesan, P.; Popov, B.N. Titania supported platinum catalyst with high electrocatalytic activity and stability for polymer electrolyte membrane fuel cell. Appl. Catal. B 2011, 102, 71–77. [Google Scholar] [CrossRef]
- Gao, Y.; Hou, M.; Shao, Z.; Zhang, C.; Qin, X.; Yi, B. Preparation and characterization of Ti0.7Sn0.3O2 as catalyst support for oxygen reduction reaction. J. Energy Chem. 2014, 23, 331–337. [Google Scholar] [CrossRef]
- Song, M.; Song, Y.; Sha, W.; Xu, B.; Guo, J.; Wu, Y. Recent advances in non-precious transition metal/nitrogen-doped carbon for Oxygen Reduction electrocatalysts in PEMFCs. Catalysts 2020, 10, 141. [Google Scholar] [CrossRef] [Green Version]
- Takuya, M.; Hitoshi, F.; Fugane, K.; Togasaki, H.; Matsumura, D.; Tamura, K.; Nishihata, Y.; Yoshikawa, H.; Kobayashi, K.; Mori, T.; et al. Role of Cerium Oxide in the Enhancement of activity for the Oxygen Reduction Reaction at Pt-CeOx Nanocomposite Electrocatalyst- An in Situ Electrochemical X-ray Absorption Fine Structure Study. J. Phys. Chem. C 2012, 116, 10098–10102. [Google Scholar]
- Goswami, C.; Hazarika, K.K.; Bharali, P. Transition metal oxide nanocatalysts for oxygen reduction reaction. Mater. Sci. Energy Technol. 2018, 1, 117–128. [Google Scholar] [CrossRef]
- Fabbri, E.; Mohamed, R.; Levecque, P.; Conrad, O.; Kötza, R.; Schmidt, T.J. Unraveling the Oxygen Reduction Reaction Mechanism and Activity of d-Band Perovskite Electrocatalysts for Low Temperature Alkaline Fuel Cells. ECS Trans. 2014, 64, 1081–1093. [Google Scholar] [CrossRef]
- Ashok, A.; Kumar, A.; Bhosale, R.R.; Almomani, F.; Malik, S.S.; Suslov, S.; Tarlochan, F. Combustion Synthesis of Bifunctional LaMO3 (M=Cr, Mn, Fe, Co, Ni) Perovskites for Oxygen Reduction and Oxygen Evolution Reaction in Alkaline Media. J. Electroanal. Chem. 2018, 809, 22–30. [Google Scholar] [CrossRef]
- Seok, J.; Villarino, A.M.; Shi, Z.; Yang, Y.; Ahmadi, M.; Muller, D.A.; DiSalvo, F.J.; Abruña, H.D. La-Based Perovskite Oxide Catalysts for Alkaline Oxygen Reduction: The Importance of Electrochemical Stability. J. Phys. Chem. C 2022, 126, 3098–3108. [Google Scholar] [CrossRef]
- Suntivich, J.; Gasteiger, H.A.; Yabuuchi, N.; Nakanishi, H.; Goodenough, J.B.; Shao-Horn, Y. Design Principles for Oxygen-Reduction Activity on Perovskite Oxide Catalysts for Fuel Cells and Metal−Air Batteries. Nat. Chem. 2011, 3, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, W.; Kamiko, M.; Yagi, S. Enhanced catalytic activity of perovskite La1−xSrxMnO3+δ for the oxygen reduction reaction. New J. Chem. 2022, 46, 13082–13088. [Google Scholar] [CrossRef]
- Aoki, Y.; Takase, K.; Kiuchi, H.; Kowalski, D.; Sato, Y.; Toriumi, H.; Kitano, S.; Habazaki, H. In Situ Activation of a Manganese Perovskite Oxygen Reduction Catalyst in Concentrated Alkaline Media. J. Am. Chem. Soc. 2021, 143, 6505–6515. [Google Scholar] [CrossRef]
- Wang, Z.; You, Y.; Yuan, J.; Yin, Y.X.; Li, Y.T.; Xin, S.; Zhang, D. Nickel-Doped La0.8Sr0.2Mn1−xNixO3 Nanoparticles Containing Abundant Oxygen Vacancies as an Optimized Bifunctional Catalyst for Oxygen Cathode in Rechargeable Lithium-Air Batteries. ACS Appl. Mater. Interfaces 2016, 8, 6520–6528. [Google Scholar] [CrossRef]
- Aoki, Y.; Tsuji, E.; Motohashi, T.; Kowalski, D.; Habazaki, H. La0.7Sr0.3Mn1−xNixO3−δ Electrocatalysts for the Four-Electron Oxygen Reduction Reaction in Concentrated Alkaline Media. J. Phys. Chem. C 2018, 122, 22301–22308. [Google Scholar] [CrossRef]
- Hong, W.T.; Risch, M.; Stoerzinger, K.A.; Grimaud, A.; Suntivich, J.; Shao-Horn, Y. Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 2015, 8, 1404–1427. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Chen, C.; Han, Z.; Yang, Y.; Li, J.; Deng, Q. Recent Advances in Oxygen Electrocatalysts Based on Perovskite Oxides. Nanomaterials 2019, 9, 1161. [Google Scholar] [CrossRef] [Green Version]
- Retuerto, M.; Pascual, L.; Calle-Vallejo, F.; Ferrer, P.; Gianolio, D.; González Pereira, A.; García, A.; Torrero, J.; Fernández-Díaz, M.T.; Bencok, P.; et al. Na-doped ruthenium perovskite electrocatalystswith improved oxygen evolution activity anddurability in acidic media. Nat. Commun. 2019, 10, 2041–2050. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Jeerh, G.; Zou, P.; Lan, R.; Wang, M.; Wang, H.; Tao, S. Recent development of perovskite oxide-based electrocatalysts and their applications in low to intermediate temperature electrochemical devices. Mater. Today 2021, 49, 351–377. [Google Scholar] [CrossRef]
- Mazzapioda, L.; Lo Vecchio, C.; Paolone, A.; Aricò, A.S.; Baglio, V.; Navarra, M.A. Enhancing Oxygen Reduction Reaction Catalytic Activity Using a sub-stoichiometric CaTiO3−δ Additive. ChemElectroChem 2019, 6, 5941–5945. [Google Scholar] [CrossRef]
- Mazzapioda, L.; Lo Vecchio, C.; Aricò, A.S.; Navarra, M.A.; Baglio, V. Performance Improvement in Direct Methanol Fuel Cells by Using CaTiO3−δ Additive at the Cathode. Catalysts 2019, 9, 1017. [Google Scholar] [CrossRef] [Green Version]
- Han, B.H.; Grimaud, A.; Giordano, L.; Hong, W.T.; Diaz-Morales, O.; Yueh-Lin, L.; Hwang, J.; Charles, N.; Stoerzinger, K.A.; Yang, W.L.; et al. Iron-Based Perovskites for Catalyzing Oxygen Evolution Reaction. J. Phys. Chem. C 2018, 122, 8445–8454. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.J.; Fabbri, E.; Castelli, I.E.; Borlaf, M.; Graule, T.; Nachtegaa, M.; Schmidt, T.J. Fe-Doping in Double Perovskite PrBaCo2(1−x)Fe2xO6−δ:Insights into Structural and Electronic Effects toEnhance Oxygen Evolution Catalyst Stability. Catalyst 2019, 9, 263. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Shi, H.; You, H.; Su, M.; Huang, L.; Zhou, Z.; Zhang, C.; Zuo, J.; Yan, J.; Xiao, T.; et al. Cu-doped CaFeO3 perovskite oxide as oxygen reduction catalyst in air cathode microbial fuel cells. Environ. Res. 2022, 214, 113968. [Google Scholar] [CrossRef]
- He, H.; Sun, D.; Zhang, Q.; Fu, F.; Tang, Y.; Guo, J.; Shao, M.; Wang, H. Iron doped cauliflower-like rutile TiO2 with superior sodium storage properties. ACS Appl. Mater. Interfaces 2017, 9, 6093–6103. [Google Scholar] [CrossRef]
- Yang, H.; Han, C.; Xue, X. Photocatalytic activity of Fe-doped CaTiO3 under UV–visible light. J. Environ. Sci. 2014, 26, 1489–1495. [Google Scholar] [CrossRef]
- Figueiredo, F.M.; Kharton, V.V.; Viskup, A.P.; Frade, J.R. Surface enhanced oxygen permeation in CaTi1−xFexO3−δ ceramic membranes. J. Membr. Sci. 2004, 236, 73–80. [Google Scholar] [CrossRef]
- Branković, G.; Vukotić, V.; Branković, Z.; Varela, J.A. Investigation on possibility of mechanochemical synthesis of CaTiO3 from different precursors. J. Eur. Ceram. Soc. 2007, 27, 729–732. [Google Scholar] [CrossRef]
- Wang, Y.; Niu, C.G.; Wang, L.; Wang, Y.; Zhang, X.G.; Zeng, G.M. Synthesis of fern-like Ag/AgCl/CaTiO3 photocatalysts and their enhanced visible-light photocatalytic properties. RSC Adv. 2016, 6, 47873–47882. [Google Scholar] [CrossRef]
- Rached, A.; Wederni, M.A.; Khirouni, K.; Alaya, S.; Martín-Palma, R.J.; Dhahri, J. Structural, optical and electrical properties of barium titanate. Mater. Chem. Phys. 2021, 267, 124600–124611. [Google Scholar] [CrossRef]
- Abdel Aal, A.; Hammad, T.R.; Zawrah, M.; Battisha, I.K.; Abou Hammad, A.B. FTIR Study of Nanostructure Perovskite BaTiO3Doped with Both Fe3+and Ni2+Ions Prepared by Sol-Gel Technique. Acta Phys. Pol. A 2014, 126, 1318–1321. [Google Scholar] [CrossRef]
- Renzi, M.; Mignini, P.; Giuli, G.; Marassi, R.; Nobili, F. Rotating disk electrode study of Pt/Cs3HPMo11VO40 composite catalysts for performing and durable PEM fuel cells. Int. J. Hydrog. Energy 2016, 41, 11163–11173. [Google Scholar] [CrossRef]
- Campbell, C.T. Catalyst–support interactions: Electronic perturbations. Nat. Chem. 2012, 4, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Wei, M.; Yu, L.; Yang, F.; Chang, R.; Liu, Z.; Fu, Q.; Bao, X. Nature of interface confinement effect in oxide/metal catalysts. J. Phys. Chem. C 2015, 119, 27556–27561. [Google Scholar] [CrossRef]
- Jia, Q.; Ghoshal, S.; Li, J.; Liang, W.; Meng, G.; Che, H.; Zhang, S.; Ma, Z.-F.; Mukerjee, S. Metal and metal oxide interactions andtheir catalytic consequences for oxygen reduction reaction. J. Am. Chem. Soc. 2017, 139, 7893–7903. [Google Scholar] [CrossRef] [PubMed]
- Ando, F.; Gunji, T.; Tanabe, T.; Fukano, I.; Abruña, H.D.; Wu, J.; Ohsaka, T.; Matsumoto, F. Enhancement of the Oxygen Reduction Reaction Activity of Pt by Tuning Its d-Band Center via Transition Metal Oxide Support Interactions. ACS Catal. 2021, 11, 9317–9332. [Google Scholar] [CrossRef]
- Ou, D.R.; Mori, T.; Togasaki, H.; Takahashi, M.; Ye, F.; Drennan, J. Microstructural and Metal-Support Interactions of the Pt-CeO2/C Catalysts for Direct Methanol Fuel Cell Application. Langmuir 2011, 27, 3859–3866. [Google Scholar] [CrossRef] [PubMed]
- Renzi, M.; Nobili, F.; Miecznikowski, K.; Kostuch, A.; Wadas, A.; Rutkowska, I.A.; Kulesza, P.J. Activation of bimetallic PtFe nanoparticles with zeolite-type cesium salts of vanadium-substituted polyoxometallates toward electroreduction of oxygen at low Pt loadings for fuel cells. J. Solid State Electrochem. 2021, 26, 3–16. [Google Scholar] [CrossRef]
- Ji, Q.; Bi, L.; Zhang, J.; Cao, H.; Zhao, X.S. The role of oxygen vacancies of ABO3 perovskite oxides in the oxygen reduction reaction. Energy Environ. Sci. 2020, 13, 1408–1428. [Google Scholar] [CrossRef]
- Santana, J.; Espinoza-Andaluz, M.; Li, T.; Andersson, M. A Detailed Analysis of Internal Resistance of a PEFC Comparing High and Low Humidification of the Reactant Gases. Front. Energy Res. 2020, 8, 217. [Google Scholar] [CrossRef]
- Brocato, S.; Serov, A.; Atanassov, P. pH dependence of catalytic activity for ORR of the non-PGM catalyst derived from heat-treated Fe–phenanthroline. Electrochim. Acta 2013, 87, 361–365. [Google Scholar] [CrossRef]
- Mazzapioda, L.; Navarra, M.A.; Panero, S. Polymer electrolyte membranes based on Nafion and a superacidic inorganic additive for fuel cell applications. Polymers 2019, 11, 914. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acronyms | Precursors | Thermal Treatment | CTFO % | CTO % | Fe2O3 % | Crystallite Size, nm |
|---|---|---|---|---|---|---|
| S1 | CaCO3, TiO2, Fe2O3 | 5 h, 1100 °C air | 66 | 22 | 12 | 107 ± 7 |
| S2 | CaCO3, TiO2, FeO(OH) | 5 h, 1100 °C air | 86 | 5 | 9 | 70 ± 2 |
| S3 | CaCO3, TiO2, FeO(OH) | 5 h, 800 °C argon | 93 | 3 | 4 | 65 ± 2 |
| Samples | ECSA/m2 g−1 |
|---|---|
| Pt/C | 71 |
| Pt/C:CTFO 1:0.5 | 65 |
| Pt/C:CTFO 1:1 | 80 |
| Samples | Rohm/mΩ | Rp/mΩ | Rct/mΩ |
|---|---|---|---|
| Pt/C | 263 | 320 | 583 |
| Pt/C:CTFO 1:1 | 286 | 545 | 831 |
| Pt/C:CTO 1:1 | 292 | 377 | 669 |
| Samples | Rohm/mΩ | Rp/mΩ | Rct/mΩ |
|---|---|---|---|
| Pt/C | 90 | 190 | 280 |
| Pt/C:CTFO 1:1 | 92 | 284 | 376 |
| Pt/C:CTO 1:1 | 93 | 173 | 266 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzapioda, L.; Renga, R.; Navarra, M.A. An Iron-Doped Calcium Titanate Cocatalyst for the Oxygen Reduction Reaction. Catalysts 2023, 13, 127. https://doi.org/10.3390/catal13010127
Mazzapioda L, Renga R, Navarra MA. An Iron-Doped Calcium Titanate Cocatalyst for the Oxygen Reduction Reaction. Catalysts. 2023; 13(1):127. https://doi.org/10.3390/catal13010127
Chicago/Turabian StyleMazzapioda, Lucia, Riccardo Renga, and Maria Assunta Navarra. 2023. "An Iron-Doped Calcium Titanate Cocatalyst for the Oxygen Reduction Reaction" Catalysts 13, no. 1: 127. https://doi.org/10.3390/catal13010127