
CO and Propane Combustion on La0.8Sr0.2CoxFe1−xO3−δ Perovskites: Effect of Fe-to-Co Ratio on Catalytic Activity



Abstract
:1. Introduction
2. Results
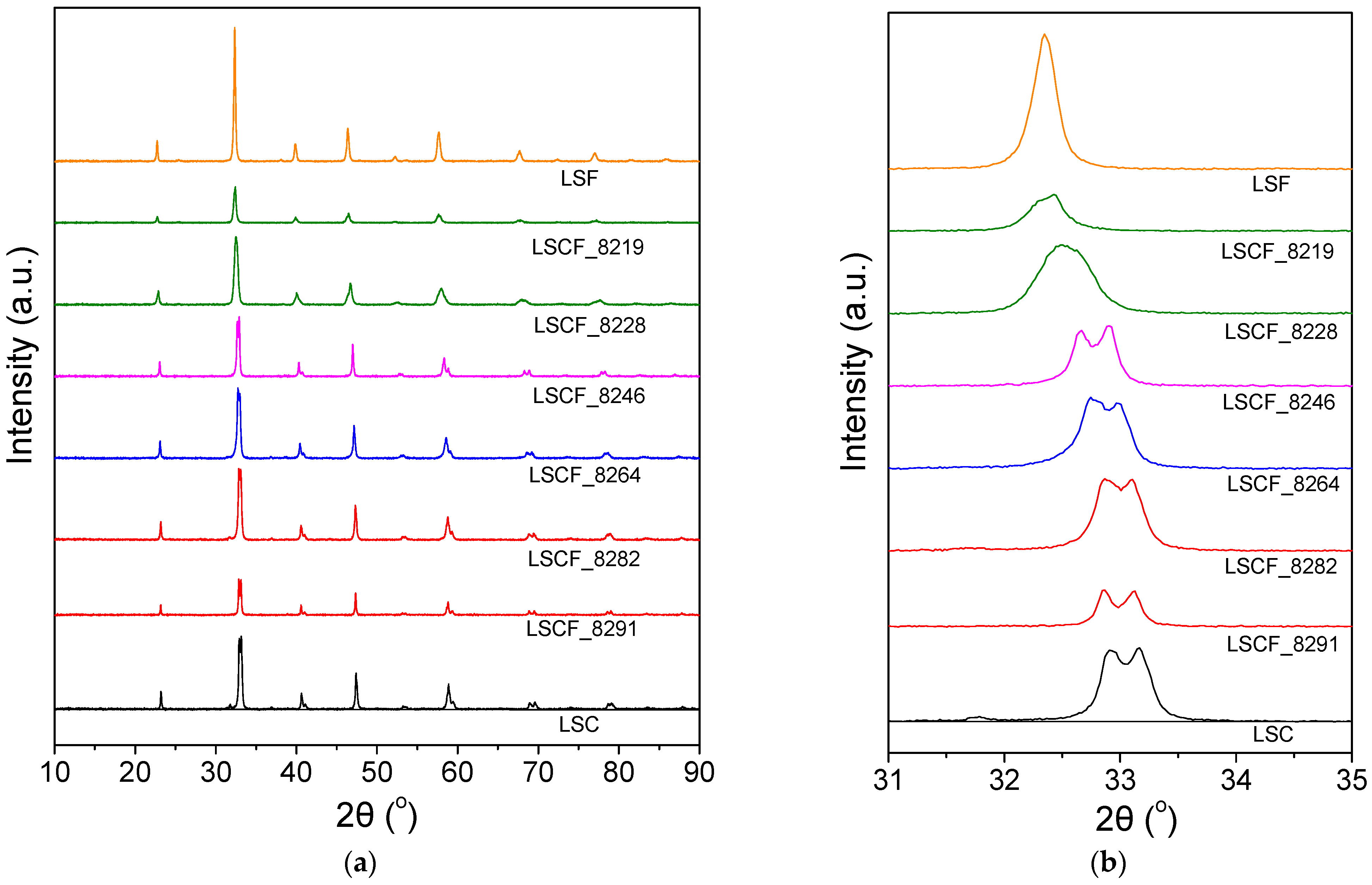
2.1. Physicochemical and Structural Characterization
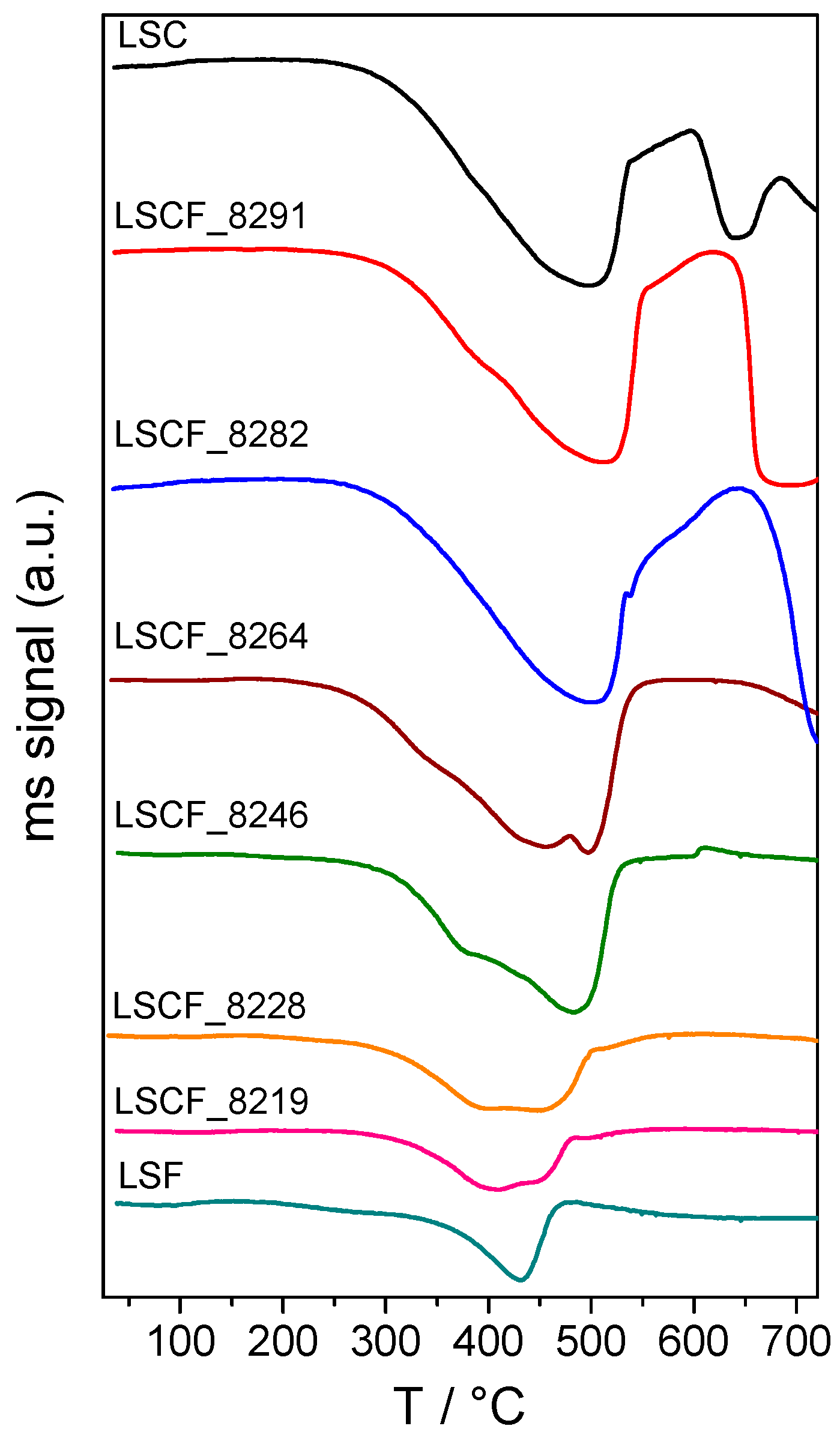
2.2. Investigation of the Perovskite Reducibility
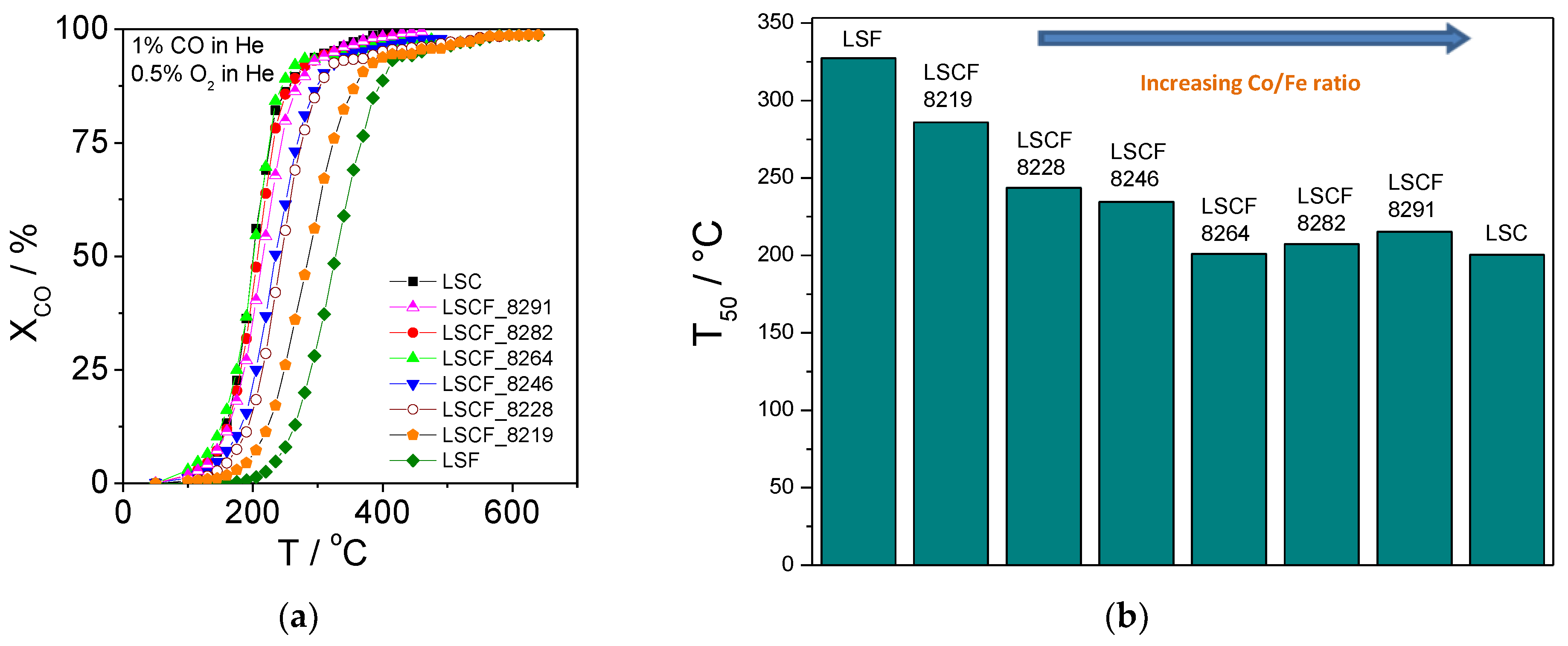
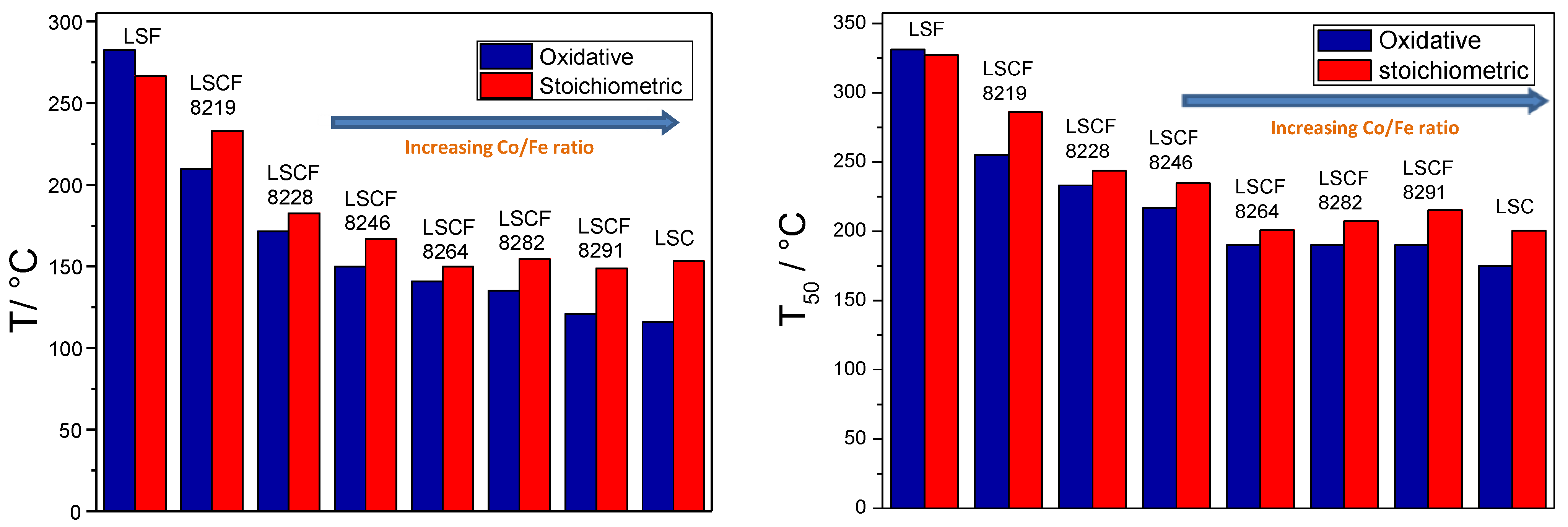
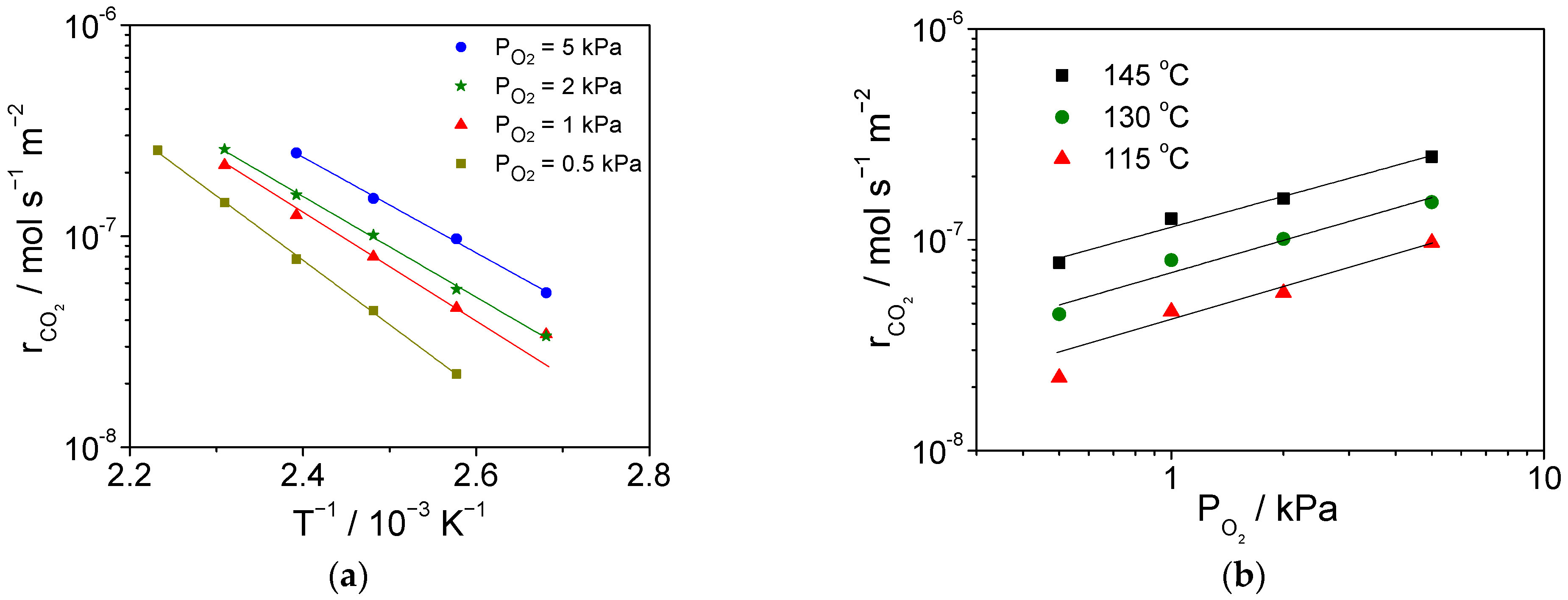
2.3. Catalytic Activity for CO Combustion
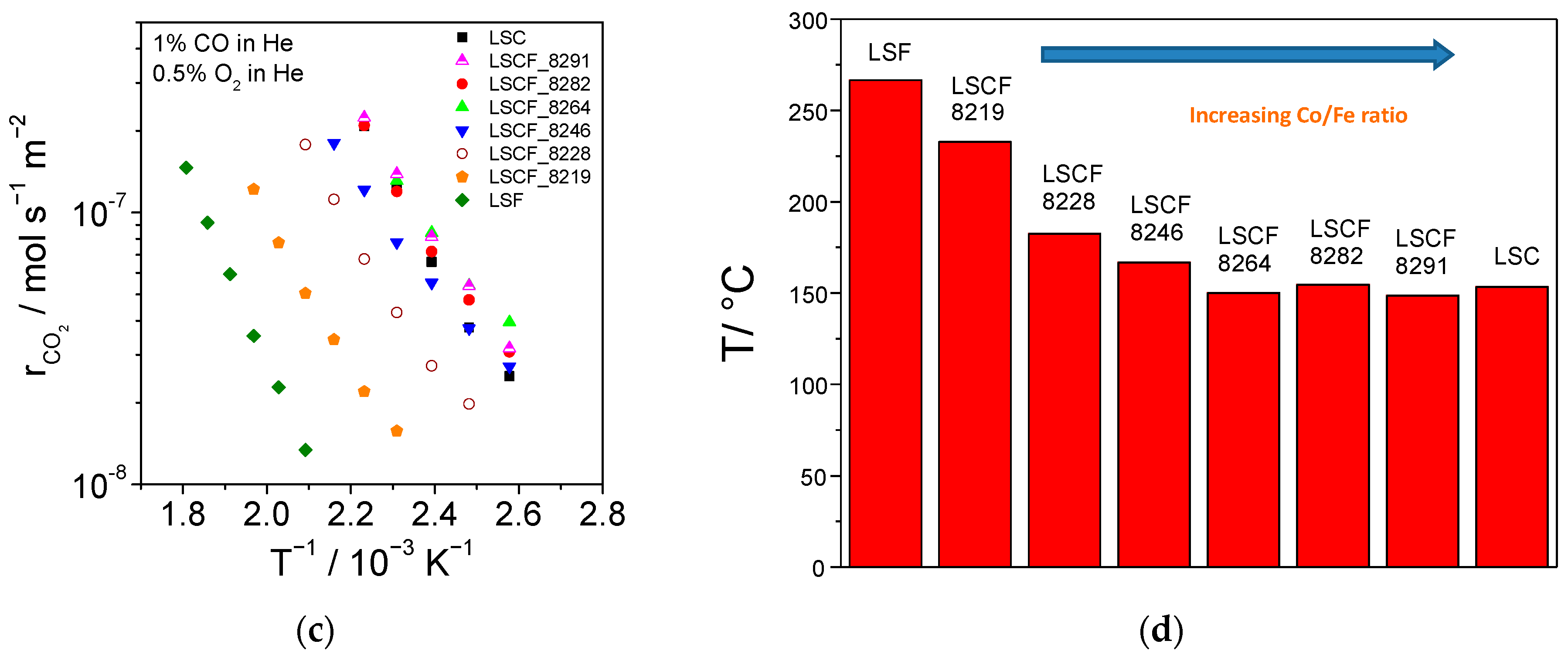
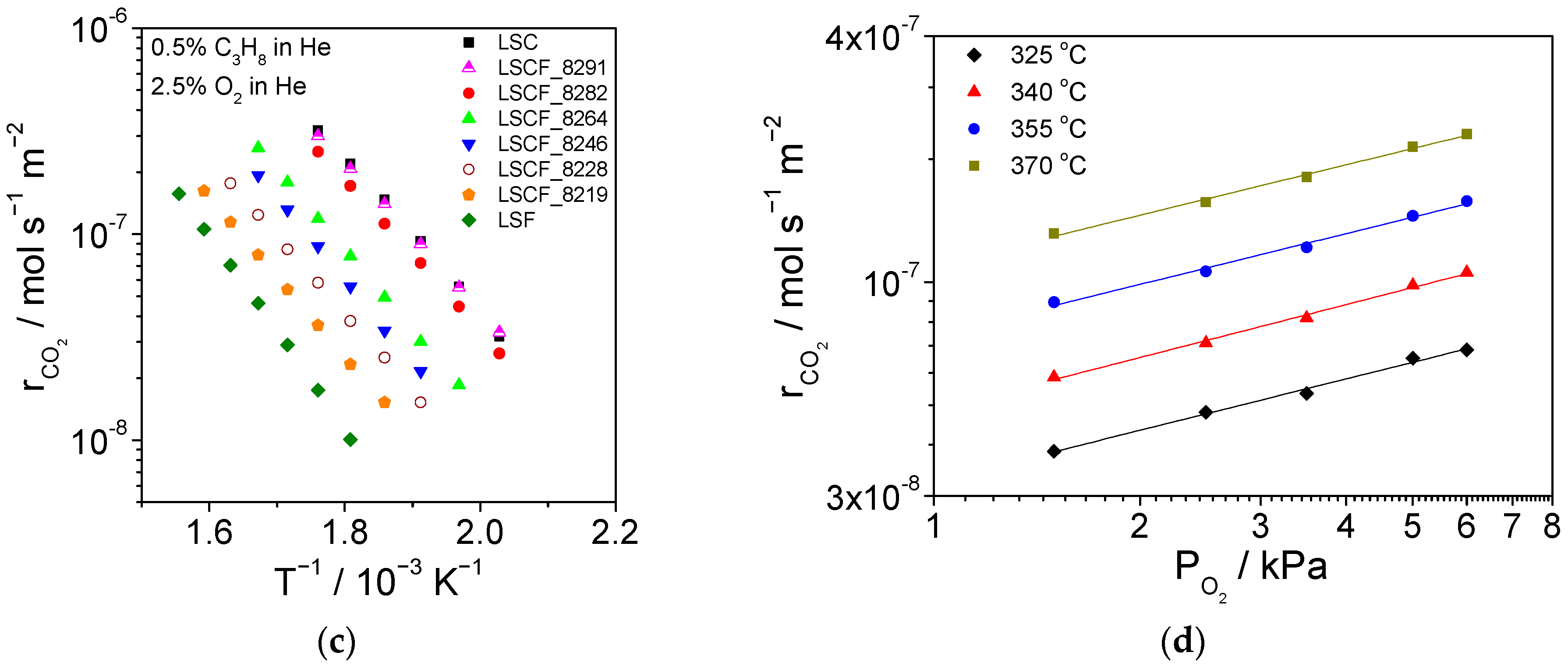
2.4. Catalytic Activity for Propane Combustion
3. Discussion
4. Materials and Methods
4.1. Synthesis of the Perovskite Oxides
4.2. Physicochemical Characterization of the Perovskite Oxides (BET, XRD, and ICP-OES)
4.3. H2 Temperature-Programmed Reduction Characterization
4.4. Catalytic Oxidation Measurements
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dey, S.; Sun, S.; Mehta, N.S. Carbon monoxide catalytic oxidation over various iron-based nanoparticles at ambient conditions: A Review. Carbon Capture Sci. Technol. 2021, 1, 100013. [Google Scholar] [CrossRef]
- Yuan, C.; Wu, H.B.; Xie, Y.; Lou, X.W. Mixed transition-metal oxides: Design, synthesis, and energy-related applications. Angew. Chem. Int. Ed. 2014, 53, 1488–1504. [Google Scholar] [CrossRef]
- Huang, W. Oxide Nanocrystal Model Catalysts. Acc. Chem. Res. 2016, 49, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Yakoumis, I.; Panou, M.; Moschovi, A.M.; Panias, D. Recovery of platinum group metals from spent automotive catalysts: A review. Clean. Eng. Technol. 2021, 3, 100112. [Google Scholar] [CrossRef]
- Laguna, O.H.; Bobadilla, L.F.; Hernández, W.Y.; Centeno, M.A. Low-Temperature CO Oxidation. In Perovskites and Related Mixed Oxides: Concepts and Applications; Granger, P., Parvulescu, V.I., Kaliaguine, S., Prellier, W., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2016; pp. 451–473. [Google Scholar] [CrossRef]
- Pramuanjaroenkij, A.; Kakaç, S. The fuel cell electric vehicles: The highlight review. Int. J. Hydrogen Energy 2023, 48, 9401–9425. [Google Scholar] [CrossRef]
- Jiao, K.; Xuan, J.; Du, Q.; Bao, Z.; Xie, B.; Wang, B.; Zhao, Y.; Fan, L.; Wang, H.; Hou, Z.; et al. Designing the next generation of proton-exchange membrane fuel cells. Nature 2021, 595, 361–369. [Google Scholar] [CrossRef]
- Bampos, G.; Bebelis, S. Performance of a Pd-Zn Cathode Electrode in a H2 Fueled Single PEM Fuel Cell. Electronics 2022, 11, 2776. [Google Scholar] [CrossRef]
- Liu, H.; Li, D.; Guo, J.; Li, Y.; Liu, A.; Bai, Y.; He, D. Recent advances on catalysts for preferential oxidation of CO. Nano Res. 2022, 16, 4399–4410. [Google Scholar] [CrossRef]
- Feng, C.; Liu, X.; Zhu, T.; Tian, M. Catalytic oxidation of CO on noble metal-based catalysts. Environ. Sci. Pollut. Res. Int. 2021, 28, 24847–24871. [Google Scholar] [CrossRef]
- Prasad, R.; Singh, P. A Review on CO Oxidation Over Copper Chromite Catalyst. Catal. Rev. 2012, 54, 224–279. [Google Scholar] [CrossRef]
- Royer, S.; Duprez, D. Catalytic Oxidation of Carbon Monoxide over Transition Metal Oxides. ChemCatChem 2011, 3, 24–65. [Google Scholar] [CrossRef]
- Peña, M.A.; Fierro, J.L.G. Chemical Structures and Performance of Perovskite Oxides. Chem. Rev. 2001, 101, 1981–2018. [Google Scholar] [CrossRef] [PubMed]
- Yamazoe, N.; Teraoka, Y. Oxidation catalysis of perovskites—relationships to bulk structure and composition (valency, defect, etc.). Catal. Today 1990, 8, 175–199. [Google Scholar] [CrossRef]
- Voorhoeve, R.J.H. Perovskite-Related Oxides as Oxidation—Reduction Catalysts. In Advanced Materials in Catalysis; Burton, J.J., Garten, R.L., Eds.; Academic Press, Inc.: New York, NY, USA, 1977; pp. 129–180. [Google Scholar] [CrossRef]
- Royer, S.; Duprez, D.; Can, F.; Courtois, X.; Batiot-Dupeyrat, C.; Laassiri, S.; Alamdari, H. Perovskites as Substitutes of Noble Metals for Heterogeneous Catalysis: Dream or Reality. Chem. Rev. 2014, 114, 10292–10368. [Google Scholar] [CrossRef]
- Giordano, L.; Akkiraju, K.; Jacobs, R.; Vivona, D.; Morgan, D.; Shao-Horn, Y. Electronic Structure-Based Descriptors for Oxide Properties and Functions. Acc. Chem. Res. 2022, 55, 298–308. [Google Scholar] [CrossRef]
- Yang, C.; Grimaud, A. Factors Controlling the Redox Activity of Oxygen in Perovskites: From Theory to Application for Catalytic Reactions. Catalysts 2017, 7, 149. [Google Scholar] [CrossRef]
- Polo-Garzon, F.; Wu, Z. Acid–base catalysis over perovskites: A review. J. Mater. Chem. A 2018, 6, 2877–2894. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, P.; Dai, S. Recent Advances of Lanthanum-Based Perovskite Oxides for Catalysis. ACS Catal. 2015, 5, 6370–6385. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, C.; Wang, J.; Li, H.; Duan, Y.; Liu, Z.; Lee, J.Y.; Hu, X.; Xi, S.; Du, Y.; et al. The interplay between the suprafacial and intrafacial mechanisms for complete methane oxidation on substituted LaCoO3 perovskite oxides. J. Catal. 2020, 390, 1–11. [Google Scholar] [CrossRef]
- Mars, P.; van Krevelen, D.W. Oxidations carried out by means of vanadium oxide catalysts. Chem. Eng. Sci. 1954, 3, 41–59. [Google Scholar] [CrossRef]
- Pecchi, G.; Jiliberto, M.G.; Buljan, A.; Delgado, E.J. Relation between defects and catalytic activity of calcium doped LaFeO3 perovskite. Solid State Ion. 2011, 187, 27–32. [Google Scholar] [CrossRef]
- Kizaki, H.; Kusakabe, K. Ab-initio study of Sr-doping effects on nitric oxide adsorption on the LaO (001) surface of LaFeO3. Surf. Sci. 2012, 606, 1783–1789. [Google Scholar] [CrossRef]
- Merino, N.A.; Barbero, B.P.; Grange, P.; Cadús, L.E. La1-xCaxCoO3 perovskite-type oxides: Preparation, characterisation, stability, and catalytic potentiality for the total oxidation of propane. J. Catal. 2005, 231, 232–244. [Google Scholar] [CrossRef]
- Zhang, H.M.; Shimizu, Y.; Teraoka, Y.; Miura, N.; Yamazoe, N. Oxygen sorption and catalytic properties of La1−xSrxCo1−yFeyO3 perovskite-type oxides. J. Catal. 1990, 121, 432–440. [Google Scholar] [CrossRef]
- Scott, S.P.; Mantzavinos, D.; Hartley, A.; Sahibzada, M.; Metcalfe, I.S. Reactivity of LSCF perovskites. Solid State Ion. 2002, 152–153, 777–781. [Google Scholar] [CrossRef]
- Lankhorst, M.H.R.; ten Elshof, J.E. Thermodynamic Quantities and Defect Structure of La0.6Sr0.4Co1−yFeyO3−δ (y=0–0.6) from High-Temperature Coulometric Titration Experiments. J. Solid State Chem. 1997, 130, 302–310. [Google Scholar] [CrossRef]
- Levasseur, B.; Kaliaguine, S. Effects of iron and cerium in La1−yCeyCo1−xFexO3 perovskites as catalysts for VOC oxidation. Appl. Catal. B Environ. 2009, 88, 305–314. [Google Scholar] [CrossRef]
- Tanaka, H.; Mizuno, N.; Misono, M. Catalytic activity and structural stability of La0.9Ce0.1Co1−xFexO3 perovskite catalysts for automotive emissions control. Appl. Catal. A Gen. 2003, 244, 371–382. [Google Scholar] [CrossRef]
- Isupova, L.A.; Sadykov, V.A.; Tsybulya, S.V.; Kryukova, G.N.; Ivanov, V.P.; Petrov, A.N.; Kononchuk, O.F. Effect of structural disorder on the catalytic activity of mixed La−Sr−Co−Fe−O perovskites. React. Kinet. Catal. Lett. 1997, 62, 129–135. [Google Scholar] [CrossRef]
- David, E.; Niculescu, V.-C. Volatile Organic Compounds (VOCs) as Environmental Pollutants: Occurrence and Mitigation Using Nanomaterials. Int. J. Environ. Res. Public Health 2021, 18, 13147. [Google Scholar] [CrossRef]
- Avila, M.S.; Vignatti, C.I.; Apesteguía, C.R.; Garetto, T.F. Effect of support on the deep oxidation of propane and propylene on Pt-based catalysts. Chem. Eng. J. 2014, 241, 52–59. [Google Scholar] [CrossRef]
- Luo, Y.; Zuo, J.; Feng, X.; Qian, Q.; Zheng, Y.; Lin, D.; Huang, B.; Chen, Q. Good interaction between well dispersed Pt and LaCoO3 nanorods achieved rapid Co3+/Co2+ redox cycle for total propane oxidation. Chem. Eng. J. 2019, 357, 395–403. [Google Scholar] [CrossRef]
- O’Brien, C.P.; Jenness, G.R.; Dong, H.; Vlachos, D.G.; Lee, I.C. Deactivation of Pt/Al2O3 during propane oxidation at low temperatures: Kinetic regimes and platinum oxide formation. J. Catal. 2016, 337, 122–132. [Google Scholar] [CrossRef]
- Hu, Z.; Wang, Z.; Guo, Y.; Wang, L.; Guo, Y.; Zhang, J.; Zhan, W. Total Oxidation of Propane over a Ru/CeO2 Catalyst at Low Temperature. Environ. Sci. Technol. 2018, 52, 9531–9541. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Liu, X.; Meng, D.; Guo, Y.; Guo, Y.; Lu, G. Effect of Ceria Crystal Plane on the Physicochemical and Catalytic Properties of Pd/Ceria for CO and Propane Oxidation. ACS Catal. 2016, 6, 2265–2279. [Google Scholar] [CrossRef]
- Wang, W.; Li, D.; Yu, H.; Liu, C.; Tang, C.; Chen, J.; Lu, J.; Luo, M. Insights into Different Reaction Behaviors of Propane and CO Oxidation over Pt/CeO2 and Pt/Nb2O5: The Crucial Roles of Support Properties. J. Phys. Chem. C 2021, 125, 19301–19310. [Google Scholar] [CrossRef]
- Ma, L.; Geng, Y.; Chen, X.; Yan, N.; Li, J.; Schwank, J.W. Reaction mechanism of propane oxidation over Co3O4 nanorods as rivals of platinum catalysts. Chem. Eng. J. 2020, 402, 125911. [Google Scholar] [CrossRef]
- Klvana, D.; Song, K.S.; Kirchnerova, J. Catalytic performance of La0.66Sr0.34Co0.2Fe0.8O3 perovskite in propane combustion: Effect of preparation and specific surface area. Korean J. Chem. Eng. 2002, 19, 932–939. [Google Scholar] [CrossRef]
- Vannice, M.A. An analysis of the Mars–van Krevelen rate expression. Catal. Today 2007, 123, 18–22. [Google Scholar] [CrossRef]
- Alifanti, M.; Kirchnerova, J.; Delmon, B.; Klvana, D. Methane and propane combustion over lanthanum transition-metal perovskites: Role of oxygen mobility. Appl. Catal. A Gen. 2004, 262, 167–176. [Google Scholar] [CrossRef]
- Nakamura, T.; Misono, M.; Yoneda, Y. Reduction-oxidation and catalytic properties of La1−xSrxCoO3. J. Catal. 1983, 83, 151–159. [Google Scholar] [CrossRef]
- Nitadori, T.; Misono, M. Catalytic properties of La1−xA′xFeO3 (A′ = Sr,Ce) and La1−xCexCoO3. J. Catal. 1985, 93, 459–466. [Google Scholar] [CrossRef]
- Merino, N.A.; Barbero, B.P.; Ruiz, P.; Cadús, L.E. Synthesis, characterisation, catalytic activity and structural stability of LaCo1−yFeyO3±λ perovskite catalysts for combustion of ethanol and propane. J. Catal. 2006, 240, 245–257. [Google Scholar] [CrossRef]
- Song, K.S.; Klvana, D.; Kirchnerova, J. Kinetics of propane combustion over La0.66Sr0.34Ni0.3Co0.7O3 perovskite. Appl. Catal. A Gen. 2001, 213, 113–121. [Google Scholar] [CrossRef]
- Safakas, A.; Bampos, G.; Bebelis, S. Oxygen reduction reaction on La0.8Sr0.2CoxFe1-xO3-δ perovskite/carbon black electrocatalysts in alkaline medium. Appl. Catal. B Environ. 2019, 244, 225–232. [Google Scholar] [CrossRef]
- Natile, M.M.; Poletto, F.; Galenda, A.; Glisenti, A.; Montini, T.; De Rogatis, L.; Fornasiero, P. La0.6Sr0.4Co1−yFeyO3−δ Perovskites: Influence of the Co/Fe Atomic Ratio on Properties and Catalytic Activity toward Alcohol Steam-Reforming. Chem. Mater. 2008, 20, 2314–2327. [Google Scholar] [CrossRef]
- Tai, L.-W.; Nasrallah, M.M.; Anderson, H.U.; Sparlin, D.M.; Sehlin, S.R. Structure and electrical properties of La1−xSrxCo1−yFeyO3. Part 1. The system La0.8Sr0.2Co1−yFeyO3. Solid State Ion. 1995, 76, 259–271. [Google Scholar] [CrossRef]
- Gholizadeh, A. The effects of A/B-site substitution on structural, redox and catalytic properties of lanthanum ferrite nanoparticles. J. Mater. Res. Technol. 2019, 8, 457–466. [Google Scholar] [CrossRef]
- Cullity, B.D.; Stock, S.R. Elements of X-ray Diffraction, 3rd ed.; Pearson Education, Ltd.: Harlow, UK, 2001; p. 174. [Google Scholar]
- Mote, V.D.; Purushotham, Y.; Dole, B.N. Williamson-Hall analysis in estimation of lattice strain in nanometer-sized ZnO particles. J. Theor. Appl. Phys. 2012, 6, 6. [Google Scholar] [CrossRef]
- Bampos, G.; Sygellou, L.; Bebelis, S. Oxygen reduction reaction activity of Pd-based bimetallic electrocatalysts in alkaline medium. Catal. Today 2020, 355, 685–697. [Google Scholar] [CrossRef]
- Deng, J.; Dai, H.; Jiang, H.; Zhang, L.; Wang, G.; He, H.; Au, C.T. Hydrothermal Fabrication and Catalytic Properties of La1-xSrxM1-yFeyO3 (M = Mn, Co) That Are Highly Active for the Removal of Toluene. Environ. Sci. Technol. 2010, 44, 2618–2623. [Google Scholar] [CrossRef] [PubMed]
- García-López, E.; Marcì, G.; Puleo, F.; La Parola, V.; Liotta, L.F. La1−xSrxCo1−yFeyO3−δ perovskites: Preparation, characterization and solar photocatalytic activity. Appl. Catal. B Environ. 2015, 178, 218–225. [Google Scholar] [CrossRef]
- Kuhn, J.N.; Ozkan, U.S. Effect of Co Content Upon the Bulk Structure of Sr- and Co-doped LaFeO3. Catal. Lett. 2008, 121, 179–188. [Google Scholar] [CrossRef]
- Siebert, E.; Roux, C.; Boréave, A.; Gaillard, F.; Vernoux, P. Oxido-reduction properties of La0.7SSr0.3Co0.8Fe0.2O3−δ perovskite oxide catalyst. Solid State Ion. 2011, 183, 40–47. [Google Scholar] [CrossRef]
- Chang, H.; Bjørgum, E.; Mihai, O.; Yang, J.; Lein, H.L.; Grande, T.; Raaen, S.; Zhu, Y.-A.; Holmen, A.; Chen, D. Effects of Oxygen Mobility in La–Fe-Based Perovskites on the Catalytic Activity and Selectivity of Methane Oxidation. ACS Catal. 2020, 10, 3707–3719. [Google Scholar] [CrossRef]
- Tascón, J.M.D.; García Fierro, J.L.; González Tejuca, L. Kinetics and Mechanism of CO Oxidation on LaCoO3. Z. Phys. Chem. 1981, 124, 249–257. [Google Scholar] [CrossRef]
- Rhee, C.K.; Lee, H.-I. CO oxidation on LaCoO3 perovskite. Korean J. Chem. Eng. 1994, 11, 48–54. [Google Scholar] [CrossRef]
- Adler, S.B.; Chen, X.Y.; Wilson, J.R. Mechanisms and rate laws for oxygen exchange on mixed-conducting oxide surfaces. J. Catal. 2007, 245, 91–109. [Google Scholar] [CrossRef]
- Campbell, I.M. Catalysis at Surfaces; Chapman and Hall: London, UK, 1988; p. 145. [Google Scholar]
- Tascón, J.M.D.; González Tejuca, L. Adsorption of CO on the Perovskite-Type Oxide LaCoO3. Z. Phys. Chem. 1980, 121, 63–78. [Google Scholar] [CrossRef]
- Luo, Y.; Zheng, Y.; Feng, X.; Lin, D.; Qian, Q.; Wang, X.; Zhang, Y.; Chen, Q.; Zhang, X. Controllable P Doping of the LaCoO3 Catalyst for Efficient Propane Oxidation: Optimized Surface Co Distribution and Enhanced Oxygen Vacancies. ACS Appl. Mater. Interfaces 2020, 12, 23789–23799. [Google Scholar] [CrossRef]
- Feng, C.; Gao, Q.; Xiong, G.; Chen, Y.; Pan, Y.; Fei, Z.; Li, Y.; Lu, Y.; Liu, C.; Liu, Y. Defect engineering technique for the fabrication of LaCoO3 perovskite catalyst via urea treatment for total oxidation of propane. Appl. Catal. B Environ. 2022, 304, 121005. [Google Scholar] [CrossRef]
- Yang, J.; Shi, L.; Li, L.; Fang, Y.; Pan, C.; Zhu, Y.; Liang, Z.; Hoang, S.; Li, Z.; Guo, Y. Surface modification of macroporous La0.8Sr0.2CoO3 perovskite oxides integrated monolithic catalysts for improved propane oxidation. Catal. Today 2021, 376, 168–176. [Google Scholar] [CrossRef]
- Nakamura, T.; Misono, M.; Yoneda, Y. Catalytic Properties of Perovskite-type Mixed Oxides, La1−xSrxCoO3. Bull. Chem. Soc. Jpn. 1982, 55, 394–399. [Google Scholar] [CrossRef]
- Itoh, T.; Shirasaki, S.; Ofuchi, H.; Hirayama, S.; Honma, T.; Nakayama, M. Oxygen partial pressure dependence of in situ X-ray absorption spectroscopy at the Co and Fe K edges for (La0.6Sr0.4)(Co0.2Fe0.8)O3−δ. Solid State Commun. 2012, 152, 278–283. [Google Scholar] [CrossRef]
- Labhasetwar, N.; Saravanan, G.; Kumar Megarajan, S.K.; Manwar, N.; Khobragade, R.; Doggali, P.; Grasset, F. Perovskite-type catalytic materials for environmental applications. Sci. Technol. Adv. Mater. 2015, 16, 036002. [Google Scholar] [CrossRef]
- Papazisi, K.M.; Balomenou, S.; Tsiplakides, D. Synthesis and characterization of La0.75Sr0.25Cr0.9M0.1O3 perovskites as anodes for CO-fuelled solid oxide fuel cells. J. Appl. Electrochem. 2010, 40, 1875–1881. [Google Scholar] [CrossRef]
- Civera, A.; Pavese, M.; Saracco, G.; Specchia, V. Combustion synthesis of perovskite-type catalysts for natural gas combustion. Catal. Today 2003, 83, 199–211. [Google Scholar] [CrossRef]
- Niu, J.; Deng, J.; Liu, W.; Zhang, L.; Wang, G.; Dai, H.; He, H.; Zi, X. Nanosized perovskite-type oxides La1−xSrxMO3−δ (M = Co, Mn; x = 0, 0.4) for the catalytic removal of ethylacetate. Catal. Today 2007, 126, 420–429. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Mechanistic Study of the Selective Methanation of CO over Ru/TiO2 Catalyst: Identification of Active Surface Species and Reaction Pathways. J. Phys. Chem. C 2011, 115, 1220–1230. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Perovskite | Notation | SSA (m2 g−1) | Crystallite Size (nm) |
|---|---|---|---|
| La0.8Sr0.2FeO3−δ | LSF | 7.2 1 | 29.1 |
| La0.8Sr0.2Co0.1Fe0.9O3−δ | LSCF_8219 | 7.9 | 19.7 |
| La0.8Sr0.2Co0.2Fe0.8O3−δ | LSCF_8228 | 5.6 1 | 16.9 |
| La0.8Sr0.2Co0.4Fe0.6O3−δ | LSCF_8246 | 4.7 1 | 50.2 |
| La0.8Sr0.2Co0.6Fe0.4O3−δ | LSCF_8264 | 6.8 1 | 32.5 |
| La0.8Sr0.2Co0.8Fe0.2O3−δ | LSCF_8282 | 5.5 1 | 32.5 |
| La0.8Sr0.2Co0.9Fe0.1O3−δ | LSCF_8291 | 4.1 | 60.2 |
| La0.8Sr0.2CoO3−δ | LSC | 5.7 1 | 34.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Safakas, A.; Kournoutis, V.C.; Bampos, G.; Bebelis, S. CO and Propane Combustion on La0.8Sr0.2CoxFe1−xO3−δ Perovskites: Effect of Fe-to-Co Ratio on Catalytic Activity. Catalysts 2023, 13, 1342. https://doi.org/10.3390/catal13101342
Safakas A, Kournoutis VC, Bampos G, Bebelis S. CO and Propane Combustion on La0.8Sr0.2CoxFe1−xO3−δ Perovskites: Effect of Fe-to-Co Ratio on Catalytic Activity. Catalysts. 2023; 13(10):1342. https://doi.org/10.3390/catal13101342
Chicago/Turabian StyleSafakas, Alexandros, Vasileios Ch. Kournoutis, Georgios Bampos, and Symeon Bebelis. 2023. "CO and Propane Combustion on La0.8Sr0.2CoxFe1−xO3−δ Perovskites: Effect of Fe-to-Co Ratio on Catalytic Activity" Catalysts 13, no. 10: 1342. https://doi.org/10.3390/catal13101342
APA StyleSafakas, A., Kournoutis, V. C., Bampos, G., & Bebelis, S. (2023). CO and Propane Combustion on La0.8Sr0.2CoxFe1−xO3−δ Perovskites: Effect of Fe-to-Co Ratio on Catalytic Activity. Catalysts, 13(10), 1342. https://doi.org/10.3390/catal13101342