
Controlling Magnesium Silicates Coprecipitation Conditions: A Tool to Tune Their Surface Acid–Base Reactivity
 , ,
, , 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthesis of Magnesium Silicates
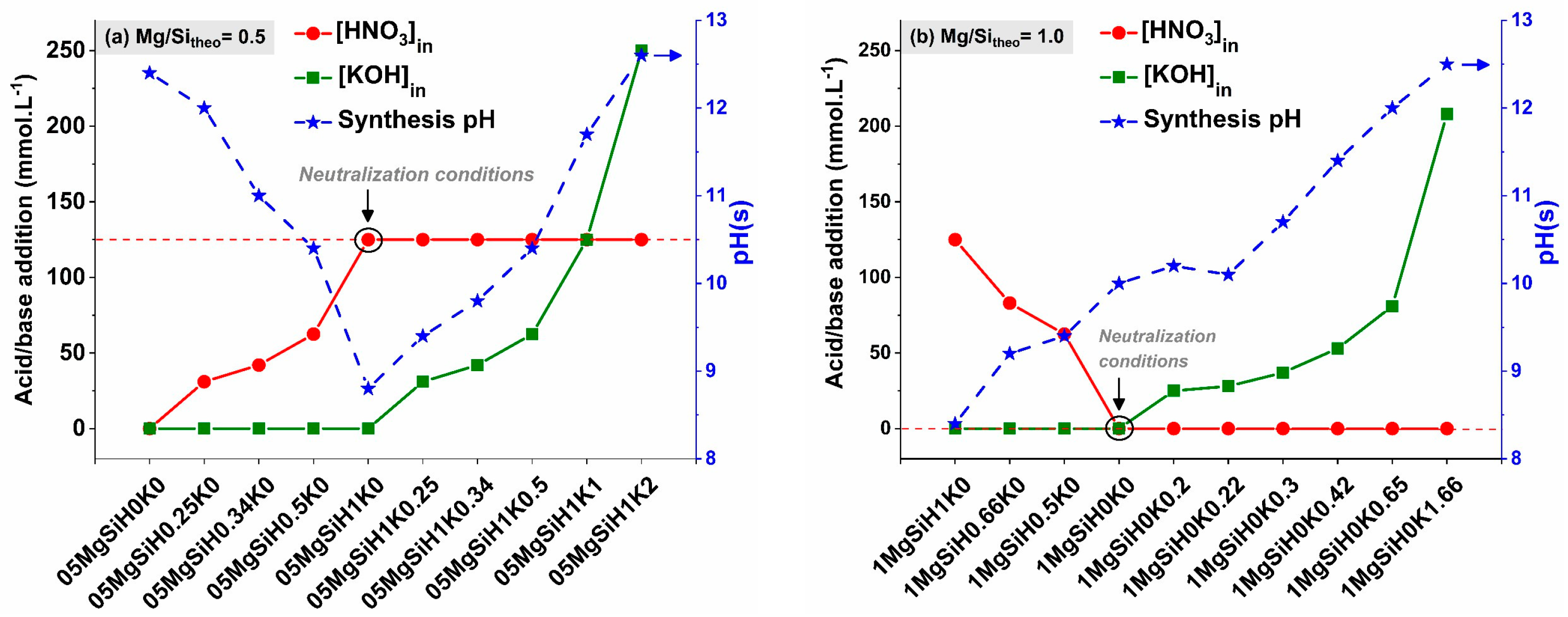
2.1.1. Synthesis pH Evolution
2.1.2. Chemical Composition of the Precipitated Solid
2.2. Thermodynamic Approach of the Precipitation
2.3. Characterization Results
2.3.1. Nitrogen Sorption Isotherms
2.3.2. Scanning Electron Microscopy (SEM)
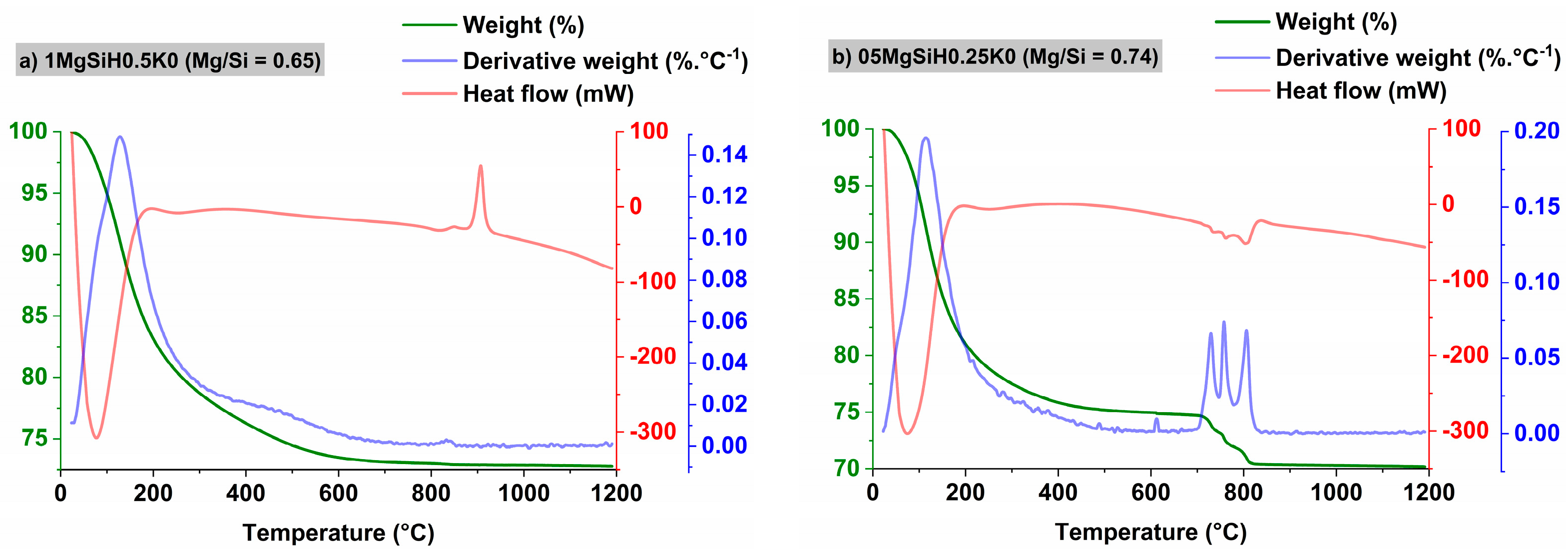
2.3.3. Thermogravimetric Analysis (TGA–DTA)
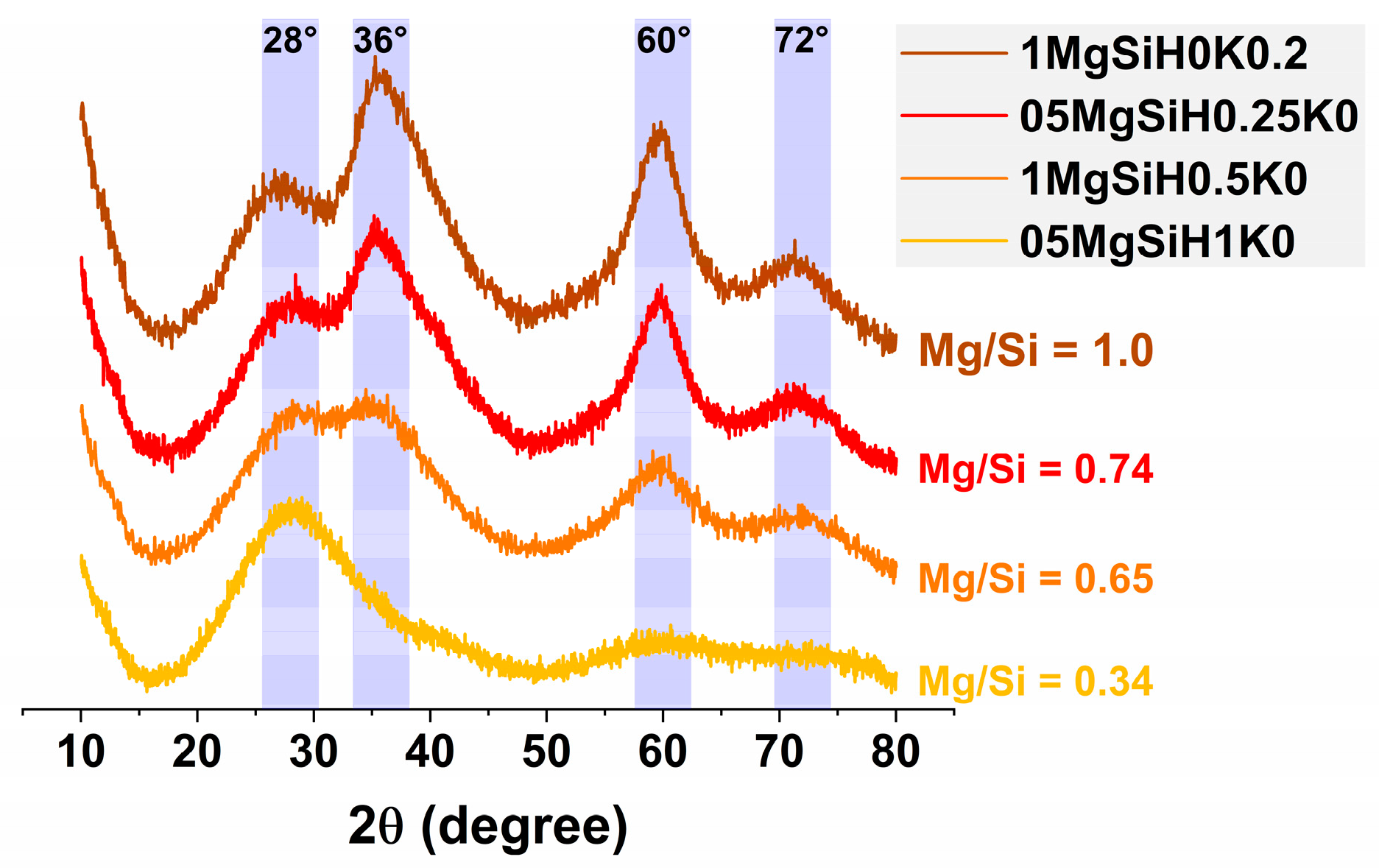
2.3.4. X-ray Diffraction
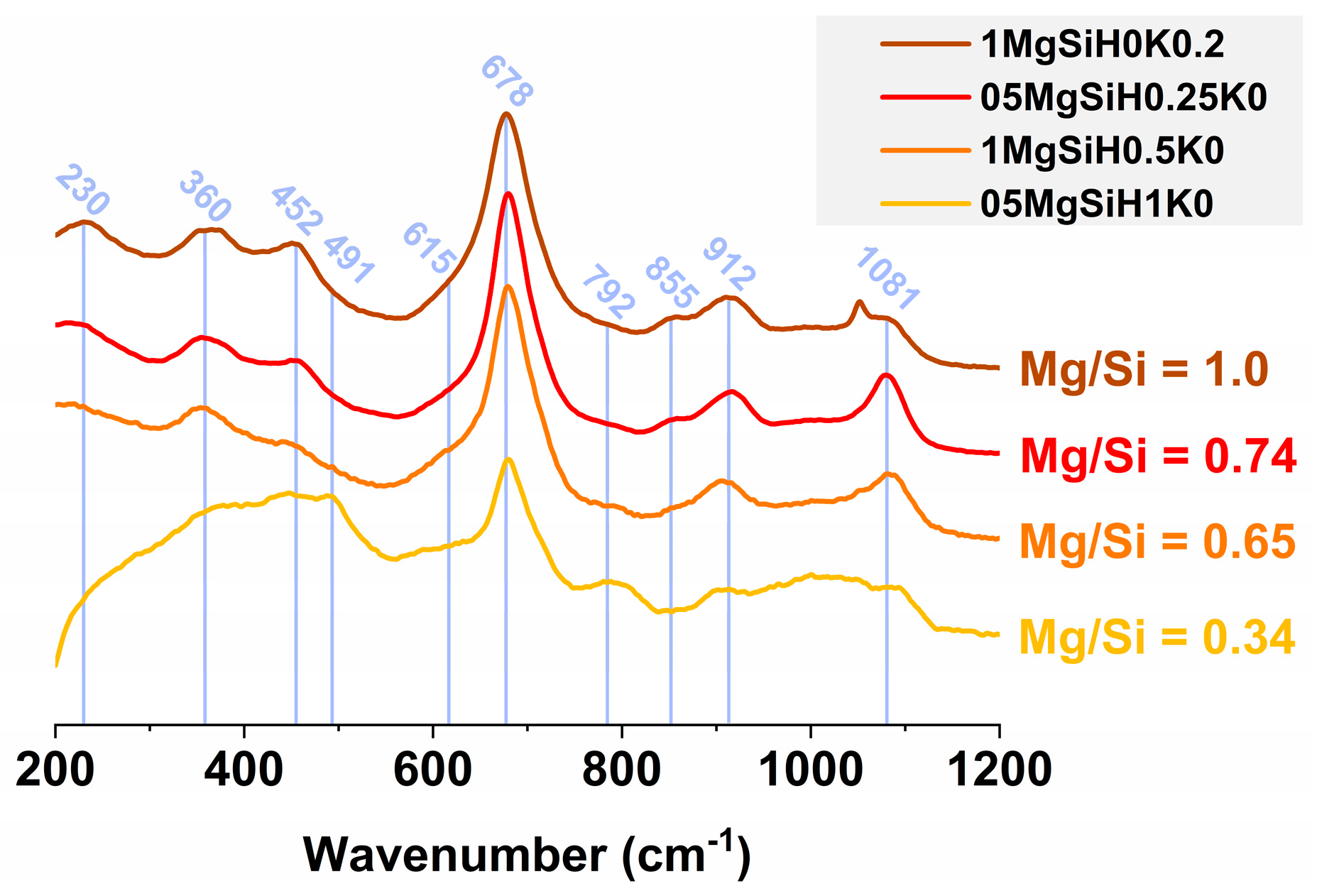
2.3.5. Raman Spectroscopy
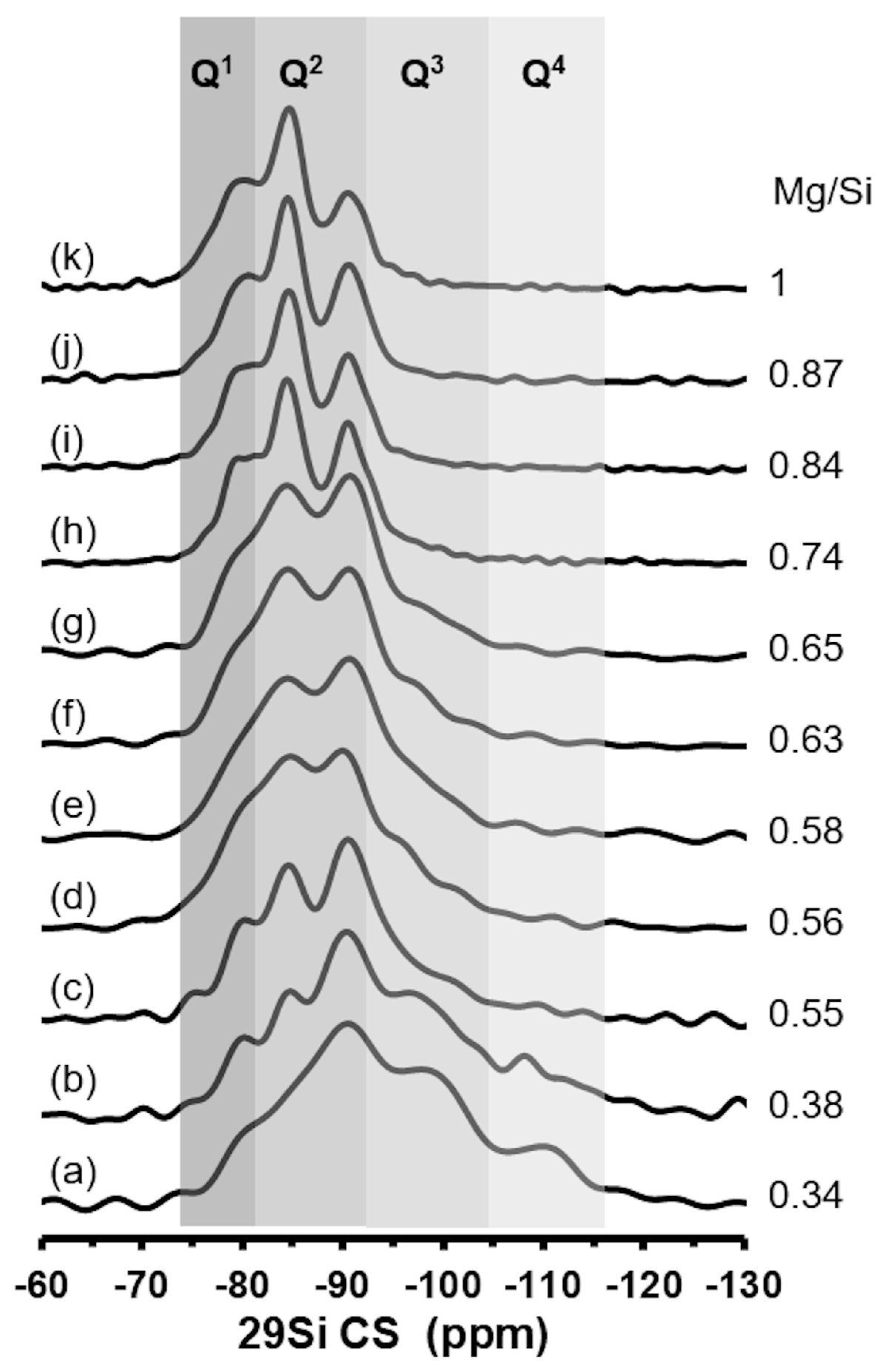
2.3.6. 29Si NMR Analysis
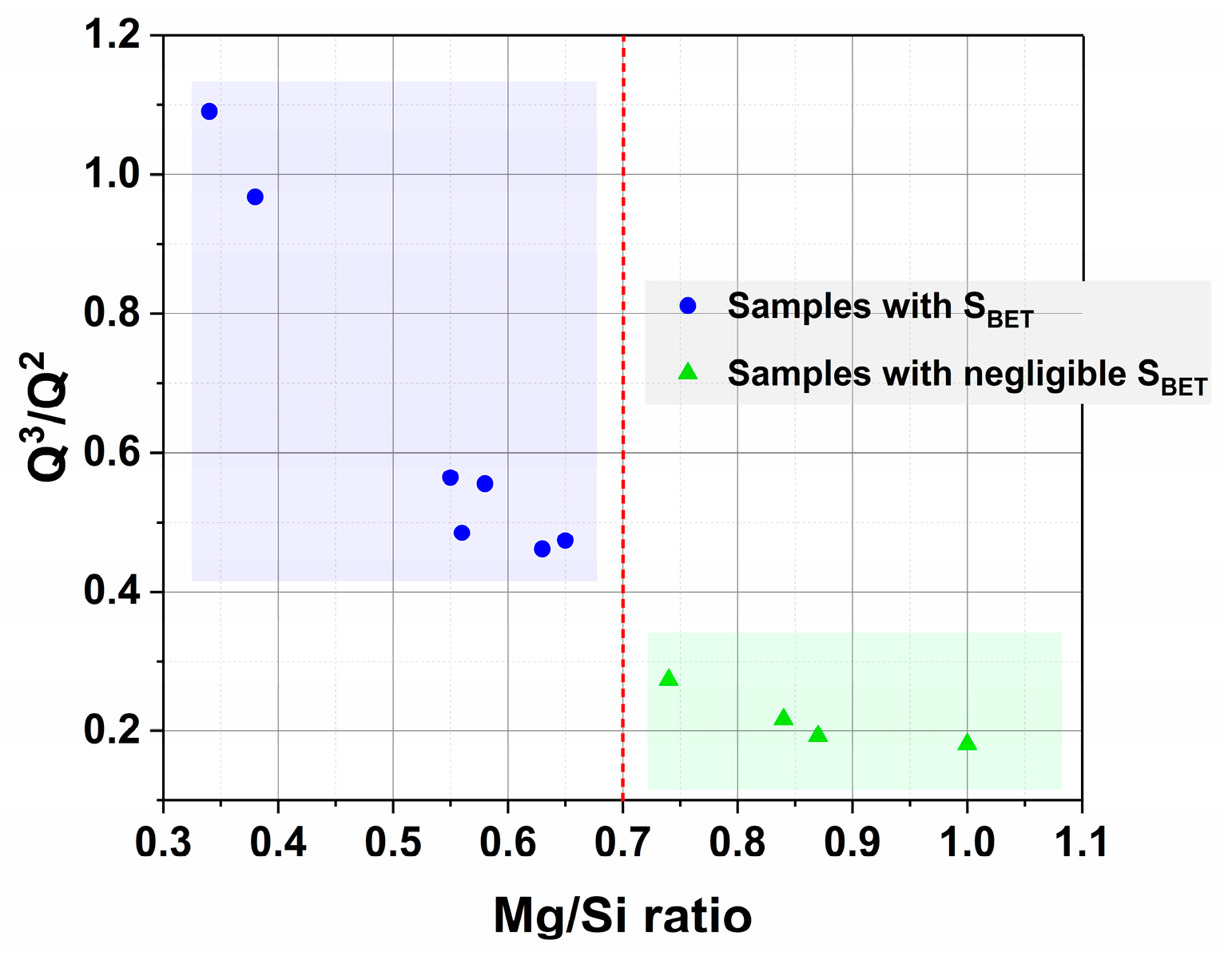
2.4. Discussion on the Existence of the Two Magnesium Silicates Families
2.5. Reactivity towards 2-Methylbut-3-yn-2-ol (MBOH)
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Magnesium Silicate
3.2.1. Experimental Device
3.2.2. Coprecipitation Experiments
3.2.3. Preparation of Magnesium and Silicate Precursor Solutions in the “Neutralization” Conditions
3.2.4. Adjustment of the Synthesis pH
3.2.5. Quantitative Analysis for the Chemical Composition of the Supernatant
3.3. Characterization
- 120 s analysis at 5 kV-60 µA without filter for the analysis of Mg, Na and Si;
- 120 s analysis at 12 kV-25 µA with a 50 µm aluminum filter for the analysis of K.
3.4. Conversion of 2-Methylbut-3-yn-2-ol (MBOH)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mackenzie, R.C. The Classification and Nomenclature of Clay Minerals. Clay Miner. 1959, 4, 52–66. [Google Scholar] [CrossRef]
- Spinthaki, A.; Petratos, G.; Matheis, J.; Hater, W.; Demadis, K.D. The Precipitation of “Magnesium Silicate” under Geothermal Stresses. Formation and Characterization. Geothermics 2018, 74, 172–180. [Google Scholar] [CrossRef]
- Jones, S.A.; Wong, S.; Burlitch, J.M.; Viswanathan, S.; Kohlstedt, D.L. Sol−Gel Synthesis and Characterization of Magnesium Silicate Thin Films. Chem. Mater. 1997, 9, 2567–2576. [Google Scholar] [CrossRef]
- Ochoa, J.V.; Bandinelli, C.; Vozniuk, O.; Chieregato, A.; Malmusi, A.; Recchi, C.; Cavani, F. An Analysis of the Chemical, Physical and Reactivity Features of MgO–SiO2 Catalysts for Butadiene Synthesis with the Lebedev Process. Green Chem. 2016, 18, 1653–1663. [Google Scholar] [CrossRef]
- Da Ros, S.; Jones, M.D.; Mattia, D.; Schwaab, M.; Barbosa-Coutinho, E.; Rabelo-Neto, R.C.; Noronha, F.B.; Pinto, J.C. Microkinetic Analysis of Ethanol to 1,3-Butadiene Reactions over MgO-SiO2 Catalysts Based on Characterization of Experimental Fluctuations. Chem. Eng. J. 2017, 308, 988–1000. [Google Scholar] [CrossRef]
- Da Ros, S.; Jones, M.D.; Mattia, D.; Schwaab, M.; Noronha, F.B.; Pinto, J.C. Modelling the Effects of Reaction Temperature and Flow Rate on the Conversion of Ethanol to 1,3-Butadiene. Appl. Catal. A Gen. 2017, 530, 37–47. [Google Scholar] [CrossRef]
- Zhu, Q.; Wang, B.; Tan, T. Conversion of Ethanol and Acetaldehyde to Butadiene over MgO–SiO2 Catalysts: Effect of Reaction Parameters and Interaction between MgO and SiO2 on Catalytic Performance. ACS Sustain. Chem. Eng. 2017, 5, 722–733. [Google Scholar] [CrossRef]
- Larina, O.V.; Kyriienko, P.I.; Trachevskii, V.V.; Vlasenko, N.V.; Soloviev, S.O. Effect of Mechanochemical Treatment on Acidic and Catalytic Properties of MgO-SiO2 Composition in the Conversion of Ethanol To 1,3-Butadiene. Theor. Exp. Chem. 2016, 51, 387–393. [Google Scholar] [CrossRef]
- Zhang, T.; Vandeperre, L.J.; Cheeseman, C.R. Magnesium-Silicate-Hydrate Cements for Encapsulating Problematic Aluminium Containing Wastes. J. Sustain. Cem. Based Mater. 2012, 1, 34–45. [Google Scholar] [CrossRef]
- Walling, S.A.; Kinoshita, H.; Bernal, S.A.; Collier, N.C.; Provis, J.L. Structure and Properties of Binder Gels Formed in the System Mg(OH)2–SiO2–H2O for Immobilisation of Magnox Sludge. Dalton Trans. 2015, 44, 8126–8137. [Google Scholar] [CrossRef]
- Zhang, T.; Cheeseman, C.R.; Vandeperre, L.J. Development of Low pH Cement Systems Forming Magnesium Silicate Hydrate (M-S-H). Cem. Concr. Res. 2011, 41, 439–442. [Google Scholar] [CrossRef]
- Ridi, F.; Fratini, E.; Baglioni, P. Cement: A Two Thousand Year Old Nano-Colloid. J. Colloid. Interface Sci. 2011, 357, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Lothenbach, B.; Rentsch, D.; Pochard, I.; Dauzères, A. Formation of Magnesium Silicate Hydrates (M-S-H). Phys. Chem. Earth 2017, 99, 142–157. [Google Scholar] [CrossRef]
- Tonelli, M.; Martini, F.; Calucci, L.; Fratini, E.; Geppi, M.; Ridi, F.; Borsacchi, S.; Baglioni, P. Structural Characterization of Magnesium Silicate Hydrate: Towards the Design of Eco-Sustainable Cements. Dalton Trans. 2016, 45, 3294–3304. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, T.; Li, X.; Chou, K.-C.; Hou, X. Some New Perspective on the Reaction Mechanism of MgO-SiO2 -H2O System. Int. J. Appl. Ceram. Technol. 2016, 13, 1164–1172. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, T.; Hu, J.; Tang, Y.; Niu, Y.; Wei, J.; Yu, Q. Characterization of Reaction Products and Reaction Process of MgO–SiO2–H2O System at Room Temperature. Constr. Build Mater. 2014, 61, 252–259. [Google Scholar] [CrossRef]
- Nied, D.; Enemark-Rasmussen, K.; L’Hopital, E.; Skibsted, J.; Lothenbach, B. Properties of Magnesium Silicate Hydrates (M-S-H). Cem. Concr. Res. 2016, 79, 323–332. [Google Scholar] [CrossRef]
- Marsiske, M.R.; Köser, R.; Bäumle, B.; Ruiz-Agudo, C. Uncovering the Early Stages of Magnesium Silicate Hydrate Formation: A Nonclassical Multistep Pathway. ACS Appl. Eng. Mater. 2023, 1, 696–707. [Google Scholar] [CrossRef]
- Bernard, E.; Lothenbach, B.; Chlique, C.; Wyrzykowski, M.; Dauzères, A.; Pochard, I.; Cau-Dit-Coumes, C. Characterization of Magnesium Silicate Hydrate (M-S-H). Cem. Concr. Res. 2019, 116, 309–330. [Google Scholar] [CrossRef]
- Brew, D.R.M.; Glasser, F.P. Synthesis and Characterisation of Magnesium Silicate Hydrate Gels. Cem. Concr. Res. 2005, 35, 85–98. [Google Scholar] [CrossRef]
- Roosz, C.; Grangeon, S.; Blanc, P.; Montouillout, V.; Lothenbach, B.; Henocq, P.; Giffaut, E.; Vieillard, P.; Gaboreau, S. Crystal Structure of Magnesium Silicate Hydrates (M-S-H): The Relation with 2:1 Mg–Si Phyllosilicates. Cem. Concr. Res. 2015, 73, 228–237. [Google Scholar] [CrossRef]
- Pedone, A.; Palazzetti, F.; Barone, V. Models of Aged Magnesium–Silicate–Hydrate Cements Based on the Lizardite and Talc Crystals: A Periodic DFT-GIPAW Investigation. J. Phys. Chem. C 2017, 121, 7319–7330. [Google Scholar] [CrossRef]
- Angelici, C.; Velthoen, M.E.Z.; Weckhuysen, B.M.; Bruijnincx, P.C.A. Effect of Preparation Method and CuO Promotion in the Conversion of Ethanol into 1,3-Butadiene over SiO2–MgO Catalysts. ChemSusChem 2014, 7, 2505–2515. [Google Scholar] [CrossRef] [PubMed]
- Angelici, C.; Velthoen, M.E.Z.; Weckhuysen, B.M.; Bruijnincx, P.C.A. Influence of Acid–Base Properties on the Lebedev Ethanol-to-Butadiene Process Catalyzed by SiO2–MgO Materials. Catal. Sci. Technol. 2015, 5, 2869–2879. [Google Scholar] [CrossRef]
- Chung, S.-H.; Angelici, C.; Hinterding, S.O.M.; Weingarth, M.; Baldus, M.; Houben, K.; Weckhuysen, B.M.; Bruijnincx, P.C.A. Role of Magnesium Silicates in Wet-Kneaded Silica–Magnesia Catalysts for the Lebedev Ethanol-to-Butadiene Process. ACS Catal. 2016, 6, 4034–4045. [Google Scholar] [CrossRef]
- Pomalaza, G.; Arango Ponton, P.; Capron, M.; Dumeignil, F. Ethanol-to-Butadiene: The Reaction and Its Catalysts. Catal. Sci. Technol. 2020, 10, 4860–4911. [Google Scholar] [CrossRef]
- Lin, L.; Cornu, D.; Mounir Daou, M.; Domingos, C.; Herledan, V.; Krafft, J.-M.; Laugel, G.; Millot, Y.; Lauron-Pernot, H. Role of Water on the Activity of Magnesium Silicate for Transesterification Reactions. ChemCatChem 2017, 9, 2399–2407. [Google Scholar] [CrossRef]
- Lin, L.; Silva Gomes, E.; Payan, F.; Jaber, M.; Krafft, J.-M.; Laugel, G.; Lauron-Pernot, H. How the Acido-Basic Properties of Mg Silicates and Clays Govern the Catalytic Mechanism of Transesterification Reactions. Catal. Sci. Technol. 2019, 9, 6072–6084. [Google Scholar] [CrossRef]
- Cornu, D.; Lin, L.; Daou, M.M.; Jaber, M.; Krafft, J.-M.; Herledan, V.; Laugel, G.; Millot, Y.; Lauron-Pernot, H. Influence of Acid–Base Properties of Mg-Based Catalysts on Transesterification: Role of Magnesium Silicate Hydrate Formation. Catal. Sci. Technol. 2017, 7, 1701–1712. [Google Scholar] [CrossRef]
- Wolf, A.; Michele, V.; Schlüter, O.F.-K.; Herbstritt, F.; Heck, J.; Mleczko, L. Precipitation in a Micromixer—From Laboratory to Industrial Scale. Chem. Eng. Technol. 2015, 38, 2017–2024. [Google Scholar] [CrossRef]
- Dietemann, M.; Baillon, F.; Espitalier, F.; Calvet, R.; Greenhill-Hooper, M. Amorphous Magnesium Silicate Ultrasound-Assisted Precipitation in a Mixing System: Population Balance Modelling and Crystallization Rates Identification. Powder Technol. 2019, 356, 83–96. [Google Scholar] [CrossRef]
- Dietemann, M.; Baillon, F.; Espitalier, F.; Calvet, R.; Accart, P.; Confetto, S.D.; Greenhill-Hooper, M. Evaluation of the Physico-Chemical Properties of an Amorphous Magnesium Silicate Synthesized by an Ultrasound-Assisted Precipitation. Chem. Eng. J. 2013, 215–216, 658–670. [Google Scholar] [CrossRef]
- Lauron-Pernot, H.; Luck, F.; Popa, J.M. Methylbutynol: A New and Simple Diagnostic Tool for Acidic and Basic Sites of Solids. Appl. Catal. 1991, 78, 213–225. [Google Scholar] [CrossRef]
- Lauron-Pernot, H. Evaluation of Surface Acido-Basic Properties of Inorganic-Based Solids by Model Catalytic Alcohol Reaction Networks. Catal. Rev. Sci. Eng. 2006, 48, 315–361. [Google Scholar] [CrossRef]
- Alexander, G.B. The Reaction of Low Molecular Weight Silicic Acids with Molybdic Acid. J. Am. Chem. Soc. 1953, 75, 5655–5657. [Google Scholar] [CrossRef]
- Sing, K.S.W.; Everett, D.H.; Haul, R.A.W.; Moscou, L.; Pierotti, R.A.; Rouquerol, J.; Siemieniewska, T. Prepared for Publication by the Subcommittee on Reporting Gas Adsorption Data Consisting. Pure Appl. Chem. 1985, 57, 603–619. [Google Scholar] [CrossRef]
- Li, C.; Wang, X.; Yang, A.; Chen, P.; Zhao, T.; Liu, F. Polyethyleneimine-Modified Amorphous Silica for the Selective Adsorption of CO2/N2 at High Temperatures. ACS Omega 2021, 6, 35389–35397. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Yu, Q.; Zhang, W.; Zhang, H. Reaction Products of MgO and Microsilica Cementitious Materials at Different Temperatures. J. Wuhan Univ. Technol. Mater. Sci. Ed. 2011, 26, 745–748. [Google Scholar] [CrossRef]
- Zhang, T.; Vandeperre, L.J.; Cheeseman, C.R. Formation of Magnesium Silicate Hydrate (M-S-H) Cement Pastes Using Sodium Hexametaphosphate. Cem. Concr. Res. 2014, 65, 8–14. [Google Scholar] [CrossRef]
- Jin, F.; Al-Tabbaa, A. Thermogravimetric Study on the Hydration of Reactive Magnesia and Silica Mixture at Room Temperature. Thermochim. Acta 2013, 566, 162–168. [Google Scholar] [CrossRef]
- Liu, X.; Liu, X.; Hu, Y. Investigation of the Thermal Decomposition of Talc. Clays Clay Miner. 2014, 62, 137–144. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Xu, Y.; Sang, S.; Jin, S. Enhanced Formation of Magnesium Silica Hydrates (M-S-H) Using Sodium Metasilicate and Caustic Magnesia in Magnesia Castables. Ceram. Int. 2017, 43, 9110–9116. [Google Scholar] [CrossRef]
- Mitsuda, T.; Taguchi, H. Formation of Magnesium Silicate Hydrate and Its Crystallization to Talc. Cem. Concr. Res. 1977, 7, 223–230. [Google Scholar] [CrossRef]
- MacKenzie, K.J.D.; Meinhold, R.H. The Thermal Reactions of Talc Studied by 29Si and 25Mg MAS NMR. Thermochim. Acta 1994, 244, 195–203. [Google Scholar] [CrossRef]
- Sabatier, G. Recherches sur la cristallisation par chauffage de mélanges amorphes de silice et des oxydes de magnésium, zinc et cuivre. Bull. Minéralogie 1952, 75, 506–526. [Google Scholar] [CrossRef]
- Tang, Q.; Wang, F.; Tang, M.; Liang, J.; Ren, C. Study on Pore Distribution and Formation Rule of Sepiolite Mineral Nanomaterials. J. Nanomater. 2012, 2012, 382603. [Google Scholar] [CrossRef]
- Mora, M.; Isabel López, M.; Ángeles Carmona, M.; Jiménez-Sanchidrián, C.; Rafael Ruiz, J. Study of the Thermal Decomposition of a Sepiolite by Mid- and near-Infrared Spectroscopies. Polyhedron 2010, 29, 3046–3051. [Google Scholar] [CrossRef]
- Viti, C. Serpentine Minerals Discrimination by Thermal Analysis. Am. Mineral. 2010, 95, 631–638. [Google Scholar] [CrossRef]
- Liao, J.; Senna, M. Thermal Behavior of Mechanically Amorphized Talc. Thermochim. Acta 1992, 197, 295–306. [Google Scholar] [CrossRef]
- Aglietti, E.F. The Effect of Dry Grinding on the Structure of Talc. Appl. Clay Sci. 1994, 9, 139–147. [Google Scholar] [CrossRef]
- Martini, F.; Tonelli, M.; Geppi, M.; Ridi, F.; Borsacchi, S.; Calucci, L. Hydration of MgO/SiO2 and Portland Cement Mixtures: A Structural Investigation of the Hydrated Phases by Means of X-ray Diffraction and Solid State NMR Spectroscopy. Cem. Concr. Res. 2017, 102, 60–67. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Dai, Y. Formation of Magnesium Silicate Hydrate in the Mg(OH)2-SiO2 Suspensions and Its Influence on the Properties of Magnesia Castables. Ceram. Int. 2018, 44, 21365–21373. [Google Scholar] [CrossRef]
- Musić, S.; Filipović-Vinceković, N.; Sekovanić, L. Precipitation of Amorphous SiO2 Particles and Their Properties. Braz. J. Chem. Eng. 2011, 28, 89–94. [Google Scholar] [CrossRef]
- Maddalena, R.; Hall, C.; Hamilton, A. Effect of Silica Particle Size on the Formation of Calcium Silicate Hydrate [C-S-H] Using Thermal Analysis. Thermochim. Acta 2019, 672, 142–149. [Google Scholar] [CrossRef]
- Chen, H.; Geng, J.; Zhang, Z.; Jiang, R.; Zhai, J.; Zhang, J. The Structure and Properties of Sepiolite with Partial Lattice Ions Substituted by Aluminum Ions. Front. Chem. 2021, 9, 721225. [Google Scholar] [CrossRef] [PubMed]
- Suárez, M.; García-Romero, E. Variability of the Surface Properties of Sepiolite. Appl. Clay Sci. 2012, 67–68, 72–82. [Google Scholar] [CrossRef]
- Rivero Crespo, M.A.; Pereira Gómez, D.; Villa García, M.V.; Gallardo Amores, J.M.; Sánchez Escribano, V. Characterization of Serpentines from Different Regions by Transmission Electron Microscopy, X-ray Diffraction, BET Specific Surface Area and Vibrational and Electronic Spectroscopy. Fibers 2019, 7, 47. [Google Scholar] [CrossRef]
- Dumas, A.; Martin, F.; Le Roux, C.; Micoud, P.; Petit, S.; Ferrage, E.; Brendlé, J.; Grauby, O.; Greenhill-Hooper, M. Phyllosilicates Synthesis: A Way of Accessing Edges Contributions in NMR and FTIR Spectroscopies. Example of Synthetic Talc. Phys. Chem. Miner. 2013, 40, 361–373. [Google Scholar] [CrossRef]
- Kogure, T.; Kameda, J.; Matsui, T.; Miyawaki, R. Stacking Structure in Disordered Talc: Interpretation of Its X-ray Diffraction Pattern by Using Pattern Simulation and High-Resolution Transmission Electron Microscopy. Am. Mineral. 2006, 91, 1363–1370. [Google Scholar] [CrossRef]
- Balek, V.; Šubrt, J.; Pérez-Maqueda, L.A.; Beneš, M.; Bountseva, I.M.; Beckman, I.N.; Pérez-Rodríguez, J.L. Thermal Behavior of Ground Talc Mineral. J. Min. Metall. B 2008, 44, 7–17. [Google Scholar] [CrossRef]
- Takeshi, E.; Billinge, S.J.L. Chapter 3—The Method of Total Scattering and Atomic Pair Distribution Function Analysis. In Pergamon Materials Series; Egami, T., Billinge, S.J.L., Eds.; Elsevier: Pergamon, Turkey, 2012; Volume 16, pp. 55–111. ISBN 1470-1804. [Google Scholar]
- Masson, O.; Thomas, P. Exact and Explicit Expression of the Atomic Pair Distribution Function as Obtained from X-ray Total Scattering Experiments. J. Appl. Crystallogr. 2013, 46, 461–465. [Google Scholar] [CrossRef]
- Billinge, S.J.L.; Kanatzidis, M.G. Beyond Crystallography: The Study of Disorder, Nanocrystallinity and Crystallographically Challenged Materials with Pair Distribution Functions. Chem. Commun. 2004, 7, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Petkov, V. Nanostructure by High-Energy X-ray Diffraction. Mat. Today 2008, 11, 28–38. [Google Scholar] [CrossRef]
- Farrow, C.L.; Billinge, S.J.L. Relationship between the Atomic Pair Distribution Function and Small-Angle Scattering: Implications for Modeling of Nanoparticles. Acta Crystallogr. A Found. Adv. 2009, 65, 232–239. [Google Scholar] [CrossRef]
- Terban, M.W.; Billinge, S.J.L. Structural Analysis of Molecular Materials Using the Pair Distribution Function. Chem. Rev. 2022, 122, 1208–1272. [Google Scholar] [CrossRef]
- Rosasco, G.J.; Blaha, J.J. Raman Microprobe Spectra and Vibrational Mode Assignments of Talc. Appl. Spectrosc. 1980, 34, 140–144. [Google Scholar] [CrossRef]
- Wang, A.; Freeman, J.J.; Jolliff, B.L. Understanding the Raman Spectral Features of Phyllosilicates. J. Raman Spectrosc. 2015, 46, 829–845. [Google Scholar] [CrossRef]
- Rinaudo, C.; Gastaldi, D.; Belluso, E. Characterization of Chrysotile, Antigorite and Lizardite by FT-Raman Spectroscopy. Can. Mineral. 2003, 41, 883–890. [Google Scholar] [CrossRef]
- Biswas, R.K.; Khan, P.; Mukherjee, S.; Mukhopadhyay, A.K.; Ghosh, J.; Muraleedharan, K. Study of Short Range Structure of Amorphous Silica from PDF Using Ag Radiation in Laboratory XRD System, RAMAN and NEXAFS. J. Non Cryst. Solids 2018, 488, 1–9. [Google Scholar] [CrossRef]
- Hong, W.-E.; Ro, J.-S. Kinetics of Solid Phase Crystallization of Amorphous Silicon Analyzed by Raman Spectroscopy. J. Appl. Phys. 2013, 114, 073511. [Google Scholar] [CrossRef]
- Alessi, A.; Agnello, S.; Buscarino, G.; Gelardi, F.M. Raman and IR Investigation of Silica Nanoparticles Structure. J. Non Cryst. Solids 2013, 362, 20–24. [Google Scholar] [CrossRef]
- Alessi, A.; Agnello, S.; Buscarino, G.; Gelardi, F.M. Structural Properties of Core and Surface of Silica Nanoparticles Investigated by Raman Spectroscopy. J. Raman Spectrosc. 2013, 44, 810–816. [Google Scholar] [CrossRef]
- Lippmaa, E.; Maegi, M.; Samoson, A.; Engelhardt, G.; Grimmer, A.R. Structural Studies of Silicates by Solid-State High-Resolution Silicon-29 NMR. J. Am. Chem. Soc. 1980, 102, 4889–4893. [Google Scholar] [CrossRef]
- Magi, M.; Lippmaa, E.; Samoson, A.; Engelhardt, G.; Grimmer, A.R. Solid-State High-Resolution Silicon-29 Chemical Shifts in Silicates. J. Phys. Chem. 1984, 88, 1518–1522. [Google Scholar] [CrossRef]
- Temuujin, J.; Okada, K.; MacKenzie, K.J.D. Role of Water in the Mechanochemical Reactions of MgO–SiO2 Systems. J. Solid State Chem. 1998, 138, 169–177. [Google Scholar] [CrossRef]
- Bernard, E.; Lothenbach, B.; Le Goff, F.; Pochard, I.; Dauzères, A. Effect of Magnesium on Calcium Silicate Hydrate (C-S-H). Cem. Concr. Res. 2017, 97, 61–72. [Google Scholar] [CrossRef]
- Zhang, T.; Zou, J.; Wang, B.; Wu, Z.; Jia, Y.; Cheeseman, C. Characterization of Magnesium Silicate Hydrate (MSH) Gel Formed by Reacting MgO and Silica Fume. Materials 2018, 11, 909. [Google Scholar] [CrossRef]
- Janssens, W.; Makshina, E.V.; Vanelderen, P.; De Clippel, F.; Houthoofd, K.; Kerkhofs, S.; Martens, J.A.; Jacobs, P.A.; Sels, B.F. Ternary Ag/MgO-SiO2 Catalysts for the Conversion of Ethanol into Butadiene. ChemSusChem 2015, 8, 994–1008. [Google Scholar] [CrossRef]
- d’Espinose de la Caillerie, J.-B.; Kermarec, M.; Clause, O. 29Si NMR Observation of an Amorphous Magnesium Silicate Formed during Impregnation of Silica with Mg(II) in Aqueous Solution. J. Phys. Chem. 1995, 99, 17273–17281. [Google Scholar] [CrossRef]
- Holland, T.J.B.; Powell, R. An Internally Consistent Thermodynamic Data Set for Phases of Petrological Interest. J. Metamorph. Geol. 1998, 16, 309–343. [Google Scholar] [CrossRef]
- Iler, R.K. The Chemistry of Silica; John Wiley and Sons: New York, NY, USA, 1979; ISBN 978-0-471-02404-0. [Google Scholar]
- Bergna, H.E. Colloid Chemistry of Silica. In The Colloid Chemistry of Silica; Advances in Chemistry; American Chemical Society: Washington, DC, USA, 1994; Volume 234, pp. 1–47. ISBN 978-0-8412-2103-1. [Google Scholar]
- Liu, H.C.; Wang, J.X.; Mao, Y.; Chen, R.S. The Preparation and Growth of Colloidal Particles of Concentrated Silica Sols. Colloids Surf. A Physicochem. Eng. Asp. 1993, 74, 7–13. [Google Scholar] [CrossRef]
- Di Patrizio, N.; Bagnaro, M.; Gaunand, A.; Hochepied, J.-F.; Horbez, D.; Pitiot, P. Hydrodynamics and Mixing Performance of Hartridge Roughton Mixers: Influence of the Mixing Chamber Design. Chem. Eng. J. 2016, 283, 375–387. [Google Scholar] [CrossRef]
- Juhás, P.; Davis, T.; Farrow, C.L.; Billinge, S.J.L. PDFgetX3: A Rapid and Highly Automatable Program for Processing Powder Diffraction Data into Total Scattering Pair Distribution Functions. J. Appl. Crystallogr. 2013, 46, 560–566. [Google Scholar] [CrossRef]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvé, S.; Alonso, B.; Durand, J.-O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modelling One- and Two-Dimensional Solid-State NMR Spectra: Modelling 1D and 2D Solid-State NMR Spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID Samples | pH(s) | Mg/Si | SBET (m2·g−1) | VP (cm3·g−1) | DP (nm) |
|---|---|---|---|---|---|
| 05MgSiH0.34K0 | 11.0 | 0.63 | 220 | 0.79 | 14.3 |
| 05MgSiH0.5K0 | 10.4 | 0.56 | 202 | 0.43 | 8.6 |
| 05MgSiH1K0 | 8.8 | 0.34 | 187 | 0.40 | 8.8 |
| 05MgSiH1K0.25 | 9.4 | 0.48 | 325 | 0.80 | 11.4 |
| 05MgSiH1K0.34 | 9.8 | 0.52 | 267 | 0.82 | 12.6 |
| 05MgSiH1K0.5 | 10.4 | 0.58 | 272 | 0.66 | 9.6 |
| 1MgSiH1K0 | 8.4 | 0.38 | 358 | 0.56 | 8.7 |
| 1MgSiH0.66K0 | 9.2 | 0.55 | 316 | 0.88 | 11.7 |
| 1MgSiH0.5K0 | 9.4 | 0.65 | 238 | 0.52 | 6.5 |
| Time on Stream (min) | Conv (%) | C Balance (%) | S(Mbyne) (%) | S(C2H2) (%) | S(acetone) (%) | Conv(BET) (%) | Y(acidic) (%) | Y(basic) (%) |
|---|---|---|---|---|---|---|---|---|
| 2 | 13 | 92 | 41 | 51 | 8 | 7 | 3 | 4 |
| 15 | 9 | 99 | 13 | 41 | 46 | 5 | 1 | 4 |
| 28 | 8 | 99 | 10 | 41 | 49 | 4 | 0 | 4 |
| 41 | 7 | 98 | 9 | 41 | 50 | 4 | 0 | 4 |
| 54 | 6 | 99 | 9 | 40 | 51 | 3 | 0 | 3 |
| Sample | Conv(BET) (%) | S(Mbyne) (%) | Y(acidic) (%) | Mg/Si | K/Si |
|---|---|---|---|---|---|
| 05MgSiH1K0 | 7 | 17 | 2 | 0.34 | 0.03 |
| 05MgSiH1K0.25 | 8 | 12 | 1 | 0.48 | 0.05 |
| 05MgSiH1K0.34 | 12 | 4 | 1 | 0.52 | 0.06 |
| 05MgSiH0.5K0 | 12 | 0 | 0 | 0.56 | 0.10 |
| 05MgSiH1K0.5 | 12 | 1 | 0 | 0.58 | 0.09 |
| 05MgSiH0.34K0 | 13 | 1 | 0 | 0.63 | 0.08 |
| 1MgSiH1K0 | 7 | 41 | 4 | 0.38 | 0.02 |
| 1MgSiH0.66K0 | 12 | 27 | 4 | 0.55 | 0.01 |
| 1MgSiH0.5K0 | 13 | 13 | 2 | 0.65 | 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Payan, F.; Issa, A.; Krafft, J.-M.; Millot, Y.; Onfroy, T.; Sassoye, C.; Hochepied, J.-F.; Laugel, G.; Lauron-Pernot, H. Controlling Magnesium Silicates Coprecipitation Conditions: A Tool to Tune Their Surface Acid–Base Reactivity. Catalysts 2023, 13, 1393. https://doi.org/10.3390/catal13111393
Payan F, Issa A, Krafft J-M, Millot Y, Onfroy T, Sassoye C, Hochepied J-F, Laugel G, Lauron-Pernot H. Controlling Magnesium Silicates Coprecipitation Conditions: A Tool to Tune Their Surface Acid–Base Reactivity. Catalysts. 2023; 13(11):1393. https://doi.org/10.3390/catal13111393
Chicago/Turabian StylePayan, François, Albert Issa, Jean-Marc Krafft, Yannick Millot, Thomas Onfroy, Capucine Sassoye, Jean-François Hochepied, Guillaume Laugel, and Hélène Lauron-Pernot. 2023. "Controlling Magnesium Silicates Coprecipitation Conditions: A Tool to Tune Their Surface Acid–Base Reactivity" Catalysts 13, no. 11: 1393. https://doi.org/10.3390/catal13111393
APA StylePayan, F., Issa, A., Krafft, J.-M., Millot, Y., Onfroy, T., Sassoye, C., Hochepied, J.-F., Laugel, G., & Lauron-Pernot, H. (2023). Controlling Magnesium Silicates Coprecipitation Conditions: A Tool to Tune Their Surface Acid–Base Reactivity. Catalysts, 13(11), 1393. https://doi.org/10.3390/catal13111393