
Peptidomimetic-Based Asymmetric Catalysts



Abstract

1. Introduction
2. Foldamers in Asymmetric Catalysis
3. Peptide–Peptoid Hybrid Catalysts
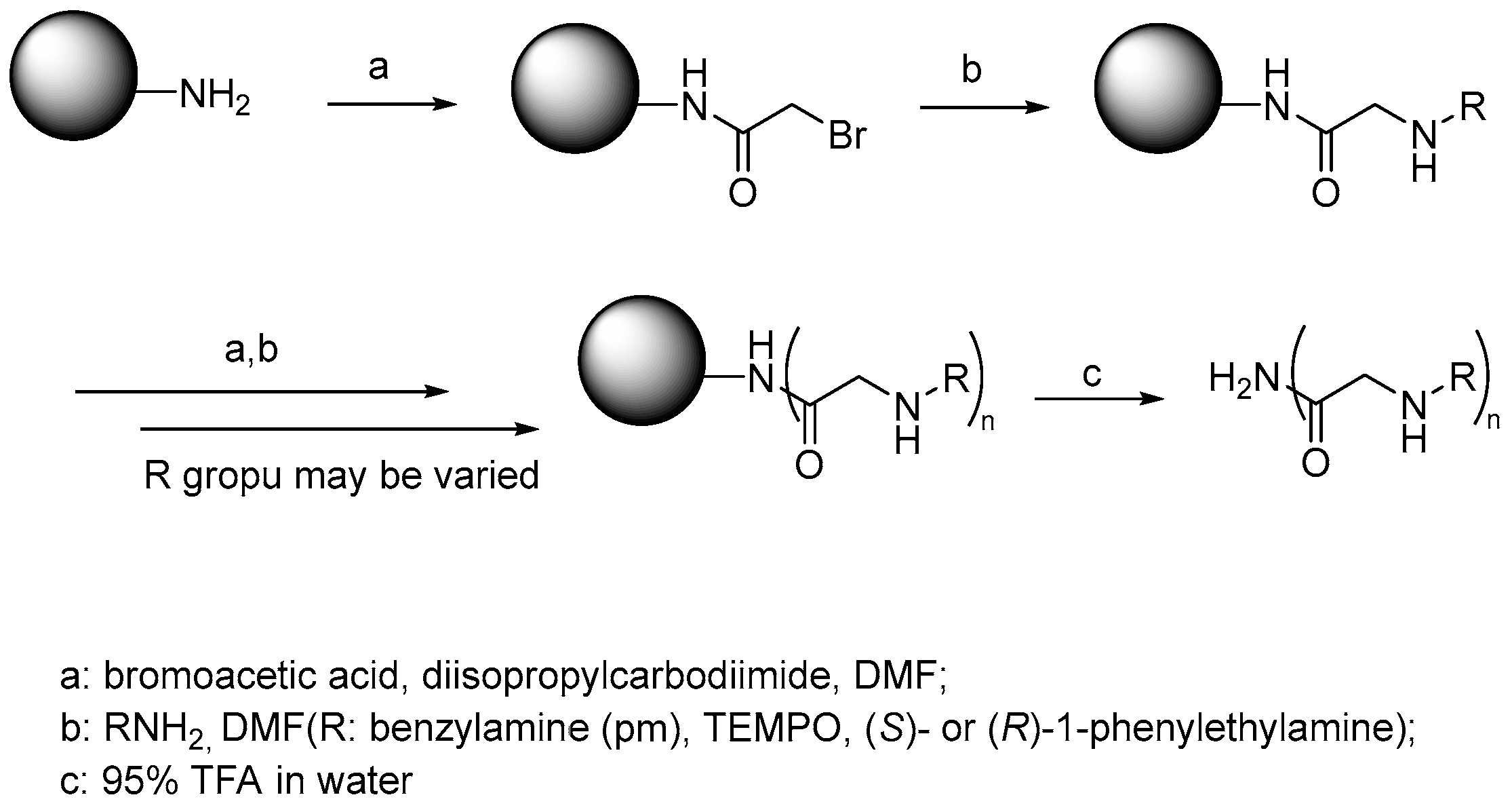
4. β-Turn Peptoid Scaffolds
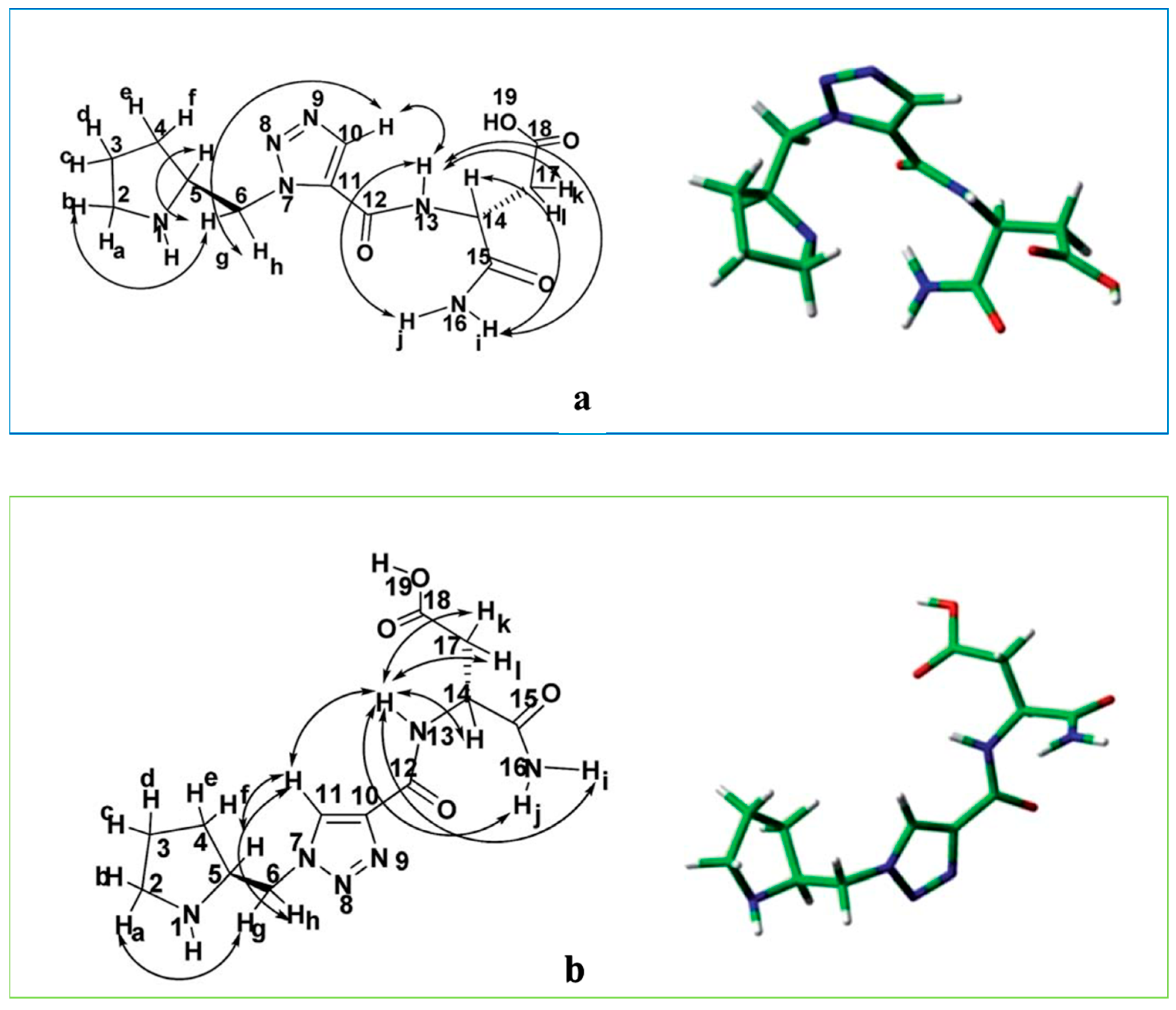
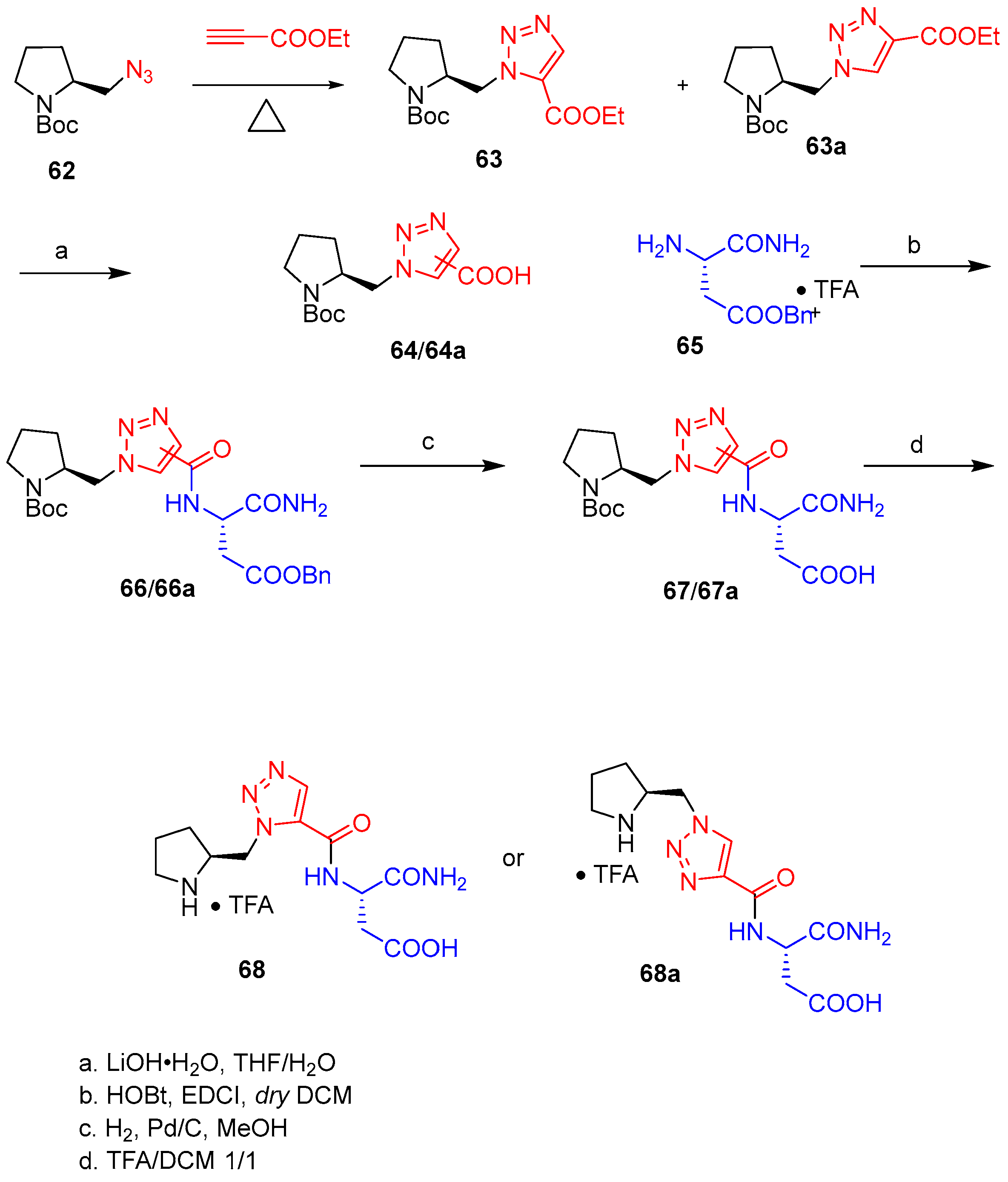
5. Peptidomimetic Triazole-Based Organocatalysts
6. Cyclic Peptoids as PTC in Asymmetric Catalysis
7. Conclusions and Outlook
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saha, D.; Kharbanda, A.; Yan, W.; Lakkaniga, N.R.; Frett, B.; Li, H.-Y. The Exploration of Chirality for Improved Druggability within the Human Kinome. J. Med. Chem. 2020, 63, 441–469. [Google Scholar] [CrossRef]
- Ceramella, J.; Iacopetta, D.; Franchini, A.; De Luca, M.; Saturnino, C.; Andreu, I.; Sinicropi, M.S.; Catalano, A. A Look at the Importance of Chirality in Drug Activity: Some Significative Examples. Appl. Sci. 2022, 12, 10909. [Google Scholar] [CrossRef]
- Cossy, J.R. The Importance of Chirality in Drugs and Agrochemicals Comprehensive. Chirality 2012, 1, 1–7. [Google Scholar] [CrossRef]
- Meskers, S.C.J. Consequences of chirality on the response of materials. Mater. Adv. 2022, 3, 2324–2336. [Google Scholar] [CrossRef]
- Han, B.; He, X.-H.; Liu, Y.-Q.; He, G.; Peng, C.; Li, J.-L. Asymmetric organocatalysis: An enabling technology for medicinal chemistry. Chem. Soc. Rev. 2021, 50, 1522–1586. [Google Scholar] [CrossRef] [PubMed]
- Noyori, R. Asymmetric Catalysis: Science and Opportunities (Nobel Lecture). Angew. Chem. Int. Ed. 2002, 41, 2008–2022. [Google Scholar] [CrossRef]
- Bell, E.L.; Finnigan, W.; France, S.P.; Green, A.P.; Hayes, M.A.; Hepworth, L.J.; Lovelock, S.L.; Niikura, H.; Osuna, S.; Romero, E.; et al. Biocatalysis. Nat. Rev. Methods Primers 2021, 1, 46. [Google Scholar] [CrossRef]
- Alemán, J.; Cabrera, S. Applications of asymmetric organocatalysis in medicinal chemistry. Chem. Soc. Rev. 2013, 42, 774–793. [Google Scholar] [CrossRef]
- Hargittai, I. The 2021 chemistry Nobel laureates and asymmetric organocatalysis. Struct. Chem. 2022, 33, 303–305. [Google Scholar] [CrossRef]
- Mancheño, O.G.; Waser, M. Recent Developments and Trends in Asymmetric Organocatalysis. Eur. J. Org. Chem. 2023, 26, e202200950. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, H.; Zhang, Q.; Zhang, P. Chirality in metal-based anticancer agents. Dalton Trans. 2018, 47, 4017–4026. [Google Scholar] [CrossRef] [PubMed]
- Wennemers, H. Asymmetric catalysis with peptides. Chem. Commun. 2011, 47, 12036–12041. [Google Scholar] [CrossRef]
- Freund, M.; Tsogoeva, S.B. Peptides for asymmetric catalysis. In Catalytic Methods in Asymmetric Synthesis: Advanced Materials, Techniques, and Applications; Gruttadauria, M., Giacalone, F., Eds.; Wiley: Hoboken, NJ, USA, 2011; pp. 529–578. [Google Scholar]
- Metrano, A.J.; Chinn, A.J.; Shugrue, C.R.; Stone, E.A.; Kim, B.; Miller, S.J. Asymmetric Catalysis Mediated by Synthetic Peptides, Version 2.0: Expansion of Scope and Mechanisms. Chem. Rev. 2020, 120, 11479–11615. [Google Scholar] [CrossRef] [PubMed]
- Revell, J.D.; Wennemers, H. Peptidic catalysts developed by combinatorial screening methods. Curr. Opin. Chem. Biol. 2007, 11, 269–278. [Google Scholar] [CrossRef]
- Davie, E.A.C.; Mennen, S.M.; Xu, Y.; Miller, S.J. Asymmetric catalysis mediated by synthetic peptides. Chem. Rev. 2007, 107, 5759–5812. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.J. In search of peptide-based catalysts for asymmetric organic synthesis. Acc. Chem. Res. 2004, 37, 601–610. [Google Scholar] [CrossRef] [PubMed]
- Wiesner, M.; Revell, J.D.; Wennemers, H. Tripeptides as efficient asymmetric catalysts for 1,4-addition reactions of aldehydes to nitroolefins—A rational approach. Angew. Chem. Int. Ed. 2008, 47, 1871–1874. [Google Scholar] [CrossRef]
- Joyce, G.F.; Visser, G.M.; van Boeckel, C.A.A.; van Boom, J.H.; Orgel, L.E.; van Westrenen, J. Chiral selection in poly(C)-directed synthesis of oligo(G). Nature 1984, 310, 602–604. [Google Scholar] [CrossRef]
- Stuhlmann, F.; Ja¨schke, A. Characterization of an RNA active site: Interactions between a diels-alderase ribozyme and its substrates and products. J. Am. Chem. Soc. 2002, 124, 3238–3244. [Google Scholar] [CrossRef]
- Coquiere, D.; Feringa, B.L.; Roelfes, G. DNA-based catalytic enantioselective Michael reactions in water. Angew. Chem. Int. Ed. 2007, 46, 9308–9311. [Google Scholar] [CrossRef]
- Boersma, A.J.; Klijin, J.E.; Feringa, B.L.; Roelfes, G. DNA-based asymmetric catalysis: Sequence-dependent rate acceleration and enantioselectivity. J. Am. Chem. Soc. 2008, 130, 11783–11790. [Google Scholar] [CrossRef]
- Zozulia, O.; Dolan, M.A.; Korendovych, I.V. Catalytic peptide assemblies. Chem. Soc. Rev. 2018, 47, 3621–3639. [Google Scholar] [CrossRef]
- Distaffen, H.H.E.; Jones, C.W.; Abraham, B.L.; Nilsson, B.L. Multivalent display of chemical signals on self-assembled peptide scaffolds. Pept. Sci. 2021, 113, e24224. [Google Scholar] [CrossRef]
- Sinibaldi, A.; Della Penna, F.; Ponzetti, M.; Fini, F.; Marchesan, S.; Baschieri, A.; Pesciaioli, F.; Carlone, A. Asymmetric Organocatalysis Accelerated via Self- Assembled Minimal Structures. Eur. J. Org. Chem. 2021, 5403–5406. [Google Scholar] [CrossRef]
- Rodríguez-Llansola, F.; Miravet, J.F.; Escuder, B. Supramolecular Catalysis with Extended Aggregates and Gels: Inversion of Stereoselectivity Caused by Self-Assembly. Chem. Eur. J. 2010, 16, 8480–8486. [Google Scholar] [CrossRef] [PubMed]
- Pelin, J.N.B.D.; Edwards-Gayle, C.J.C.; Castelletto, V.; Aguilar, A.M.; Alves, W.A.; Seitsonen, J.; Ruokolainen, J.; Hamley, I.W. Self-Assembly, Nematic Phase Formation, and Organocatalytic Behavior of a Proline-Functionalized Lipopeptide. ACS Appl. Mater. Interfaces 2020, 12, 13671–13679. [Google Scholar] [CrossRef]
- Vagner, H.J.; Qu, H.; Hruby, V.J. Peptidomimetics, a synthetic tool of drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Lenci, E.; Trabocchi, A. Peptidomimetic toolbox for drug discovery. Chem. Soc. Rev. 2020, 49, 3262–3277. [Google Scholar] [CrossRef]
- Maayan, G.; Ward, M.D.; Kirshenbaum, K. Folded biomimetic oligomers for enantioselective catalysis. Proc. Natl. Acad. Sci. USA 2009, 106, 13679–13684. [Google Scholar] [CrossRef] [PubMed]
- Gellman, S.H. Foldamers: A manifesto. Acc. Chem. Res. 1998, 31, 173–180. [Google Scholar] [CrossRef]
- Pasco, M.; Dolain, C.; Guichard, G. Foldamers in Medicinal Chemistry. In Comprehensive Supramolecular Chemistry II; Atwood, J.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 89–120. [Google Scholar]
- Lichtor, P.A.; Miller, S.J. Combinatorial evolution of site- and enantioselective catalysts for polyene epoxidation. Nat. Chem. 2012, 4, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Lichtor, P.A.; Miller, S. One-Bead-One-Catalyst Approach to Aspartic Acid-Based Oxidation Catalyst Discovery. J. ACS Comb. Sci. 2011, 13, 321–326. [Google Scholar] [CrossRef]
- Berkessel, A. The discovery of catalytically active peptides through combinatorial chemistry. Curr. Opin. Chem. Biol. 2003, 7, 409–419. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef]
- Maeda, Y.; Makhlynets, O.V.; Matsui, H.; Korendovych, I.V. Design of Catalytic Peptides and Proteins Through Rational and Combinatorial Approaches. Annu. Rev. Biomed. Eng. 2016, 18, 311–328. [Google Scholar] [CrossRef]
- de la Torre, A.F.; Rivera, D.G.; Ferreira, M.A.B.; Corrêa, A.G.; Paixão, M.W. Multicomponent Combinatorial Development and Conformational Analysis of Prolyl Peptide–Peptoid Hybrid Catalysts: Application in the Direct Asymmetric Michael Addition. J. Org. Chem 2013, 78, 10221–10232. [Google Scholar] [CrossRef] [PubMed]
- Metrano, A.J.; Abascal, N.C.; Mercado, B.Q.; Paulson, E.K.; Hurtley, A.E.; Miller, S.J. Diversity of Secondary Structure in Catalytic Peptides with β-Turn-Biased Sequences. J. Am. Chem. Soc. 2017, 139, 492–516. [Google Scholar] [CrossRef]
- Rainaldi, M.; Moretto, V.; Crisma, M.; Peggion, E.; Mammi, S.; Toniolo, C.; Cavicchioni, G. Peptoid residues and β-turn formation. J. Pept. Sci. 2002, 8, 241–252. [Google Scholar] [CrossRef]
- Darapaneni, C.M.; Ghosh, P.; Ghosh, T.; Maayan, G. Unique β-Turn Peptoid Structures and Their Application as Asymmetric Catalysts. Chem. Eur. J. 2020, 26, 9573–9579. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.B.; Simmons, B.; Mastracchio, A.; MacMillan, D.W.C. Collective synthesis of natural products by means of organocascade catalysis. Nature 2011, 475, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Antilla, J.C. Enantioselective Construction of Pyrroloindolines Catalyzed by Chiral Phosphoric Acids: Total Synthesis of (−)-Debromoflustramine B. Angew. Chem. Int. Ed. 2012, 51, 11778–11782. [Google Scholar] [CrossRef]
- Hui, C.; Pu, F.; Xu, J. Metal-Catalyzed Asymmetric Michael Addition in Natural Product Synthesis. Chem. Eur. J. 2017, 23, 4023–4036. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, W.; Guzei, I.A.; Spencer, L.C.; Gellman, S.H. New Preorganized γ-Amino Acids as Foldamer Building Blocks. Org. Lett. 2012, 14, 2582–2585. [Google Scholar] [CrossRef]
- Giuliano, M.W.; Maynard, S.J.; Almeida, A.M.; Guo, L.; Guzei, I.A.; Spencer, L.C.; Gellman, S.H. A γ-Amino Acid That Favors 12/10-Helical Secondary Structure in α/γ-Peptides. J. Am. Chem. Soc. 2014, 136, 15046–15053. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.A.; Estevez, A.M.; Campos, M.; Estevez, J.C.; Estevez, R.J. Protocol for the Incorporation of γ-Amino Acids into Peptides: Application to (−)-Shikimic Acid Based 2-Amino-Methylcyclohexanecarboxylic Acids. J. Org. Chem. 2018, 83, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Sukhorukov, A.Y.; Sukhanova, A.; Zlotin, S. Stereoselective reactions of nitro compounds in the synthesis of natural compound analogs and active pharmaceutical ingredients. Tetrahedron 2016, 72, 6191–6281. [Google Scholar] [CrossRef]
- Holub, J.M.; Kirshenbaum, K. Tricks with clicks: Modification of peptidomimetic oligomers via copper-catalyzed azide-alkyne [3 + 2] cycloaddition. Chem. Soc. Rev. 2010, 39, 1325–1337. [Google Scholar] [CrossRef]
- Araszczuk, A.M.; D’Amato, A.; Schettini, R.; Costabile, C.; Della Sala, G.; Pierri, G.; Tedesco, C.; De Riccardis, F.; Izzo, I. Macrocyclic Triazolopeptoids: A Promising Class of Extended Cyclic Peptoids. Org. Lett. 2022, 24, 7752–7756. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Kumar, C.P.; Kumar, T.P.; Haribabu, K.; Jagadeesh, B.; Lakshmi, J.K.; Mainkar, P.S. Peptidomimetic organocatalysts: Efficient Michael addition of ketones onto nitroolefins with very low catalyst loading. RSC Adv. 2014, 4, 30325–30331. [Google Scholar] [CrossRef]
- Webster, A.M.; Cobb, S.L. Recent Advances in the Synthesis of Peptoid Macrocycles. Chem. Eur. J. 2018, 24, 7560–7573. [Google Scholar] [CrossRef]
- Yoo, B.; Shin, S.B.Y.; Huang, M.L.; Kirshenbaum, K. Peptoid Macrocycles: Making the Rounds with Peptidomimetic Oligomers. Chem. Eur. J. 2010, 16, 5528–5537. [Google Scholar] [CrossRef]
- Driggers, E.M.; Hale, S.P.; Lee, J.; Terrett, N.K. The exploration of macrocycles for drug discovery—An underexploited structural class. Nat. Rev. Drug Discov. 2008, 7, 608–624. [Google Scholar] [CrossRef]
- Shin, S.B.Y.; Yoo, B.; Todaro, L.J.; Kirshenbaum, K. Cyclic Peptoids. J. Am. Chem. Soc. 2007, 129, 3218–3225. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, A. Bioactivity Relationship in Cyclic Peptoids: An Overview. Eur. J. Org. Chem. 2022, e202200665. [Google Scholar] [CrossRef]
- Schneider, A.; Craven, T.W.; Kasper, A.C.; Yun, C.; Haugbro, M.; Briggs, E.M.; Svetlov, V.; Nudler, E.; Knaut, H.; Bonneau, R.; et al. Design of Peptoid-peptide Macrocycles to Inhibit the β-catenin TCF Interaction in Prostate Cancer. Nat. Commun. 2018, 9, 4396. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.; Lee, J.H.; Moon, H.; Hyun, Y.-J.; Lim, H.-S. A Chemical Inhibitor of the Skp2/p300 Interaction that Promotes p53- Mediated Apoptosis. Angew. Chem. Int. Ed. 2016, 55, 602–606. [Google Scholar] [CrossRef]
- DuBose, M.B.; Sartawi, T.; Sawatzky, T.M.; Causey, C.P.; Rehman, F.K.; Knuckley, B. A peptoid-based inhibitor of protein arginine methyltransferase 1 (PRMT1) induces apoptosis and autophagy in cancer cells. J. Biol. Chem. 2022, 298, 102205. [Google Scholar] [CrossRef]
- Comegna, D.; Benincasa, M.; Gennaro, R.; Izzo, I.; De Riccardis, F. Design, synthesis and antimicrobial properties of non-hemolytic cationic α-cyclopeptoids. Bioorg. Med. Chem. 2010, 18, 2010–2018. [Google Scholar] [CrossRef]
- Huang, M.L.; Shin, S.B.Y.; Benson, M.A.; Torres, V.J.; Kirshenbaum, K. A Comparison of Linear and Cyclic Peptoid Oligomers as Potent Antimicrobial Agents. ChemMedChem 2012, 7, 114–122. [Google Scholar] [CrossRef]
- Huang, M.L.; Benson, M.A.; Shin, S.B.Y.; Torres, V.J.; Kirshenbaum, K. Amphiphilic Cyclic Peptoids That Exhibit Antimicrobial Activity by Disrupting Staphylococcus aureus Membranes. Eur. J. Org. Chem. 2013, 2013, 3560–3566. [Google Scholar] [CrossRef]
- Nam, H.Y.; Choi, J.; Kumar, S.D.; Nielsen, J.E.; Kyeong, M.; Wang, S.; Kang, D.; Lee, Y.; Lee, J.; Yoon, M.-H.; et al. Helicity Modulation Improves the Selectivity of Antimicrobial Peptoids. ACS Infect. Dis. 2020, 6, 2732–2744. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.E.; Alford, M.A.; Yung, D.B.Y.; Molchanova, N.; Fortkort, J.A.; Lin, J.S.; Diamond, G.; Hancock, R.E.W.; Jenssen, H.; Pletzer, D.; et al. Self-Assembly of Antimicrobial Peptoids Impacts Their Biological Effects on ESKAPE Bacterial Pathogens. ACS Infect. Dis. 2022, 8, 533–545. [Google Scholar] [CrossRef]
- Caumes, C.; Gillon, E.; Legeret, B.; Taillefumier, C.; Imberty, A.; Faure, S. Multivalent thioglycopeptoids via photoclick chemistry: Potent affinities towards LecA and BC2L-A lectins. Chem. Comm. 2015, 51, 12301–12304. [Google Scholar] [CrossRef] [PubMed]
- Lepage, M.L.; Meli, A.; Bodlenner, A.; Tarnus, C.; De Riccardis, F.; Izzo, I.; Compain, P. Synthesis of the first examples of iminosugar clusters based on cyclopeptoid cores. Beilstein J. Org. Chem. 2014, 10, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Lepage, M.L.; Schneider, J.P.; Bodlenner, A.; Meli, A.; De Riccardis, F.; Schmitt, M.; Tarnus, C.; Nguyen-Huynh, N.-T.; Francois, Y.-N.; Leize-Wagner, E.; et al. Iminosugar-Cyclopeptoid Conjugates Raise Multivalent Effect in Glycosidase Inhibition at Unprecedented High Levels. Chem. Eur. J. 2016, 22, 5151–5155. [Google Scholar] [CrossRef]
- D’Amato, A.; Volpe, R.; Vaccaro, M.C.; Terracciano, S.; Bruno, I.; Tosolini, M.; Tedesco, C.; Pierri, G.; Tecilla, P.; Costabile, C.; et al. Cyclic Peptoids as Mycotoxin Mimics: An Exploration of Their Structural and Biological Properties. J. Org. Chem. 2017, 82, 8848–8863. [Google Scholar] [CrossRef]
- Schettini, R.; Tosolini, M.; ur Rehman, J.; Shah, M.R.; Pierri, G.; Tedesco, C.; Della Sala, G.; De Riccardis, F.; Tecilla, P.; Izzo, I. Role of Lipophilicity in the Activity of Hexameric Cyclic Peptoid Ion Carriers. Eur. J. Org. Chem. 2021, 2021, 464–472. [Google Scholar] [CrossRef]
- Schettini, R.; Costabile, C.; Della Sala, G.; Buirey, J.; Tosolini, M.; Tecilla, P.; Vaccaro, M.C.; Bruno, I.; De Riccardis, F.; Izzo, I. Tuning the biomimetic performances of 4-hydroxyproline-containing cyclic peptoids. Org. Biomol. Chem. 2018, 16, 6708–6717. [Google Scholar] [CrossRef]
- Maulucci, N.; Izzo, I.; Bifulco, G.; Aliberti, A.; De Cola, C.; Comegna, D.; Gaeta, C.; Napolitano, A.; Pizza, C.; Tedesco, C.; et al. Synthesis, structures, and properties of nine-, twelve-, and eighteen-membered N-benzyloxyethyl cyclic α-peptoids. Chem. Commun. 2008, 3927–3929. [Google Scholar] [CrossRef]
- De Cola, C.; Licen, S.; Comegna, D.; Cafaro, E.; Bifulco, G.; Izzo, I.; Tecilla, P.; De Riccardis, F. Size-dependent cation transport by cyclic α-peptoid ion carriers. Org. Biomol. Chem. 2009, 7, 2851–2854. [Google Scholar] [CrossRef] [PubMed]
- Hurley, M.F.D.; Northrup, J.D.; Ge, Y.; Schafmeister, C.E.; Voelz, V.A. Metal Cation-Binding Mechanisms of Q-Proline Peptoid Macrocycles in Solution. J. Chem. Inf. Model. 2021, 61, 2818–2828. [Google Scholar] [CrossRef]
- Izzo, I.; Ianniello, G.; De Cola, C.; Nardone, B.; Erra, L.; Vaughan, G.; Tedesco, C.; De Riccardis, F. Structural Effects of Proline Substitution and Metal Binding on Hexameric Cyclic Peptoids. Org. Lett. 2013, 15, 598–601. [Google Scholar] [CrossRef]
- Baskina, M.; Maayan, G. A rationally designed metal-binding helical peptoid for selective recognition processes. Chem. Sci. 2016, 7, 2809–2820. [Google Scholar] [CrossRef]
- De Santis, E.; Edwards, A.A.; Alexander, B.D.; Holder, S.J.; Biesse-Martin, A.-S.; Nielsen, B.V.; Mistry, D.; Waters, L.; Siligardi, G.; Hussain, R.; et al. Selective complexation of divalent cations by a cyclic α,β-peptoid hexamer: A spectroscopic and computational study. Org. Biomol. Chem. 2016, 14, 11371–11380. [Google Scholar] [CrossRef]
- Della Sala, G.; Nardone, B.; De Riccardis, F.; Izzo, I. Cyclopeptoids: A novel class of phase-transfer catalysts. Org. Biomol. Chem. 2013, 11, 726–731. [Google Scholar] [CrossRef]
- Schettini, R.; Nardone, B.; De Riccardis, F.; Della Sala, G.; Izzo, I. Cyclopeptoids as Phase-Transfer Catalysts for the Enantioselective Synthesis of α-Amino Acids. Eur. J. Org. Chem. 2014, 2014, 7793–7797. [Google Scholar] [CrossRef]
- Schettini, R.; De Riccardis, F.; Della Sala, G.; Izzo, I. Enantioselective Alkylation of Amino Acid Derivatives Promoted by Cyclic Peptoids under Phase-Transfer Conditions. J. Org. Chem. 2016, 81, 2494–2505. [Google Scholar] [CrossRef]
- Schettini, R.; D’Amato, A.; De Riccardis, F.; Della Sala, G.; Izzo, I. Catalytic Alkylation of 2-Aryl-2-oxazoline-4-carboxylic Acid Esters Using Cyclopeptoids; Newly Designed Phase-Transfer Catalysts. Synthesis 2017, 49, 1319–1326. [Google Scholar] [CrossRef]
- Schettini, R.; Sicignano, M.; De Riccardis, F.; Izzo, I.; Della Sala, G. Macrocyclic Hosts in Asymmetric Phase-Transfer Catalyzed Reactions. Synthesis 2018, 50, 4777–4795. [Google Scholar] [CrossRef]
- Frązczak, O.; Lasota, A.; Leśniak, A.; Lipkowski, A.W.; Olma, A. The Biological Consequences of Replacing D-Ala in Biphalin with Amphiphilic α-Alkylserines. Chem. Biol. Drug Des. 2014, 84, 199–206. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptoid Catalyst | Oligomer Sequence |
|---|---|
| 1S | Ntempo(Nspe)6 |
| 1R | Ntempo(Nrpe)6 |
| 1Ac | Ntempo(Nspe)6 |
| 2S | NspeNtempo(Nspe)5 |
| 3S | (Nspe)2Ntempo(Nspe)4 |
| 4S | (Nspe)3Ntempo(Nspe)3 |
| 5S-A | Ntempo(Nspe)5 |
| 5S-B | Ntempo(Nspe)4 |
| 5S-C | Ntempo(Nspe)3 |
| 5S-D | Ntempo(Nspe)2 |
| 6 | (Nspe)2 NpmNtempoNspeNpmNspe |
| 7 | Nspe(Npm)2Ntempo(Npm)2Nspe |
| 8 | NtempoNspeNpm(Nspe)2NpmNspe |
| 9 | Ntempo(Npm)2 Nspe(Npm)2Nspe |
| Catalytic System | Conversion (%) a | Selectivity (%) b | ee % |
|---|---|---|---|
| 1S | 84 | 60 (S) | >99 (R) |
| 1R | 85 | 59 (R) | >99 (S) |
| 2S | 26 | None | None |
| 3S | 25 | None | None |
| 4S | 56 | 52 (R) | 5 (S) |

| Entry | Acid | R1 | R2 | R3 | Catalyst | Yield (%) c |
|---|---|---|---|---|---|---|
| 1 | L-Pro | Gly-OMe a | H | Cy | 17 | 78 |
| 2 | L-Pro | Val-OMe b | H | Cy | 18 | 81 |
| 3 | L-Pro | Leu-OMe b | H | Cy | 19 | 85 |
| 4 | L-Pro | Ile-OMe b | H | Cy | 20 | 77 |
| 5 | L-Pro | Phe-OMe b | H | Cy | 21 | 83 |
| 6 | L-Pro | tBuGly-OMe b | H | Cy | 22 | 61 |
| 7 | L-Pro | (S)-α-MeBn a | H | Cy | 23 | 91 |
| 8 | L-Pro | Bn a | H | Cy | 24 | 93 |
| 9 | L-Pro | (S)-α-MeBn a | Me | Cy | 25 | 77 |
| 10 | L-Pro | Bn a | Me | Cy | 26 | 73 |
| 11 | L-Pro | (S)-α-MeBn a | H | tBu | 27 | 88 |
| 12 | L-Pro | (S)-α-MeBn a | H | Gly-OMe | 28 | 82 |
| 13 | D-Pro | (S)-α-MeBn a | Me | Cy | 29 | 79 |

| Entry | Catalyst | Yield (%) a | dr (syn/anti) b | ee (%) c |
|---|---|---|---|---|
| 1 | 17 | 87 | 96:4 | 90 |
| 2 | 18 | 92 | 92:8 | 91 |
| 3 | 19 | 83 | 97:3 | 79 |
| 4 | 20 | 89 | 97:3 | 90 |
| 5 | 21 | 84 | 93:7 | 64 |
| 6 | 22 | 94 | 90:10 | 82 |
| 7 | 23 | 74 | 96:4 | 89 |
| 8 | 24 | 91 | 93:7 | 92 |
| 9 | 25 | 85 | 94:6 | 98 |
| 10 | 26 | 84 | 94:6 | 87 |
| 11 | 27 | 93 | 94:6 | 91 |
| 12 | 28 | 77 | 89:11 | 85 |
| 13 | 29 | 93 | 84:16 | –86 |

| Entry | Peptoid Catalyst | Conv (%) a | syn/antia | ee (%) b |
|---|---|---|---|---|
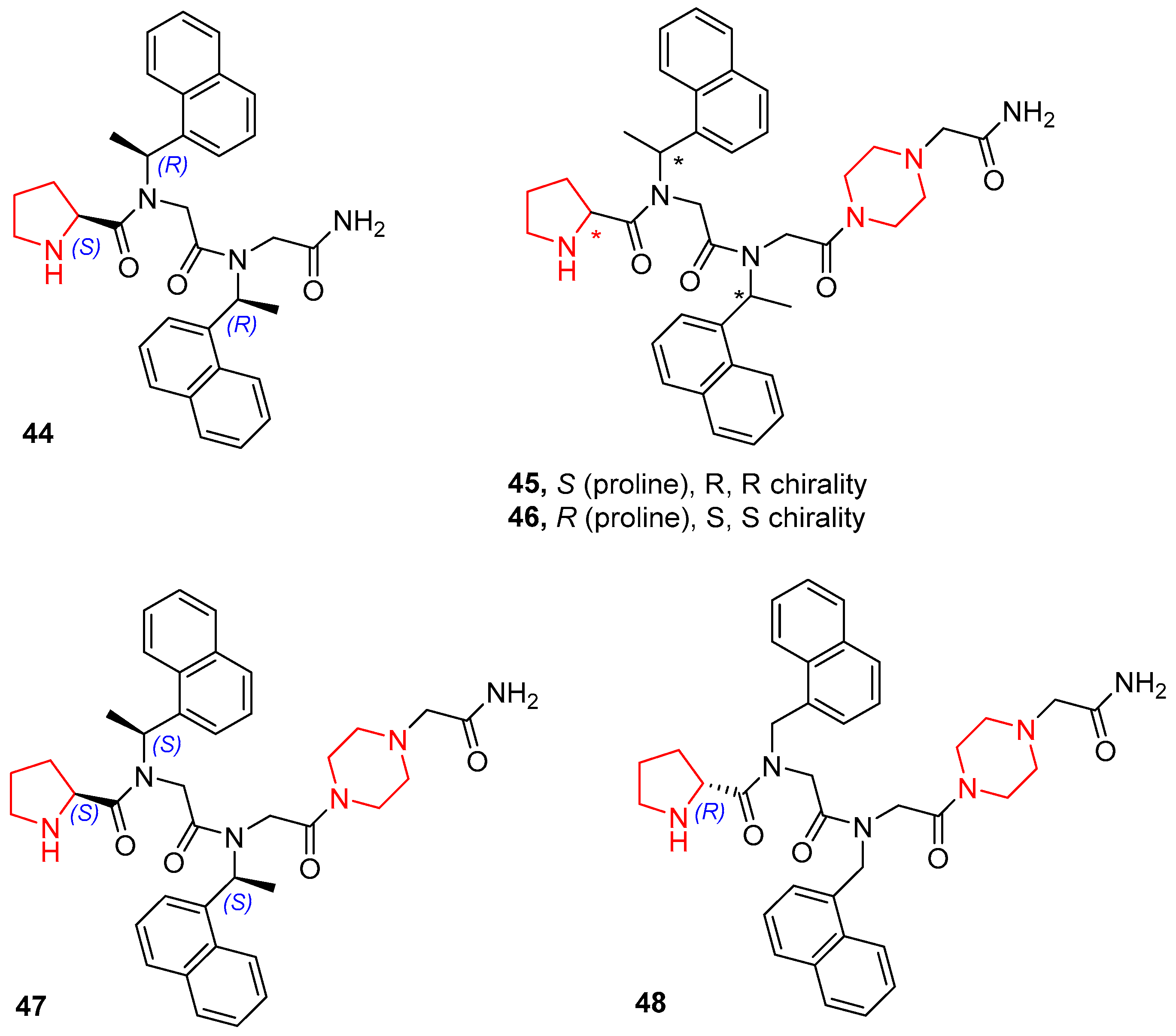
| 1 | 44 | 96 | 91:9 | 87 |
| 2 | 45 | 99 | 92:8 | 95 |
| 3 | 46 | 99 | 92:8 | 95 |
| 4 | 47 | 99 | 88:12 | 56 |
| 5 | 48 | 90 | 88:12 | 41 |
| 6 | L-proline | nr | - | - |

| Entry | Catalyst (mol%) a | Solvent | Time (h) | Yield b (%) | dr c | ee d |
|---|---|---|---|---|---|---|
| 1 | 68 | DCM | 30 | 55 | 90:10 | >99 |
| 2 | 68a | DCM | 32 | 50 | 93:7 | 91:9 |
| 3 | 68 | H2O | 30 | 72 | 97:3 | 99:1 |
| 4 | 68a | H2O | 35 | 70 | 95:5 | 99:1 |
| 5 | 68 | CH3CN | 16 | 93 | 92:8 | 96:4 |
| 6 | 68a | CH3CN | 24 | 90 | 93:7 | 95:5 |
| 7 | 68 | - | 30 | 92 | 92:8 | 97:3 |
| 8 | 68a | - | 30 | 90 | 94:6 | 96:4 |
| 9 | 68 | MeOH | 30 | 95 | 96:4 | 97:3 |
| 10 | 68a | MeOH | 35 | 91 | 94:6 | 96:4 |
| 11 | 68 | iPrOH | 30 | 96 | 97:3 | 97:3 |
| 12 | 68a | iPrOH | 35 | 94 | 95:5 | 91:9 |
| 13 | 68 | MeOH | 35 | 82 | 97:3 | 97:3 |
| 14 | 68a | MeOH | 39 | 79 | 92:8 | 91:9 |
| 15 | 68 | MeOH | 40 | 82 | 97:3 | 97:3 |
| 16 | 68a | MeOH | 45 | 78 | 91:9 | 86:14 |
| 17 | 68 | MeOH | 48 | 80 | 96:4 | 98:2 |
| 18 | 68a | MeOH | 52 | 78 | 90:10 | 96:4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khettar, I.; Araszczuk, A.M.; Schettini, R. Peptidomimetic-Based Asymmetric Catalysts. Catalysts 2023, 13, 244. https://doi.org/10.3390/catal13020244
Khettar I, Araszczuk AM, Schettini R. Peptidomimetic-Based Asymmetric Catalysts. Catalysts. 2023; 13(2):244. https://doi.org/10.3390/catal13020244
Chicago/Turabian StyleKhettar, Ibrahim, Alicja Malgorzata Araszczuk, and Rosaria Schettini. 2023. "Peptidomimetic-Based Asymmetric Catalysts" Catalysts 13, no. 2: 244. https://doi.org/10.3390/catal13020244
APA StyleKhettar, I., Araszczuk, A. M., & Schettini, R. (2023). Peptidomimetic-Based Asymmetric Catalysts. Catalysts, 13(2), 244. https://doi.org/10.3390/catal13020244