
Phase-Transfer Catalyzed Microfluidic Glycosylation: A Small Change in Concentration Results in a Dramatic Increase in Stereoselectivity


Abstract
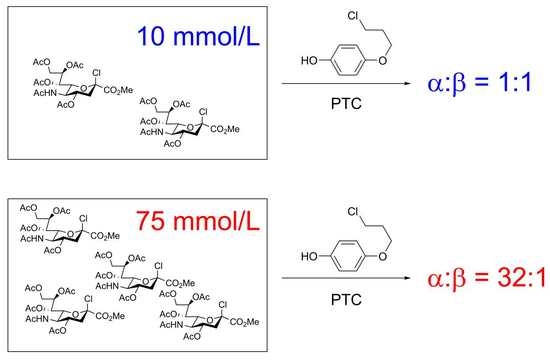
:
1. Introduction



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry 1 | C, mmol/L | Yield of 2, % 2 | α/β 2 | Yield of 3, % 2,3 | Conversion of 1, % 2 |
|---|---|---|---|---|---|
| 1 4 | 5 | 15 | 0.9 | 5 | 75 |
| 2 4 | 10 | 12 | 1.0 | 8 | 66 |
| 3 | 15 | 10 | 2.5 | 2 | 69 |
| 4 5 | 25 | 21 | 2.3 | 4 | 57 |
| 5 6 | 35 | 14 | 4.3 | n. d. 7 | 58 |
| 6 8 | 50 | 36 | 6.2 | 12 | 100 |
| 7 | 60 | 29 | 17.0 | 5 | 66 |
| 8 | 75 | 47 | 31.6 | 11 | 100 |
| 9 | 100 | 43 | 20.7 | 9 | 79 |
| 10 | 200 | 66 | 17.6 | 12 | 100 |


2. Results
3. Discussion
3.1. Flow in Microfluidic System and the Choice of Flow Rate
3.2. Concentration-Dependent Stereoselectivity of the Sialylation
3.3. Supramer Approach for the Explanation of the Phenomenon of Bimodality of Glycosylation
- Type I supramers: both sides of a glycosyl cation formed from N-acetylsialyl chloride 1 along the SN1-like pathway are accessible for the attack of nucleophile leading to unselective reaction (see Figure 5a).
- Type II supramers: only one side of a glycosyl cation formed from N-acetylsialyl chloride 1 along the SN1-like pathway is accessible for the attack of a nucleophile leading to formation of α-anomer of glycoside 2 only (see Figure 5b).
- Type III supramers: only one side of a glycosyl cation formed from N-acetylsialyl chloride 1 along the SN1-like pathway is accessible for the attack of a nucleophile leading to formation of β-anomer of glycoside 2 only (see Figure 5c).
3.4. Bimodality of Glycosylation in Glycochemistry
4. Materials and Methods
4.1. General Methods
4.2. Flow Reactor
4.3. General Glycosylation Procedure
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Magnusson, G.; Chernyak, A.Y.; Kihlberg, J.; Kononov, L.O. Synthesis of Neoglycoconjugates. In Neoglycoconjugates: Preparation and Application; Lee, Y.C., Lee, R.T., Eds.; Academic Press Inc.: San Diego, CA, USA, 1994; pp. 53–143. [Google Scholar]
- Larsen, K.; Thygesen, M.B.; Guillaumie, F.; Willats, W.G.T.; Jensen, K.J. Solid-phase Chemical Tools for Glycobiology. Carbohydr. Res. 2006, 341, 1209–1234. [Google Scholar] [CrossRef]
- Villadsen, K.; Martos-Maldonado, M.C.; Jensen, K.J.; Thygesen, M.B. Chemoselective Reactions for the Synthesis of Glycoconjugates from Unprotected Carbohydrates. ChemBioChem 2017, 18, 574–612. [Google Scholar] [CrossRef]
- Seeberger, P.H. Chemical Glycobiology: Why Now? Nat. Chem. Biol. 2009, 5, 368–372. [Google Scholar] [CrossRef]
- Solís, D.; Bovin, N.V.; Davis, A.P.; Jiménez-Barbero, J.; Romero, A.; Roy, R.; Smetana, K., Jr.; Gabius, H.-J. A Guide into Glycosciences: How Chemistry, Biochemistry and Biology Cooperate to Crack the Sugar Code. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 186–235. [Google Scholar] [CrossRef]
- Stallforth, P.; Lepenies, B.; Adibekian, A.; Seeberger, P.H. Carbohydrates: A Frontier in Medicinal Chemistry. J. Med. Chem. 2009, 52, 5561–5577. [Google Scholar] [CrossRef]
- Bhatia, S.; Dimde, M.; Haag, R. Multivalent Glycoconjugates as Vaccines and Potential Drug Candidates. Med. Chem. Commun. 2014, 5, 862–878. [Google Scholar] [CrossRef]
- Rademacher, C.; Seeberger, P.H. (Eds.) Carbohydrates as Drugs, 1st ed.; Springer International Publishing: Cham, Switzerland, 2014. [Google Scholar] [CrossRef]
- Abronina, P.I.; Zinin, A.I.; Romashin, D.A.; Tereshina, V.V.; Chizhov, A.O.; Kononov, L.O. Application of a Janus Aglycon with Dual Function in Benzyl-Free Synthesis of Spacer-Armed Oligosaccharide Fragments of Polysaccharides from Rhizobacterium Azospirillum brasilense sp7. Carbohydr. Res. 2018, 464, 28–43. [Google Scholar] [CrossRef]
- Abronina, P.I.; Shvyrkina, J.S.; Zinin, A.I.; Chizhov, A.O.; Kononov, L.O. Synthesis of Selectively Protected α-(1-3)-, α-(1-5)-Linked Octasaccharide Fragment, Containing Janus Aglycon, Related to Branching Region of Mycobacterial Polysaccharides. Russ. Chem. Bull. 2022, 71, 2740–2750. [Google Scholar]
- Abronina, P.I.; Zinin, A.I.; Malysheva, N.N.; Stepanova, E.V.; Chizhov, A.O.; Torgov, V.I.; Kononov, L.O. A Novel Glycosyl Donor with a Triisopropylsilyl Nonparticipating Group in Benzyl-Free Stereoselective 1,2-cis-Galactosylation. Synlett 2017, 28, 1608–1613. [Google Scholar] [CrossRef]
- Stepanova, E.V.; Podvalnyy, N.M.; Abronina, P.I.; Kononov, L.O. Length Matters: One Additional Methylene Group in a Reactant is Able to Affect the Reactivity Pattern and Significantly Increase the Product Yield. Synlett 2018, 29, 2043–2045. [Google Scholar] [CrossRef]
- Stepanova, E.V.; Abronina, P.I.; Zinin, A.I.; Chizhov, A.O.; Kononov, L.O. Janus Glycosides of Next Generation: Synthesis of 4-(3-Chloropropoxy)Phenyl and 4-(3-Azidopropoxy)Phenyl Glycosides. Carbohydr. Res. 2019, 471, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, E.V.; Zinin, A.I.; Abronina, P.I.; Chizhov, A.O.; Kononov, L.O. Azidation of Partially Protected Carbohydrate Derivatives: Efficient Suppression of Acyl Migration. Synlett 2020, 31, 1491–1496. [Google Scholar] [CrossRef]
- Abronina, P.I.; Malysheva, N.N.; Zinin, A.I.; Karpenko, M.Y.; Kolotyrkina, N.G.; Kononov, L.O. Trifluoroacetic Acid-Promoted Ring Contraction in 2,3-Di-O-silylated O-Galactopyranosides and Hemiacetals. Synlett 2022, 33, 473–477. [Google Scholar] [CrossRef]
- Abronina, P.I.; Malysheva, N.N.; Stepanova, E.V.; Shvyrkina, J.S.; Zinin, A.I.; Kononov, L.O. Five Triisopropylsilyl Substituents in Ara-β-(1→2)-Ara Disaccharide Glycosyl Donor Make Unselective Glycosylation Reaction Stereoselective. Eur. J. Org. Chem. 2022, 2022, e202201110. [Google Scholar] [CrossRef]
- Dehmlow, E.V.; Dehmlow, S.S. Phase Transfer Catalysis, 3rd ed.; Wiley VCH: Weinheim, Germany, 1993; p. 499. [Google Scholar]
- Otevrel, J.; Waser, M. Asymmetric Phase-Transfer Catalysis—From Classical Applications to New Concepts. In Asymmetric Organocatalysis: New Strategies, Catalysts, and Opportunities; Albrecht, Ł., Albrecht, A., Dell’Amico, L., Eds.; WILEY-VCH GmbH: Weinheim, Germany, 2023; Volume 2, pp. 71–120. [Google Scholar] [CrossRef]
- Roy, R. Phase Transfer Catalysis in Carbohydrate Chemistry. In Handbook of Phase Transfer Catalysis; Sasson, Y., Neumann, R., Eds.; Springer: Dordrecht, The Netherland, 1997; pp. 244–275. [Google Scholar] [CrossRef]
- Roy, R.; Tropper, F.D.; Cao, S.; Kim, J.M. Anomeric Group Transformations Under Phase-Transfer Catalysis. In Phase-Transfer Catalysis; Halpern, M., Ed.; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 1997; Volume 659, pp. 163–180. [Google Scholar] [CrossRef]
- Yang, Y.; Yu, B. Recent Advances in the Chemical Synthesis of C-Glycosides. Chem. Rev. 2017, 117, 12281–12356. [Google Scholar] [CrossRef]
- Dimakos, V.; Taylor, M.S. Recent Advances in the Direct O-Arylation of Carbohydrates. Org. Biomol. Chem. 2021, 19, 514–524. [Google Scholar] [CrossRef]
- Demchenko, A.V. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar] [CrossRef]
- Ágoston, K.; Watt, G.M. 2.04—Methods for O-Glycoside Synthesis. In Comprehensive Glycoscience, 2nd ed.; Barchi, J.J., Vidal, S., Eds.; Elsevier: Oxford, UK, 2021; Volume 2, pp. 103–159. [Google Scholar] [CrossRef]
- Singh, Y.; Geringer, S.A.; Demchenko, A.V. Synthesis and Glycosidation of Anomeric Halides: Evolution from Early Studies to Modern Methods of the 21st Century. Chem. Rev. 2022, 122, 11701–11758. [Google Scholar] [CrossRef]
- Roy, R.; Tropper, F.D.; Romanowska, A.; Letellier, M.; Cousineau, L.; Meunier, S.J.; Boratynski, J. Expedient Syntheses of Neoglycoproteins Using Phase-Transfer Catalysis and Reductive Amination as Key Reactions. Glycoconj. J. 1991, 8, 75–81. [Google Scholar] [CrossRef]
- Rothermel, J.; Faillard, H. Phase-transfer-catalyzed Synthesis of Aryl α-Ketosides of N-Acetylneuraminic Acid. A 2-Methylfluoran-6-yl Glycoside of N-Acetylneuraminic Acid, 2-Methyl-6-(5-acetamido-3,5-dideoxy-α-d-glycero-d-galacto-nonulopyranosylonic acid)xanthene-9-spiro-1′-isobenzofuran-3′-one, a New Substrate for Neuraminidase Assay. Carbohydr. Res. 1990, 196, 29–40. [Google Scholar] [CrossRef]
- Roy, R.; Tropper, F. Stereospecific Synthesis of Aryl β-D-N-Acetylglucopyranosides by Phase Transfer Catalysis. Synth. Commun. 1990, 20, 2097–2102. [Google Scholar] [CrossRef]
- Roy, R.; Andersson, F.O.; Harms, G.; Kelm, S.; Schauer, R. Synthesis of Esterase-Resistant 9-O-Acetylated Polysialoside as Inhibitor of Influenza C Virus Hemagglutinin. Angew. Chem. Int. Ed. Engl. 1992, 31, 1478–1481. [Google Scholar] [CrossRef]
- Gan, Z.H.; Roy, R. Facile Preparation of Divalent Sialoside Derivatives by Olefin Metathesis Reaction. Tetrahedron 2000, 56, 1423–1428. [Google Scholar] [CrossRef]
- Gan, Z.; Roy, R. Sialoside Clusters as Potential Ligands for Siglecs (Sialoadhesins). Can. J. Chem. 2002, 80, 908–916. [Google Scholar] [CrossRef]
- Carlescu, I.; Osborn, H.M.I.; Desbrieres, J.; Scutaru, D.; Popa, M. Synthesis of Poly(aspartimide)-Based Bio-Glycoconjugates. Carbohydr. Res. 2010, 345, 33–40. [Google Scholar] [CrossRef]
- Myachin, I.V.; Mamirgova, Z.Z.; Stepanova, E.V.; Zinin, A.I.; Chizhov, A.O.; Kononov, L.O. Black Swan in Phase Transfer Catalysis: Influence of Mixing Mode on the Stereoselectivity of Glycosylation. Eur. J. Org. Chem. 2022, 2022, e202101377. [Google Scholar] [CrossRef]
- Adero, P.O.; Amarasekara, H.; Wen, P.; Bohé, L.; Crich, D. The Experimental Evidence in Support of Glycosylation Mechanisms at the SN1–SN2 Interface. Chem. Rev. 2018, 118, 8242–8284. [Google Scholar] [CrossRef]
- Crich, D. En Route to the Transformation of Glycoscience: A Chemist’s Perspective on Internal and External Crossroads in Glycochemistry. J. Am. Chem. Soc. 2021, 143, 17–34. [Google Scholar] [CrossRef]
- Andreana, P.R.; Crich, D. Guidelines for O-Glycoside Formation from First Principles. ACS Cent. Sci. 2021, 7, 1454–1462. [Google Scholar] [CrossRef]
- Tanaka, K.; Mori, Y.; Fukase, K. Practical Synthesis of a Manβ(1-4)GlcNTroc Fragment via Microfluidic β-Mannosylation. J. Carbohydr. Chem. 2009, 28, 1–11. [Google Scholar] [CrossRef]
- Tanaka, K.; Fukase, K. Acid-mediated Reactions under Microfluidic Conditions: A New Strategy for Practical Synthesis of Biofunctional Natural Products. Beilstein J. Org. Chem. 2009, 5, 40. [Google Scholar] [CrossRef]
- Tanaka, K.; Fukase, K. Renaissance of Traditional Organic Reactions under Microfluidic Conditions: A New Paradigm for Natural Products Synthesis. Org. Process Res. Dev. 2009, 13, 983–990. [Google Scholar] [CrossRef]
- Tanaka, K.; Miyagawa, T.; Fukase, K. Chemical N-Glycosylation by Asparagine under Integrated Microfluidic/Batch Conditions. Synlett 2009, 1571–1574. [Google Scholar] [CrossRef]
- Uchinashi, Y.; Nagasaki, M.; Zhou, J.; Tanaka, K.; Fukase, K. Reinvestigation of the C5-Acetamide Sialic Acid Donor for α-Selective Sialylation: Practical Procedure under Microfluidic Conditions. Org. Biomol. Chem. 2011, 9, 7243–7248. [Google Scholar] [CrossRef]
- Uchinashi, Y.; Tanaka, K.; Manabe, Y.; Fujimoto, Y.; Fukase, K. Practical and Efficient Method for α-Sialylation with an Azide Sialyl Donor Using a Microreactor. J. Carbohydr. Chem. 2014, 33, 55–67. [Google Scholar] [CrossRef]
- Nagasaki, M.; Manabe, Y.; Minamoto, N.; Tanaka, K.; Silipo, A.; Molinaro, A.; Fukase, K. Chemical Synthesis of a Complex-Type N-Glycan Containing a Core Fucose. J. Org. Chem. 2016, 81, 10600–10616. [Google Scholar] [CrossRef] [PubMed]
- Fukase, K.; Tanaka, K.; Fujimoto, Y.; Shimoyama, A.; Manabe, Y. Sugar Synthesis by Microfluidic Techniques. In Glycochemical Synthesis; Hung, S.-C., Zulueta, M.M.L., Eds.; Wiley Online Books; Wiley: Hoboken, NJ, USA, 2016; pp. 205–219. [Google Scholar] [CrossRef]
- Myachin, I.V.; Orlova, A.V.; Kononov, L.O. Glycosylation in Flow: Effect of the Flow Rate and Type of the Mixer. Russ. Chem. Bull. 2019, 68, 2126–2129. [Google Scholar] [CrossRef]
- Boons, G.J.; Demchenko, A.V. Recent Advances in O-Sialylation. Chem. Rev. 2000, 100, 4539–4566. [Google Scholar] [CrossRef] [PubMed]
- De Meo, C.; Jones, B.T. Chapter Two—Chemical Synthesis of Glycosides of N-Acetylneuraminic Acid. Adv. Carbohydr. Chem. Biochem. 2018, 75, 215–316. [Google Scholar] [CrossRef]
- De Meo, C.; Goeckner, N. 2.07—Synthesis of Glycosides of Sialic Acid. In Comprehensive Glycoscience, 2nd ed.; Barchi, J.J., Vidal, S., Eds.; Elsevier: Amsterdam, The Netherland, 2021; Volume 2, pp. 228–266. [Google Scholar] [CrossRef]
- Kononov, L.O. Chemical Reactivity and Solution Structure: On the Way to a Paradigm Shift? RSC Adv. 2015, 5, 46718–46734. [Google Scholar] [CrossRef]
- Ahmed-Omer, B.; Barrow, D.A.; Wirth, T. Heck Reactions using Segmented Flow Conditions. Tetrahedron Lett. 2009, 50, 3352–3355. [Google Scholar] [CrossRef]
- Kononov, L.O.; Malysheva, N.N.; Kononova, E.G.; Orlova, A.V. Intermolecular Hydrogen-bonding Pattern of a Glycosyl Donor: The Key to Understanding the Outcome of Sialylation. Eur. J. Org. Chem. 2008, 2008, 3251–3255. [Google Scholar] [CrossRef]
- Kononov, L.O.; Malysheva, N.N.; Orlova, A.V.; Zinin, A.I.; Laptinskaya, T.V.; Kononova, E.G.; Kolotyrkina, N.G. Concentration Dependence of Glycosylation Outcome: A Clue to Reproducibility and Understanding the Reasons Behind. Eur. J. Org. Chem. 2012, 2012, 1926–1934. [Google Scholar] [CrossRef]
- Kononov, L.O. Modulation of Stereoselectivity of Glycosylation: A Supramer Approach. In Advances in Chemistry Research; Taylor, J.C., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2013; Volume 18, pp. 143–178. [Google Scholar]
- Ahiadorme, D.A.; Podvalnyy, N.M.; Orlova, A.V.; Chizhov, A.O.; Kononov, L.O. Glycosylation of Dibutyl Phosphate Anion with Arabinofuranosyl Bromide: Unusual Influence of Concentration of the Reagents on the Ratio of Anomeric Glycosyl Phosphates Formed. Russ. Chem. Bull. 2016, 65, 2776–2778. [Google Scholar] [CrossRef]
- Kononov, L.O.; Fedina, K.G.; Orlova, A.V.; Kondakov, N.N.; Abronina, P.I.; Podvalnyy, N.M.; Chizhov, A.O. Bimodal Concentration-dependent Reactivity Pattern of a Glycosyl Donor: Is the Solution Structure Involved? Carbohydr. Res. 2017, 437, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Nagornaya, M.O.; Orlova, A.V.; Stepanova, E.V.; Zinin, A.I.; Laptinskaya, T.V.; Kononov, L.O. The Use of the Novel Glycosyl Acceptor and Supramer Analysis in the Synthesis of Sialyl-α(2–3)-Galactose Building Block. Carbohydr. Res. 2018, 470, 27–35. [Google Scholar] [CrossRef]
- Orlova, A.V.; Laptinskaya, T.V.; Malysheva, N.N.; Kononov, L.O. Light Scattering in Non-Aqueous Solutions of Low-Molecular-Mass Compounds: Application for Supramer Analysis of Reaction Solutions. J. Solut. Chem. 2020, 49, 629–644. [Google Scholar] [CrossRef]
- Orlova, A.V.; Ahiadorme, D.A.; Laptinskaya, T.V.; Kononov, L.O. Supramer Analysis of 2,3,5-tri-O-Benzoyl-α-d-Arabinofuranosyl Bromide Solutions in Different Solvents: Supramolecular Aggregation of Solute Molecules in 1,2-Dichloroethane Mediated by Halogen Bonds. Russ. Chem. Bull. 2021, 70, 2214–2219. [Google Scholar] [CrossRef]
- Baker, A.; Graz, M.; Saunders, R.; Evans, G.J.S.; Kaul, S.; Wirth, T. Flow Synthesis of Symmetrical Di- and Trisulfides Using Phase-Transfer Catalysis. J. Flow Chem. 2013, 3, 118–121. [Google Scholar] [CrossRef]
- Hisamoto, H.; Saito, T.; Tokeshi, M.; Hibara, A.; Kitamori, T. Fast And High Conversion Phase-Transfer Synthesis Exploiting the Liquid–Liquid Interface Formed in a Microchannel Chip. Chem. Commun. 2001, 1, 2662–2663. [Google Scholar] [CrossRef]
- Ueno, M.; Hisamoto, H.; Kitamori, T.; Kobayashi, S. Phase-Transfer Alkylation Reactions using Microreactors. Chem. Commun. 2003, 3, 936–937. [Google Scholar] [CrossRef]
- Orlova, A.V.; Andrade, R.R.; da Silva, C.O.; Zinin, A.I.; Kononov, L.O. Polarimetry as a Tool for the Study of Solutions of Chiral Solutes. ChemPhysChem 2014, 15, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Kononov, L.O.; Magnusson, G. Synthesis of Methyl and Allyl α-Glycosides of N-Acetylneuraminic Acid in the Absence of Added Promoter. Acta Chem. Scand. 1998, 52, 141–144. [Google Scholar] [CrossRef]
- Kulikova, N.Y.; Shpirt, A.M.; Chinarev, A.; Kononov, L.O. Synthesis of O-Acetylated N-Acetylneuraminic Acid Glycal. In Carbohydrate Chemistry: Proven Synthetic Methods; Kovac, P., Ed.; CRC Press-Taylor & Francis Group: Boca Raton, FL, USA, 2012; Volume 1, pp. 245–250. [Google Scholar] [CrossRef]
- Reynolds, O. XXIX. An Experimental Investigation of the Circumstances which Determine whether the Motion of Water Shall be Direct or Sinuous, and of the Law of Resistance in Parallel Channels. Philos. Trans. R. Soc. Lond. 1883, 174, 935–982. [Google Scholar] [CrossRef]
- Jahnisch, K.; Hessel, V.; Löwe, H.; Baerns, M. Chemistry in Microstructured Reactors. Angew. Chem. Int. Ed. 2004, 43, 406. [Google Scholar] [CrossRef] [PubMed]
- Elvira, K.S.; I Solvas, X.C.; Wootton, R.C.R.; Demello, A.J. The Past, Present and Potential for Microfluidic Reactor Technology in Chemical Synthesis. Nat. Chem. 2013, 5, 905–915. [Google Scholar] [CrossRef]
- Wang, Q.; Steinbock, O. Materials Synthesis and Catalysis in Microfluidic Devices: Prebiotic Chemistry in Mineral Membranes. ChemCatChem 2020, 12, 63–74. [Google Scholar] [CrossRef]
- Fang, W.-F.; Yang, J.-T. A Novel Microreactor with 3D Rotating Flow to Boost Fluid Reaction and Mixing of Viscous Fluids. Sens. Actuators B 2009, 140, 629–642. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Fang, W.-F.; Liu, Y.-C.; Yang, J.-T. Analysis of Chaos and FRET Reaction in Split-and-Recombine Microreactors. Microfluid. Nanofluid. 2011, 11, 339–352. [Google Scholar] [CrossRef]
- Chen, Y.T.; Chen, K.H.; Fang, W.F.; Tsai, S.H.; Fang, J.M.; Yang, J.T. Flash Synthesis of Carbohydrate Derivatives in Chaotic Microreactors. Chem. Eng. J. 2011, 174, 421–424. [Google Scholar] [CrossRef]
- deMello, A.J. Control and Detection of Chemical Reactions in Microfluidic Systems. Nature 2006, 442, 394–402. [Google Scholar] [CrossRef]
- Bothe, D.; Stemich, C.; Warnecke, H.-J. Fluid Mixing in a T-Shaped Micro-Mixer. Chem. Eng. Sci. 2006, 61, 2950–2958. [Google Scholar] [CrossRef]
- Kockmann, N.; Kiefer, T.; Engler, M.; Woias, P. Convective Mixing and Chemical Reactions in Microchannels with High Flow Rates. Sens. Actuators B 2006, 117, 495–508. [Google Scholar] [CrossRef]
- Rahimi, M.; Azimi, N.; Parsamogadam, M.A.; Rahimi, A.; Masahy, M.M. Mixing Performance of T, Y, and Oriented Y-Micromixers with Spatially Arranged Outlet Channel: Evaluation with Villermaux/Dushman Test Reaction. Microsyst. Technol. 2017, 23, 3117–3130. [Google Scholar] [CrossRef]
- Zhang, H.; Kopfmüller, T.; Achermann, R.; Zhang, J.; Teixeira, A.; Shen, Y.; Jensen, K.F. Accessing Multidimensional Mixing via 3D Printing and Showerhead Micromixer Design. AIChE J. 2020, 66, e16873. [Google Scholar] [CrossRef]
- Kononov, L.O.; Tsvetkov, D.E.; Orlova, A.V. Conceivably the First Example of a Phase Transition in Aqueous Solutions Of Oligosaccharide Glycosides. Evidence from Variable-Temperature 1H NMR and Optical Rotation Measurements for a Solution of Allyl Lactoside. Russ. Chem. Bull. 2002, 51, 1337–1338. [Google Scholar] [CrossRef]
- Orlova, A.V.; Zinin, A.I.; Kononov, L.O. Mutarotation in Aqueous Solutions of d-Levoglucosan: A Supramer Approach. Russ. Chem. Bull. 2014, 63, 295–297. [Google Scholar] [CrossRef]
- Orlova, A.V.; Kononov, L.O. Polarimetry as a Method for Studying the Structure of Aqueous Carbohydrate Solutions: Correlation with Other Methods. RENSIT 2020, 12, 95–106. [Google Scholar] [CrossRef]
- Orlova, A.V.; Laptinskaya, T.V.; Bovin, N.V.; Kononov, L.O. Differences in Reactivity of N-Acetyl- and N,N-Diacetylsialyl Chlorides, Caused by Their Different Supramolecular Organization in Solutions. Russ. Chem. Bull. 2017, 66, 2173–2179. [Google Scholar] [CrossRef]
- Sedlák, M. Large-scale Supramolecular Structure in Solutions of Low Molar Mass Compounds and Mixtures of Liquids: I. Light Scattering Characterization. J. Phys. Chem. B 2006, 110, 4329–4338. [Google Scholar] [CrossRef]
- Sedlák, M. Large-scale Supramolecular Structure in Solutions of Low Molar Mass Compounds and Mixtures of liquids: II. Kinetics of the Formation and Long-time Stability. J. Phys. Chem. B 2006, 110, 4339–4345. [Google Scholar] [CrossRef]
- Sedlák, M. Large-scale Supramolecular Structure in Solutions of Low Molar Mass Compounds and Mixtures of Liquids. III. Correlation with Molecular Properties and Interactions. J. Phys. Chem. B 2006, 110, 13976–13984. [Google Scholar] [CrossRef] [PubMed]
- Sedlák, M.; Rak, D. Large-Scale Inhomogeneities in Solutions of Low Molar Mass Compounds and Mixtures of Liquids: Supramolecular Structures or Nanobubbles? J. Phys. Chem. B 2013, 117, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Sedlák, M.; Rak, D. On the Origin of Mesoscale Structures in Aqueous Solutions of Tertiary Butyl Alcohol: The Mystery Resolved. J. Phys. Chem. B 2014, 118, 2726–2737. [Google Scholar] [CrossRef] [PubMed]
- Rak, D.; Ovadová, M.; Sedlák, M. (Non)existence of Bulk Nanobubbles: The Role of Ultrasonic Cavitation and Organic Solutes in Water. J. Phys. Chem. Lett. 2019, 10, 4215–4221. [Google Scholar] [CrossRef] [PubMed]
- Rak, D.; Sedlák, M. On the Mesoscale Solubility in Liquid Solutions and Mixtures. J. Phys. Chem. B 2019, 123, 1365–1374. [Google Scholar] [CrossRef]
- Rak, D.; Sedlák, M. Solvophobicity-Driven Mesoscale Structures: Stabilizer-Free Nanodispersions. Langmuir 2023, in press. [Google Scholar] [CrossRef]
- Subramanian, D.; Anisimov, M.A. Phase Behavior and Mesoscale Solubilization in Aqueous Solutions of Hydrotropes. Fluid Phase Equilib. 2014, 362, 170–176. [Google Scholar] [CrossRef]
- Zemb, T.; Kunz, W. Weak Aggregation: State of the Art, Expectations and Open Questions. Curr. Opin. Colloid Interface Sci. 2016, 22, 113–119. [Google Scholar] [CrossRef]
- Mostafa, S.; Behafarid, F.; Croy, J.R.; Ono, L.K.; Li, L.; Yang, J.C.; Frenkel, A.I.; Cuenya, B.R. Shape-dependent Catalytic Properties of Pt Nanoparticles. J. Am. Chem. Soc. 2010, 132, 15714–15719. [Google Scholar] [CrossRef]
- Roldan Cuenya, B.; Behafarid, F. Nanocatalysis: Size- and Shape-Dependent Chemisorption and Catalytic Reactivity. Surf. Sci. Rep. 2015, 70, 135–187. [Google Scholar] [CrossRef]
- Buchecker, T.; Krickl, S.; Winkler, R.; Grillo, I.; Bauduin, P.; Touraud, D.; Pfitzner, A.; Kunz, W. The Impact of the Structuring of Hydrotropes in Water on the Mesoscale Solubilisation of a Third Hydrophobic Component. Phys. Chem. Chem. Phys. 2017, 19, 1806–1816. [Google Scholar] [CrossRef] [PubMed]
- Krickl, S.; Touraud, D.; Bauduin, P.; Zinn, T.; Kunz, W. Enzyme Activity of Horseradish Peroxidase in Surfactant-Free Microemulsions. J. Colloid Interface Sci. 2018, 516, 466–475. [Google Scholar] [CrossRef]
- Hahn, M.; Krickl, S.; Buchecker, T.; Jost, G.; Touraud, D.; Bauduin, P.; Pfitzner, A.; Klamt, A.; Kunz, W. Ab Initio Prediction of Structuring/Mesoscale Inhomogeneities in Surfactant-Free Microemulsions And Hydrogen-Bonding-Free Microemulsions. Phys. Chem. Chem. Phys. 2019, 21, 8054–8066. [Google Scholar] [CrossRef] [PubMed]
- Svard, M.; Renuka Devi, K.; Khamar, D.; Mealey, D.; Cheuk, D.; Zeglinski, J.; Rasmuson, A.C. Solute Clustering in Undersaturated Solutions—Systematic Dependence on Time, Temperature and Concentration. Phys. Chem. Chem. Phys. 2018, 20, 15550–15559. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Ishiwata, A.; Ito, Y. Recent Advances of the Stereoselective Bimodal Glycosylations for the Synthesis of Various Glucans. In Studies in Natural Products Chemistry; Elsevier B.V.: Amsterdam, The Netherlands, 2022; Volume 74, pp. 1–40. [Google Scholar] [CrossRef]
- Ishiwata, A.; Tanaka, K.; Ao, J.; Ding, F.; Ito, Y. Recent Advances in Stereoselective 1,2-cis-O-Glycosylations. Front. Chem. 2022, 10, 972429. [Google Scholar] [CrossRef] [PubMed]
- Zinin, A.I.; Stepanova, E.V.; Jost, U.; Kondakov, N.N.; Shpirt, A.M.; Chizhov, A.O.; Torgov, V.I.; Kononov, L.O. An Efficient Multigram-Scale Synthesis of 4-(ω-Chloroalkoxy)phenols. Russ. Chem. Bull. 2017, 66, 304–312. [Google Scholar] [CrossRef]
- Kulikova, N.Y.; Shpirt, A.M.; Kononov, L.O. A Facile Synthesis of N-Acetylneuraminic Acid Glycal. Synthesis 2006, 4113–4114. [Google Scholar] [CrossRef]
- Armarego, W.L.F. Purification of Laboratory Chemicals, 8th ed.; Butterworth-Heinemann: Oxford, UK, 2017. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Myachin, I.V.; Kononov, L.O. Phase-Transfer Catalyzed Microfluidic Glycosylation: A Small Change in Concentration Results in a Dramatic Increase in Stereoselectivity. Catalysts 2023, 13, 313. https://doi.org/10.3390/catal13020313
Myachin IV, Kononov LO. Phase-Transfer Catalyzed Microfluidic Glycosylation: A Small Change in Concentration Results in a Dramatic Increase in Stereoselectivity. Catalysts. 2023; 13(2):313. https://doi.org/10.3390/catal13020313
Chicago/Turabian StyleMyachin, Ilya V., and Leonid O. Kononov. 2023. "Phase-Transfer Catalyzed Microfluidic Glycosylation: A Small Change in Concentration Results in a Dramatic Increase in Stereoselectivity" Catalysts 13, no. 2: 313. https://doi.org/10.3390/catal13020313