
Active Sites in H-Mordenite Catalysts Probed by NMR and FTIR

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Elemental Analysis
2.2. X-ray Diffraction Studies
2.3. SEM Studies
2.4. Thermogravimetric Analysis
2.5. NMR Characterization
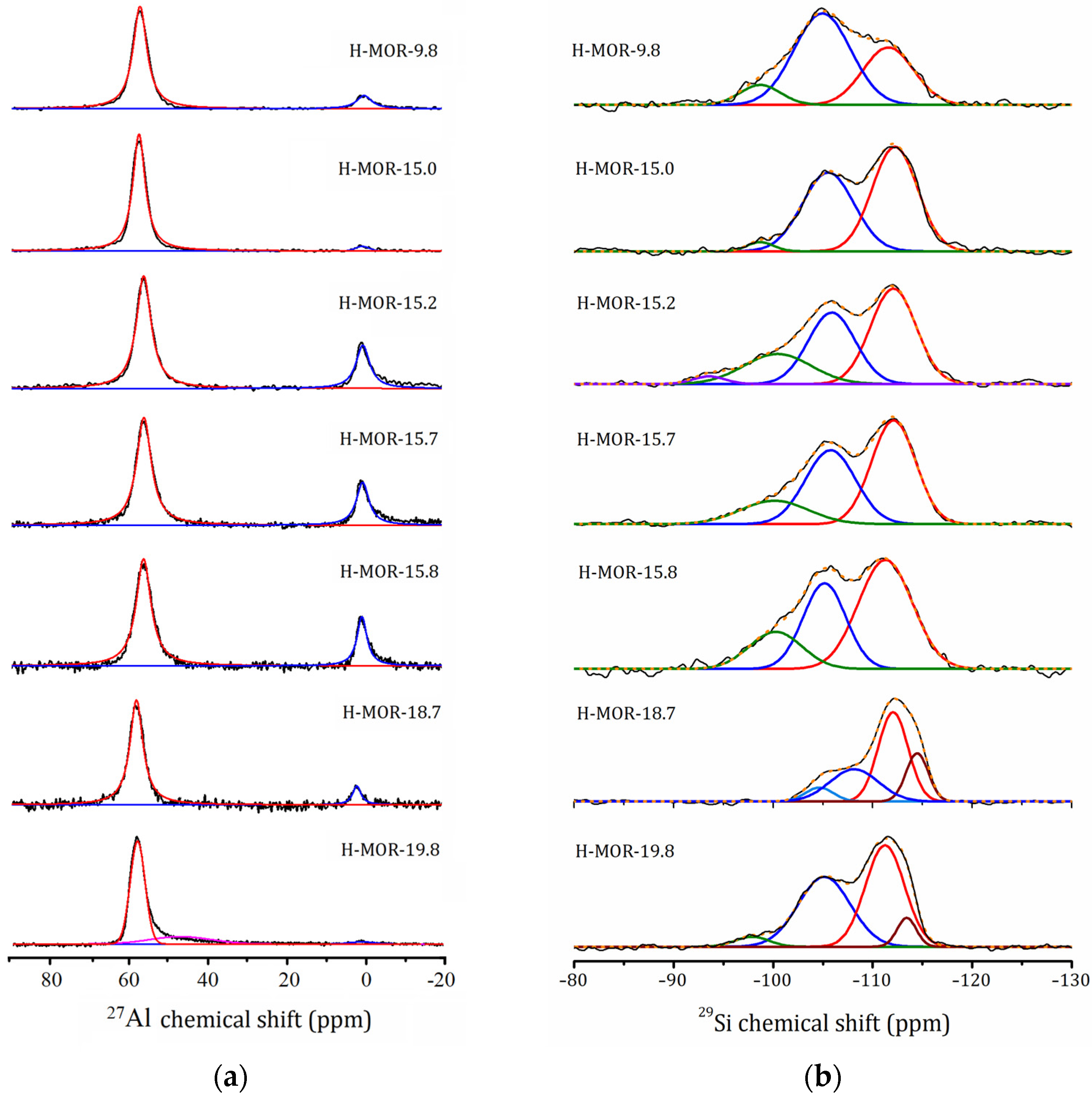
2.5.1. 27Al and 29Si MAS NMR
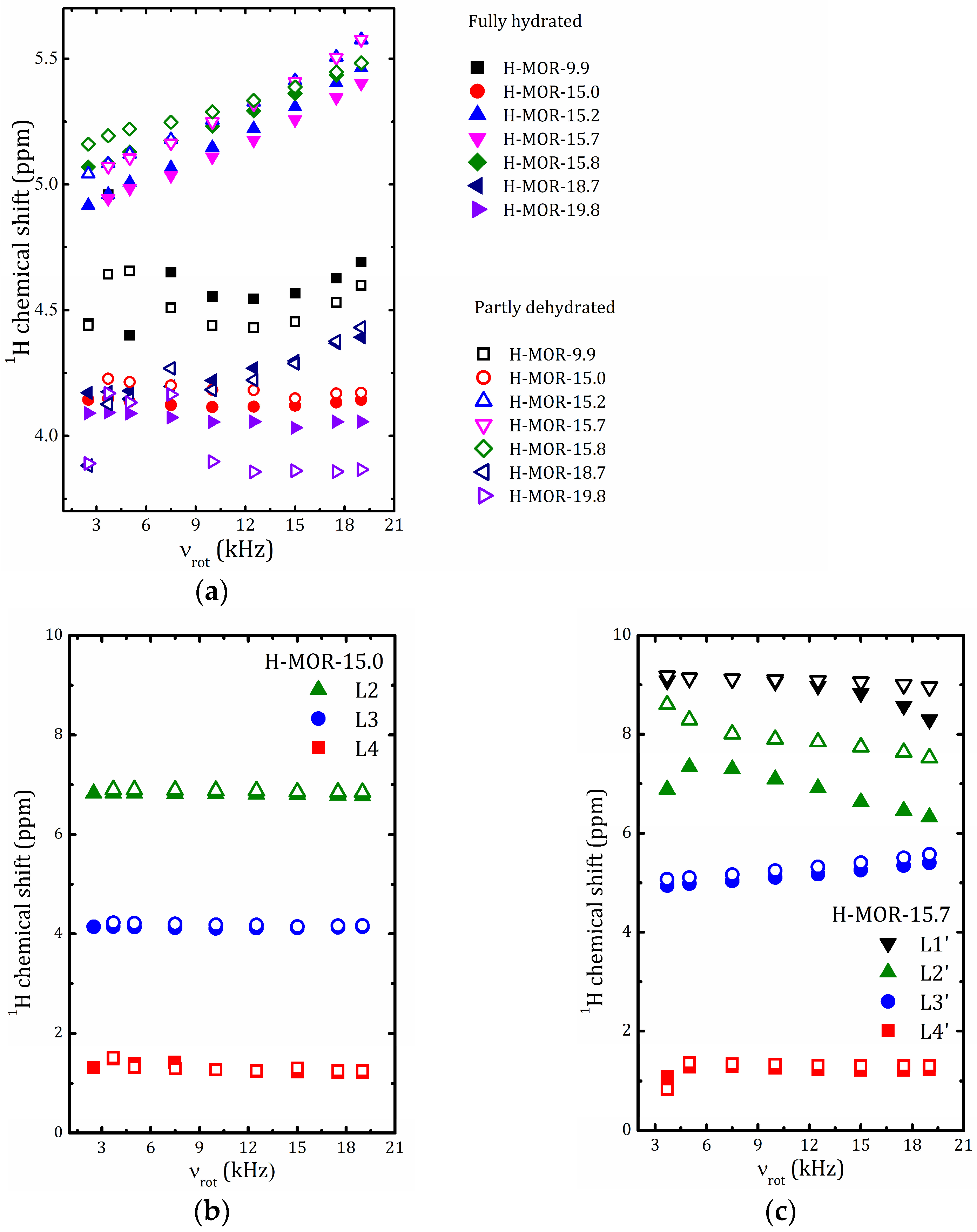
2.5.2. 1H MAS NMR
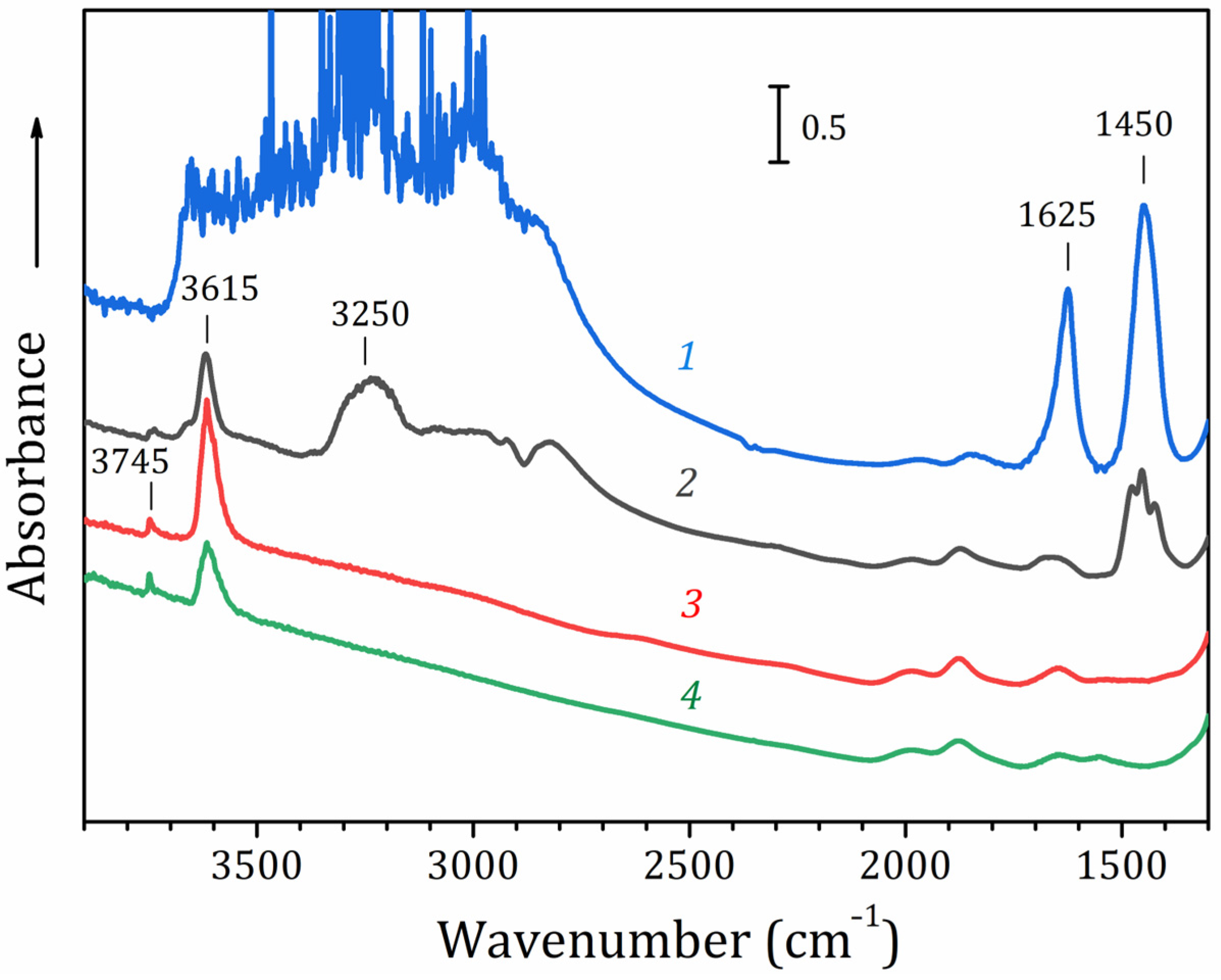
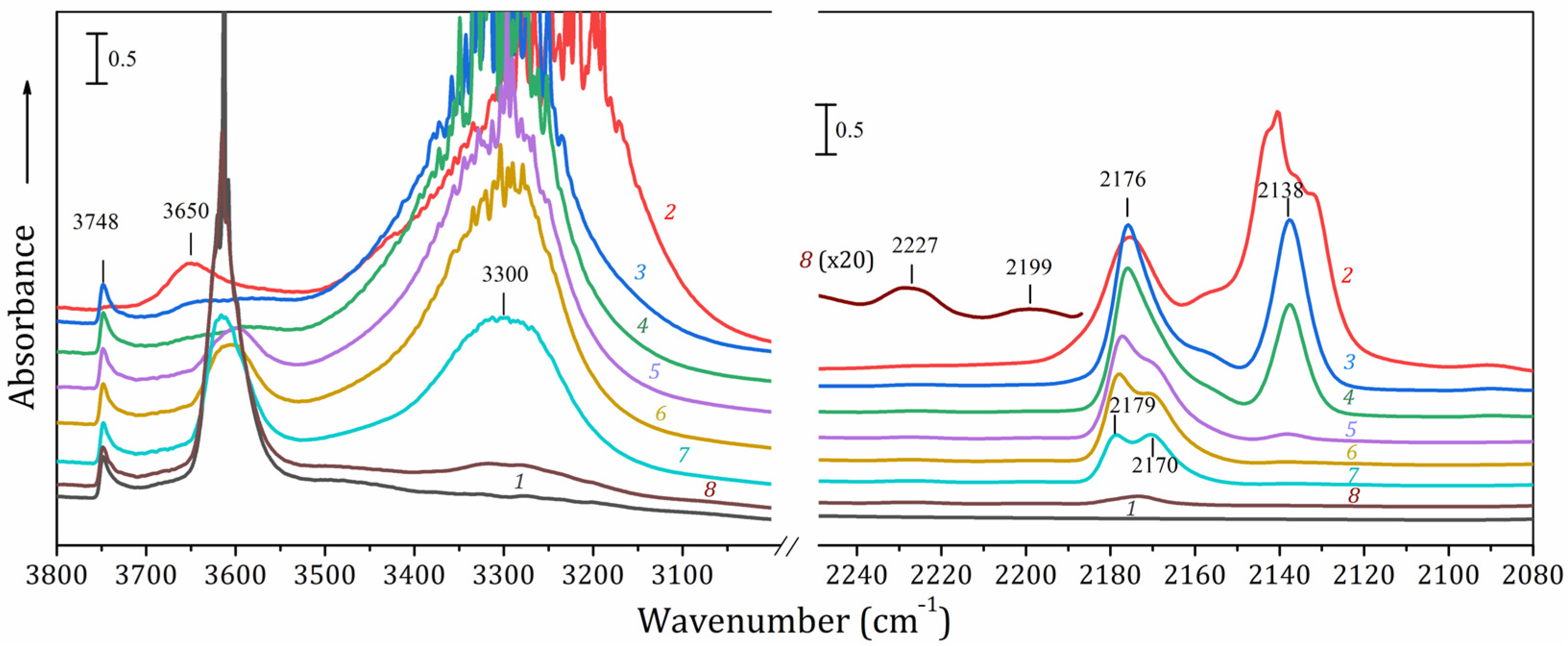
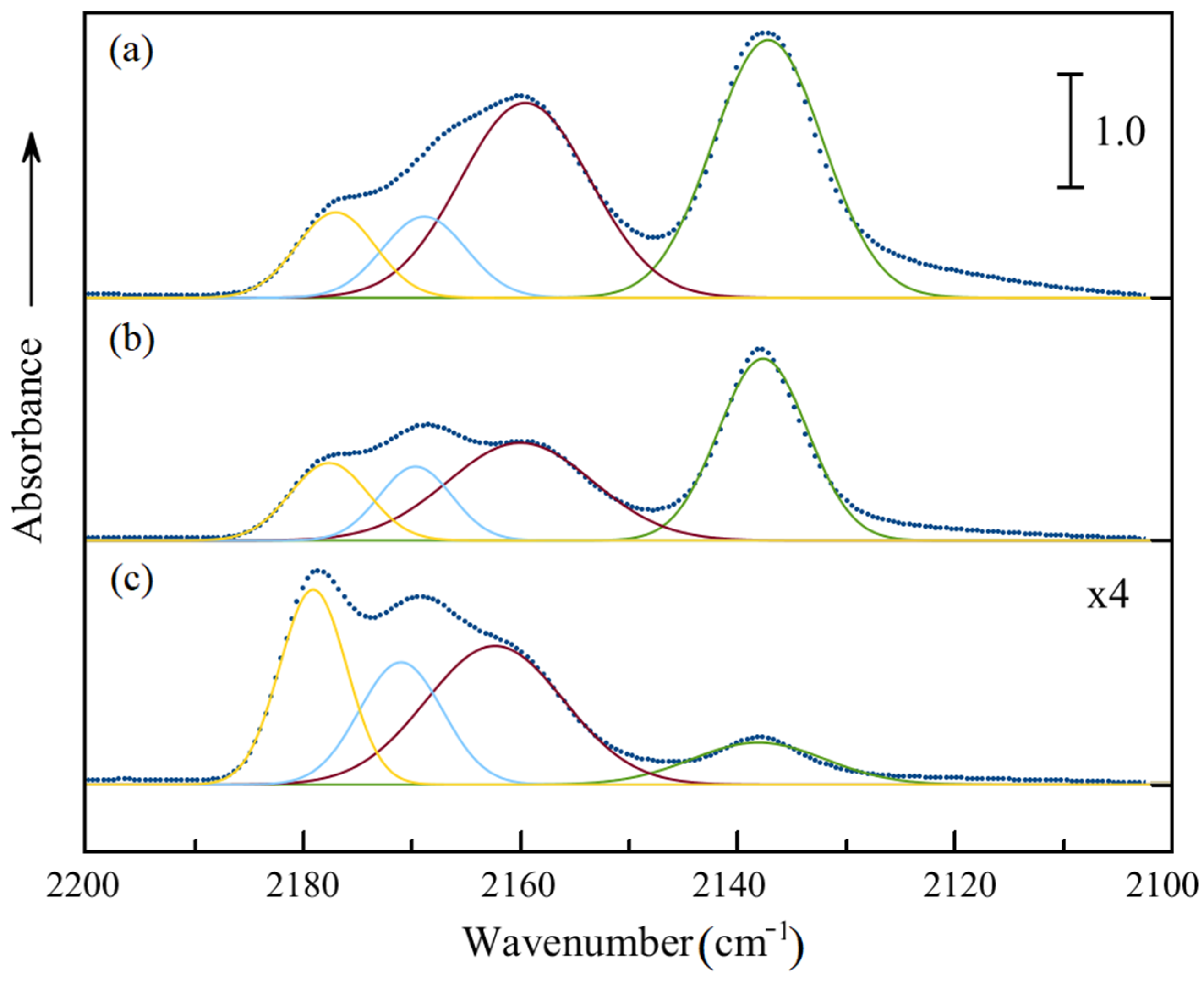
2.6. FTIR Spectroscopy Study
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Breck, D.W. Zeolite Molecular Sieves: Structure, Chemistry, and Use; John Wiley and Sons: New York, NY, USA; London, UK; Syndey, Australia; Toronto, ON, Canada, 1974; ISBN 0471099856. [Google Scholar]
- Baerlocher, C.; McCusker, L.B. Database of Zeolite Structures. Available online: http://www.iza-structure.org/databases/ (accessed on 11 November 2022).
- Van Speybroeck, V.; Hemelsoet, K.; Joos, L.; Waroquier, M.; Bell, R.G.; Catlow, C.R.A. Advances in theory and their application within the field of zeolite chemistry. Chem. Soc. Rev. 2015, 44, 7044–7111. [Google Scholar] [CrossRef]
- Rodríguez-Iznaga, I.; Shelyapina, M.G.; Petranovskii, V. Ion Exchange in Natural Clinoptilolite: Aspects Related to Its Structure and Applications. Minerals 2022, 12, 1628. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, N.; Yu, J. Advances in Catalytic Applications of Zeolite-Supported Metal Catalysts. Adv. Mater. 2021, 33, 2104442. [Google Scholar] [CrossRef]
- Sánchez-López, P.; Kotolevich, Y.; Yocupicio-Gaxiola, R.I.; Antúnez-García, J.; Chowdari, R.K.; Petranovskii, V.; Fuentes-Moyado, S. Recent Advances in Catalysis Based on Transition Metals Supported on Zeolites. Front. Chem. 2021, 9, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Tahraoui, Z.; Nouali, H.; Marichal, C.; Forler, P.; Klein, J.; Daou, T.J. Influence of the Compensating Cation Nature on the Water Adsorption Properties of Zeolites. Molecules 2020, 25, 944. [Google Scholar] [CrossRef] [PubMed]
- Antúnez-García, J.; Galván, D.; Petranovskii, V.; Murrieta-Rico, F.N.; Yocupicio-Gaxiola, R.I.; Shelyapina, M.G.; Fuentes-Moyado, S. The effect of chemical composition on the properties of LTA zeolite: A theoretical study. Comput. Mater. Sci. 2021, 196, 110557. [Google Scholar] [CrossRef]
- Valtchev, V.; Majano, G.; Mintova, S.; Pérez-Ramírez, J. Tailored crystalline microporous materials by post-synthesis modification. Chem. Soc. Rev. 2012, 42, 263–290. [Google Scholar] [CrossRef] [PubMed]
- Wheatley, P.S.; Chlubná-Eliášová, P.; Greer, H.; Zhou, W.; Seymour, V.R.; Dawson, D.M.; Ashbrook, S.E.; Pinar, A.B.; McCusker, L.B.; Opanasenko, M.; et al. Zeolites with Continuously Tuneable Porosity. Angew. Chem. Int. Ed. 2014, 53, 13210–13214. [Google Scholar] [CrossRef]
- Zvereva, I.A.; Shelyapina, M.G.; Chislov, M.; Novakowski, V.; Malygina, E.; Rodríguez-Iznaga, I.; Hernández, M.-A.; Petranovskii, V. A comparative analysis of natural zeolites from various Cuban and Mexican deposits: Structure, composition, thermal properties and hierarchical porosity. J. Therm. Anal. Calorim. 2021, 147, 6147–6159. [Google Scholar] [CrossRef]
- Barthomeuf, D. Zeolite acidity dependence on structure and chemical environment. Correlations with catalysis. Mater. Chem. Phys. 1987, 17, 49–71. [Google Scholar] [CrossRef]
- Mihályi, R.; Lázár, K.; Kollár, M.; Lónyi, F.; Pál-Borbély, G.; Szegedi, Á. Structure, acidity and redox properties of MCM-22 ferrisilicate. Microporous Mesoporous Mater. 2008, 110, 51–63. [Google Scholar] [CrossRef]
- Catizzone, E.; Migliori, M.; Mineva, T.; van Daele, S.; Valtchev, V.; Giordano, G. New synthesis routes and catalytic applications of ferrierite crystals. Part 2: The effect of OSDA type on zeolite properties and catalysis. Microporous Mesoporous Mater. 2020, 296, 109988. [Google Scholar] [CrossRef]
- Derbe, T.; Temesgen, S.; Bitew, M. A Short Review on Synthesis, Characterization, and Applications of Zeolites. Adv. Mater. Sci. Eng. 2021, 2021, 6637898. [Google Scholar] [CrossRef]
- Xu, H.; Wu, P. New progress in zeolite synthesis and catalysis. Natl. Sci. Rev. 2022, 9, nwac045. [Google Scholar] [CrossRef]
- Sharma, V.; Javed, B.; Byrne, H.; Curtin, J.; Tian, F. Zeolites as Carriers of Nano-Fertilizers: From Structures and Principles to Prospects and Challenges. Appl. Nano 2022, 3, 163–186. [Google Scholar] [CrossRef]
- Ivankovic, T.; Dikic, J.; du Roscoat, S.R.; Dekic, S.; Hrenovic, J.; Ganjto, M. Removal of emerging pathogenic bacteria using metal-exchanged natural zeolite bead filter. Water Sci. Technol. 2019, 80, 1085–1098. [Google Scholar] [CrossRef]
- Nizam, M.K.; Azhari, S.; Jaya, M.A.T. Modified Zeolite as Purification Material in Wastewater Treatment: A Review. Sci. Res. J. 2021, 18, 177–213. [Google Scholar] [CrossRef]
- Vogt, E.T.; Whiting, G.T.; Chowdhury, A.D.; Weckhuysen, B.M. Zeolites and Zeotypes for Oil and Gas Conversion. In Advances in Catalysis; Academic Press: Cambridge, MA, USA, 2015; Volume 58, pp. 143–314. [Google Scholar] [CrossRef]
- Weitkamp, J.; Puppe, L. (Eds.) Catalysis and Zeolites: Fundamentals and Applications; Springer: Berlin/Heidelberg, Germany, 1999. [Google Scholar]
- Weissenberger, T.; Machoke, A.G.F.; Reiprich, B.; Schwieger, W. Preparation and Potential Catalytic Applications of Hierarchically Structured Zeolites with Macropores. Adv. Mater. Interfaces 2021, 8, 2001653. [Google Scholar] [CrossRef]
- Petranovskii, V.; Marzke, R.; Diaz, G.; Gómez, A.; Bogdanchikova, N.; Fuentes, S.; Katada, N.; Pestryakov, A.; Gurin, V. Characterization of H and Cu mordenites with varying SiO2/Al2O3 ratios, by optical spectroscopy, MAS NMR of 29Si, 27Al and 1H, temperature programmed desorption and catalytic activity for nitrogen oxide reduction. Stud. Surf. Sci. Catal. 2002, 142, 815–822. [Google Scholar] [CrossRef]
- Wang, M.; Huang, S.; Lü, J.; Cheng, Z.; Li, Y.; Wang, S.; Ma, X. Modifying the acidity of H-MOR and its catalytic carbonylation of dimethyl ether. Chin. J. Catal. 2016, 37, 1530–1537. [Google Scholar] [CrossRef]
- Lu, K.; Huang, J.; Ren, L.; Li, C.; Guan, Y.; Hu, B.; Xu, H.; Jiang, J.; Ma, Y.; Wu, P. High Ethylene Selectivity in Methanol-to-Olefin (MTO) Reaction over MOR-Zeolite Nanosheets. Angew. Chem. Int. Ed. 2020, 59, 6258–6262. [Google Scholar] [CrossRef] [PubMed]
- Hancsók, J.; Kasza, T.; Visnyei, O. Isomerization of n-C5/C6 Bioparaffins to Gasoline Components with High Octane Number. Energies 2020, 13, 1672. [Google Scholar] [CrossRef]
- Bateni, H.; Able, C. Development of Heterogeneous Catalysts for Dehydration of Methanol to Dimethyl Ether: A Review. Catal. Ind. 2019, 11, 7–33. [Google Scholar] [CrossRef]
- Sánchez-López, P.; Kotolevich, Y.; Khramov, E.; Chowdari, R.K.; Estrada, M.A.; Berlier, G.; Zubavichus, Y.; Fuentes, S.; Petranovskii, V.; Chávez-Rivas, F. Properties of Iron-Modified-by-Silver Supported on Mordenite as Catalysts for NOx Reduction. Catalysts 2020, 10, 1156. [Google Scholar] [CrossRef]
- Campa, M.C.; Pietrogiacomi, D.; Occhiuzzi, M. The simultaneous selective catalytic reduction of N2O and NOX with CH4 on Co- and Ni-exchanged mordenite. Appl. Catal. B: Environ. 2015, 168–169, 293–302. [Google Scholar] [CrossRef]
- Sugi, Y. Shape-selective alkylation of biphenyl catalyzed by H-Mordenites. Korean J. Chem. Eng. 2000, 17, 1–11. [Google Scholar] [CrossRef]
- Sugi, Y.; Anand, C.; Subramaniam, V.P.; Stalin, J.; Choy, J.-H.; Cha, W.S.; Elzatahry, A.A.; Tamada, H.; Komura, K.; Vinu, A. The isopropylation of naphthalene with propene over H-mordenite: The catalysis at the internal and external acid sites. J. Mol. Catal. A: Chem. 2014, 395, 543–552. [Google Scholar] [CrossRef]
- Petranovskii, V.; Pestryakov, A.; Hernández, M.; Rivas, F.C.; Claverie, A.L.; Fuentes, S. Hydrophilicity of Mordenites with Different SiO2/Al2O3 Molar Ratio. Procedia Chem. 2015, 15, 72–78. [Google Scholar] [CrossRef]
- Xie, Z.; Liu, Z.; Wang, Y.; Yang, Q.; Xu, L.; Ding, W. An Overview of Recent Development in Composite Catalysts from Porous Materials for Various Reactions and Processes. Int. J. Mol. Sci. 2010, 11, 2152–2187. [Google Scholar] [CrossRef] [Green Version]
- Lukyanov, D.B.; Vazhnova, T.; Cherkasov, N.; Casci, J.L.; Birtill, J.J. Insights into Brønsted Acid Sites in the Zeolite Mordenite. J. Phys. Chem. C 2014, 118, 23918–23929. [Google Scholar] [CrossRef]
- Zhukov, Y.; Efimov, A.Y.; Shelyapina, M.; Petranovskii, V.; Zhizhin, E.; Burovikhina, A.; Zvereva, I. Effect of preparation method on the valence state and encirclement of copper exchange ions in mordenites. Microporous Mesoporous Mater. 2016, 224, 415–419. [Google Scholar] [CrossRef]
- Zhukov, Y.; Shelyapina, M.; Zvereva, I.; Efimov, A.; Petranovskii, V. Microwave assisted versus convention Cu2+ exchange in mordenite. Microporous Mesoporous Mater. 2018, 259, 220–228. [Google Scholar] [CrossRef]
- Shelyapina, M.; Gurgul, J.; Łątka, K.; Sánchez-López, P.; Bogdanov, D.; Kotolevich, Y.; Petranovskii, V.; Fuentes, S. Mechanism of formation of framework Fe3+ in bimetallic Ag-Fe mordenites—Effective catalytic centers for deNOx reaction. Microporous Mesoporous Mater. 2019, 299, 109841. [Google Scholar] [CrossRef]
- Kalvachev, Y.; Todorova, T.; Popov, C. Recent Progress in Synthesis and Application of Nanosized and Hierarchical Mordenite—A Short Review. Catalysts 2021, 11, 308. [Google Scholar] [CrossRef]
- Krylova, E.A.; Shelyapina, M.G.; Mazur, A.; Baranov, D.A.; Tsyganenko, A.A.; Petranovskii, V.P. Local Structure of Protonated Mordenites with SiO2/Al2O3 ≈ 15 Probed by Multinuclear NMR. J. Struct. Chem. 2022, 63, 930–943. [Google Scholar] [CrossRef]
- Zhukov, Y.M.; Shelyapina, M.G.; Efimov, A.Y.; Zhizhin, E.V.; Petranovskii, V. Recognition of depth composition profiles of copper-exchanged mordenites applying analytical methods with different depth vision. Mater. Chem. Phys. 2019, 236, 121787. [Google Scholar] [CrossRef]
- Vicanek, M. Electron transport processes in reflection electron energy loss spectroscopy (REELS) and X-ray photoelectron spectroscopy (XPS). Surf. Sci. 1999, 440, 1–40. [Google Scholar] [CrossRef]
- Goldstein, J.; Newbury, D.E.; Joy, D.C.; Lyman, C.E.; Echlin, P.; Lifshin, E.; Sawyer, L.; Michael, J.R. Scanning Electron Microscopy and X-ray Microanalysis, 3rd ed.; Springer: New York, NY, USA, 2003. [Google Scholar]
- Shelyapina, M.G.; Krylova, E.A.; Zhukov, Y.M.; Zvereva, I.A.; Rodriguez-Iznaga, I.; Petranovskii, V.; Fuentes-Moyado, S. Comprehensive Analysis of the Copper Exchange Implemented in Ammonia and Protonated Forms of Mordenite Using Microwave and Conventional Methods. Molecules 2019, 24, 4216. [Google Scholar] [CrossRef]
- Krylova, E.; Shelyapina, M.; Nowak, P.; Harańczyk, H.; Chislov, M.; Zvereva, I.; Privalov, A.; Becher, M.; Vogel, M.; Petranovskii, V. Mobility of water molecules in sodium- and copper-exchanged mordenites: Thermal analysis and 1H NMR study. Microporous Mesoporous Mater. 2018, 265, 132–142. [Google Scholar] [CrossRef]
- Zhukov, Y.; Kovalyov, A.N.; Kultaeva, A.Y.; Shelyapina, M.; Petranovskii, V. A comparative analysis of the protonated and copper exchanged mordenites with SiO2/Al2O3 molar ratio equal to 10. Int. J. Nanotechnol. 2016, 13, 136. [Google Scholar] [CrossRef]
- Chen, K.; Gan, Z.; Horstmeier, S.; White, J.L. Distribution of Aluminum Species in Zeolite Catalysts: 27Al NMR of Framework, Partially-Coordinated Framework, and Non-Framework Moieties. J. Am. Chem. Soc. 2021, 143, 6669–6680. [Google Scholar] [CrossRef] [PubMed]
- Shelyapina, M.; Kasperovich, V.; Wolfers, P. Electronic structure and electric-field-gradients distribution in Y3Al5O12: An ab initio study. J. Phys. Chem. Solids 2006, 67, 720–724. [Google Scholar] [CrossRef]
- Kolodziejski, W.; Barrie, P.J.; He, H.; Klinowski, J. Two-dimensional J-scaled 29Si NMR COSY of highly siliceous mordenite. J. Chem. Soc. Chem. Commun. 1991, 961–962. [Google Scholar] [CrossRef]
- Fyfe, C.A.; Feng, Y.; Grondey, H.; Kokotailo, G.T.; Gies, H. One- and two-dimensional high-resolution solid-state NMR studies of zeolite lattice structures. Chem. Rev. 1991, 91, 1525–1543. [Google Scholar] [CrossRef]
- Loewenstein, W. The distribution of aluminum in the tetrahedra of silicates and aluminates. Am. Mineral. 1954, 39, 92–96. [Google Scholar]
- Antúnez-García, J.; Galván, D.; Petranovskii, V.; Murrieta-Rico, F.N.; Yocupicio-Gaxiola, R.I.; Shelyapina, M.G.; Fuentes-Moyado, S. Aluminum distribution in mordenite-zeolite framework: A new outlook based on density functional theory calculations. J. Solid State Chem. 2021, 306, 122725. [Google Scholar] [CrossRef]
- Glaser, R.H.; Wilkes, G.L.; Bronnimann, C.E. Solid-state 29Si NMR of TEOS-based multifunctional sol-gel materials. J. Non-Cryst. Solids 1989, 113, 73–87. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Yocupicio-Gaxiola, R.I.; Zhelezniak, I.V.; Chislov, M.V.; Antúnez-García, J.; Murrieta-Rico, F.N.; Galván, D.H.; Petranovskii, V.; Fuentes-Moyado, S. Local Structures of Two-Dimensional Zeolites—Mordenite and ZSM-5—Probed by Multinuclear NMR. Molecules 2020, 25, 4678. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Silyukov, O.I.; Lushpinskaia, I.P.; Kurnosenko, S.A.; Mazur, A.S.; Shenderovich, I.G.; Zvereva, I.A. NMR Study of Intercalates and Grafted Organic Derivatives of H2La2Ti3O10. Molecules 2020, 25, 5229. [Google Scholar] [CrossRef]
- Xu, M.; Harris, K.D.; Thomas, J.M. In situ solid-state 1H NMR studies of hydration of the solid acid catalyst ZSM-5 in its ammonium form. Solid State Nucl. Magn. Reson. 2009, 35, 93–99. [Google Scholar] [CrossRef]
- Heeribout, L.; Dorémieux-Morin, C.; Nogier, J.-P.; Vincent, R.; Fraissard, J. Study of high-silica H-ZSM-5 acidity by 1H NMR techniques using water as base. Microporous Mesoporous Mater. 1998, 24, 101–112. [Google Scholar] [CrossRef]
- Huo, H.; Peng, L.; Grey, C.P. Low Temperature 1H MAS NMR Spectroscopy Studies of Proton Motion in Zeolite HZSM-5. J. Phys. Chem. C 2009, 113, 8211–8219. [Google Scholar] [CrossRef]
- Shelyapina, M.G.; Silyukov, O.I.; Andronova, E.A.; Nefedov, D.Y.; Antonenko, A.O.; Missyul, A.; Kurnosenko, S.A.; Zvereva, I.A. 1H NMR Study of the HCa2Nb3O10 Photocatalyst with Different Hydration Levels. Molecules 2021, 26, 5943. [Google Scholar] [CrossRef] [PubMed]
- Shelyapina, M.G.; Nefedov, D.Y.; Kostromin, A.V.; Silyukov, O.I.; Zvereva, I.A. Proton mobility in Ruddlesden–Popper phase H2La2Ti3O10 studied by 1H NMR. Ceram. Int. 2018, 45, 5788–5795. [Google Scholar] [CrossRef]
- Cattaneo, A.S.; Ferrara, C.; Marculescu, A.M.; Giannici, F.; Martorana, A.; Mustarelli, P.; Tealdi, C. Solid-state NMR characterization of the structure and thermal stability of hybrid organic–inorganic compounds based on a HLaNb2O7 Dion–Jacobson layered perovskite. Phys. Chem. Chem. Phys. 2016, 18, 21903–21912. [Google Scholar] [CrossRef]
- Essayem, N.; Tong, Y.Y.; Jobic, H.; Vedrine, J.C. Characterization of protonic sites in H3PW12O40 and Cs1.9H1.1PW12O40: A solid-state 1H, 2H, 31P MAS-NMR and inelastic neutron scattering study on. Appl. Catal. A Gen. 2000, 194–195, 109–122. [Google Scholar] [CrossRef]
- Hronský, V. Measurement of Sample Temperatures and Temperature Gradients in Magic-Angle Spinning Nmr. Acta Electrotech. et Inform. 2013, 13, 95–98. [Google Scholar] [CrossRef]
- Yesinowski, J.P.; Ladouceur, H.D.; Purdy, A.P.; Miller, J. Electrical and ionic conductivity effects on magic-angle spinning nuclear magnetic resonance parameters of CuI. J. Chem. Phys. 2010, 133, 234509. [Google Scholar] [CrossRef]
- Losch, P.; Joshi, H.; Stegmann, N.; Vozniuk, O.; Schmidt, W. Studying Proton Mobility in Zeolites by Varying Temperature Infrared Spectroscopy. Molecules 2019, 24, 3199. [Google Scholar] [CrossRef]
- Katsiotis, M.S.; Fardis, M.; Al Wahedi, Y.; Stephen, S.; Tzitzios, V.; Boukos, N.; Kim, H.J.; Alhassan, S.M.; Papavassiliou, G. Water Coordination, Proton Mobility, and Lewis Acidity in HY Nanozeolites: A High-Temperature 1H and 27Al NMR Study. J. Phys. Chem. C 2015, 119, 3428–3438. [Google Scholar] [CrossRef]
- Tsyganenko, A.A.; Kondratieva, E.V.; Yanko, V.S.; Storozhev, P.Y. FTIR study of CO adsorption on basic zeolites. J. Mater. Chem. 2006, 16, 2358–2363. [Google Scholar] [CrossRef]
- Morterra, C.; Magnacca, G. A case study: Surface chemistry and surface structure of catalytic aluminas, as studied by vibrational spectroscopy of adsorbed species. Catal. Today 1996, 27, 497–532. [Google Scholar] [CrossRef]
- Hadjiivanov, K.I.; Vayssilov, G.N. Characterization of oxide surfaces and zeolites by carbon monoxide as an IR probe molecule. Adv. Catal. 2002, 47, 307–511. [Google Scholar]
- Kondratieva, E.V.; Manoilova, O.V.; Tsyganenko, A.A. Integrated absorption coefficient of adsorbed CO. Kinet. Catal. 2008, 49, 451–456. [Google Scholar] [CrossRef]
- Bulanin, K.M.; Mikheleva, A.Y.; Shchepkin, D.N.; Rudakova, A.V. Determination of the Extinction Coefficient of Carbon Monoxide Adsorbed on Titanium Dioxide. Opt. Spectrosc. 2022, 130, 248–256. [Google Scholar] [CrossRef]
- Gabrienko, A.A.; Danilova, I.G.; Arzumanov, S.S.; Pirutko, L.V.; Freude, D.; Stepanov, A.G. Direct Measurement of Zeolite Brønsted Acidity by FTIR Spectroscopy: Solid-State 1H MAS NMR Approach for Reliable Determination of the Integrated Molar Absorption Coefficients. J. Phys. Chem. C 2018, 122, 25386–25395. [Google Scholar] [CrossRef]
- Henriques, C.; Marie, O.; Thibault-Starzyk, F.; Lavalley, J.-C. NO+ ions as IR probes for the location of OH groups and Na+ ions in main channels and side pockets of mordenite. Microporous Mesoporous Mater. 2001, 50, 167–171. [Google Scholar] [CrossRef]
- Marie, O.; Massiani, P.; Thibault-Starzyk, F. Infrared Evidence of a Third Brønsted Site in Mordenites. J. Phys. Chem. B 2004, 108, 5073–5081. [Google Scholar] [CrossRef]
- Tsyganenko, A.A. Variable Temperature IR Spectroscopy in the Studies of Oxide Catalysts. Top. Catal. 2013, 56, 905–913. [Google Scholar] [CrossRef]
- Levantovsky, A. MagicPlot 3.0.0; Magicplot Systems, LLC.: St. Petersburg, Russia, 2021; Available online: https://magicplot.com/ (accessed on 23 December 2022).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | XPS | EDS | ICP-OES | NMR (27Al) 1 | NMR (29Si) 1 |
|---|---|---|---|---|---|
| H-MOR-9.9 | 10.8 ± 0.3 | 9.3 ± 0.5 | 8.72 ± 0.09 | 9.9 ± 0.1 | 10.4 ± 0.2 |
| H-MOR-15.0 | 14.4 ± 0.2 | 14.3 ± 0.9 | 13.94 ± 0.14 | 14.3 ± 0.2 | 16.0 ± 0.3 |
| H-MOR-15.2 | 21.4 ± 0.4 | 14.4 ± 0.7 | 13.38 ± 0.14 | 18.1 ± 0.2 | 17.8 ± 1.0 |
| H-MOR-15.7 | 24.9 ± 0.2 | 15.1 ± 0.9 | 13.63 ± 0.16 | 18.2 ± 0.2 | 21.0 ± 1.0 |
| H-MOR-15.8 | 21.2 ± 0.3 | 14.5 ± 0.8 | 14.06 ± 0.14 | 18.2 ± 0.2 | 23.0 ± 1.0 |
| H-MOR-18.7 | 16.5 ± 0.4 | 17.6 ± 0.9 | 16.67 ± 0.17 | 18.5 ± 0.2 | 20.5 ± 0.8 |
| H-MOR-19.8 | 21.6 ± 0.4 | 19.3 ± 0.9 | 17.61 ± 0.18 | 18.2 ± 0.2 | 15.8 ± 0.5 |
| Sample | Lattice Parameters (Å) | Unit Cell Volume (Å3) | K = a/b | ||
|---|---|---|---|---|---|
| a | b | c | |||
| H-MOR-9.9 | 18.194 ± 0.003 | 20.425 ± 0.003 | 7.521 ± 0.001 | 2794.8 ± 0.8 | 0.8907 ± 0.3 |
| H-MOR-15.0 | 18.152 ± 0.002 | 20.368 ± 0.002 | 7.491 ± 0.001 | 2769.7 ± 0.5 | 0.8912 ± 0.2 |
| H-MOR-15.2 | 18.161 ± 0.002 | 20.292 ± 0.002 | 7.493 ± 0.001 | 2761.4 ± 0.4 | 0.8950 ± 0.2 |
| H-MOR-15.7 | 18.149 ± 0.002 | 20.267 ± 0.002 | 7.487 ± 0.001 | 2754.1 ± 0.3 | 0.8955 ± 0.2 |
| H-MOR-15.8 | 18.182 ± 0.004 | 20.245 ± 0.004 | 7.489 ± 0.002 | 2756.5 ± 0.9 | 0.8981 ± 0.4 |
| H-MOR-18.7 | 18.112 ± 0.002 | 20.327 ± 0.002 | 7.481 ± 0.001 | 2754.2 ± 0.4 | 0.8910 ± 0.2 |
| H-MOR-19.8 | 18.133 ± 0.002 | 20.346 ± 0.002 | 7.481 ± 0.001 | 2760.1 ± 0.4 | 0.8912 ± 0.2 |
| Site | Parameter | Sample | ||||||
|---|---|---|---|---|---|---|---|---|
| H-MOR-9.9 | H-MOR-15.0 | H-MOR-15.2 | H-MOR-15.7 | H-MOR-15.8 | H-MOR-18.7 | H-MOR-19.8 | ||
| Altetra-I | I (%) | 88 ± 1 | 97 ± 1 | 74 ± 1 | 75 ± 1 | 77 ± 1 | 90 ± 1 | 75 ± 1 |
| Altetra-II | I (%) | – | – | – | – | – | – | 22 ± 1 |
| Alocta | I (%) | 12 ± 1 | 3 ± 1 | 26 ± 1 | 23 ± 1 | 23 ± 1 | 10 ± 1 | 3 ± 1 |
| Q4(0Al) 1 | δiso (ppm) | – | – | – | – | – | −114.5 ± 0.1 | −113.4 ± 0.1 |
| I (%) | – | – | – | – | – | 16 ± 1 | 6 ± 1 | |
| Q4(0Al) | δiso (ppm) | −111.6 ± 0.1 | −112.2 ± 0.1 | −112.1 ± 0.1 | −112.1 ± 0.1 | −111.3 ± 0.1 | −111.6 ± 0.1 | −111.6 ± 0.1 |
| I (%) | 33 ± 2 | 52 ± 2 | 42 ± 2 | 46 ± 2 | 51 ± 2 | 45 ± 1 | 53 ± 1 | |
| Q4(1Al) | δiso (ppm) | – | – | – | – | – | −108.1 ± 0.1 | – |
| I (%) | – | – | – | – | – | 28 ± 1 | – | |
| Q4(1Al) | δiso (ppm) | −104.9 ± 0.1 | −105.5 ± 0.1 | −105.9 ± 0.1 | −105.8 ± 0.1 | −105.2 ± 0.1 | −104.6 ± 0.1 | −105.1 ± 0.1 |
| I (%) | 58 ± 2 | 45 ± 2 | 36 ± 2 | 38 ± 2 | 34 ± 2 | 11 ± 1 | 43 ± 1 | |
| Q4(2Al) | δiso (ppm) | −98.7 ± 0.1 | −98.7 ± 0.1 | – | – | – | – | −97.9 ± 0.1 |
| I (%) | 9 ± 3 | 3 ± 1 | – | – | – | – | 4 ± 1 | |
| Q4(3Al) | δiso (ppm) | – | – | −93.5 ± 0.1 | – | – | – | – |
| I (%) | – | – | 3 ± 1 | – | – | – | – | |
| Q3(0Al) | δiso (ppm) | – | – | −100.4 ± 0.1 | −100.1 ± 0.1 | −100.2 ± 0.1 | – | – |
| I (%) | – | – | 19 ± 1 | 16 ± 1 | 15 ± 2 | – | – | |
| Sample | L1 (L1′) | L2 (L2′) | L3 (L3′) | L4 (L4′) | ||||
|---|---|---|---|---|---|---|---|---|
| δiso (ppm) | I (%) | δiso (ppm) | I (%) | δiso (ppm) | I (%) | δiso (ppm) | I (%) | |
| Fully hydrated | ||||||||
| H-MOR-9.9 | 9.0 ± 0.1 | 7.0 ± 0.1 | 6.8 ± 0.1 | 33 ± 2 | 4.6 ± 0.1 | 54 ± 2 | 1.3 ± 0.2 | 6.0 ± 0.5 |
| H-MOR-15.0 | – | – | 6.8 ± 0.1 | 32 ± 2 | 4.1 ± 0.1 | 63 ± 2 | 1.2 ± 0.2 | 5.0 ± 0.5 |
| H-MOR-15.2 | 8.9 ± 0.1 | 5.0 ± 0.5 | 6.9 ± 0.1 | 13 ± 1 | 5.3 ± 0.1 | 80 ± 2 | 1.2 ± 0.2 | 2.0 ± 0.5 |
| H-MOR-15.7 | 8.9 ± 0.1 | 5.0 ± 0.5 | 6.6 ± 0.1 | 15 ± 1 | 5.3 ± 0.1 | 76 ± 2 | 1.2 ± 0.2 | 4.0 ± 0.5 |
| H-MOR-15.8 | 8.8 ± 0.1 | 6.0 ± 0.5 | 6.7 ± 0.1 | 14 ± 1 | 5.3 ± 0.1 | 78 ± 2 | 1.2 ± 0.2 | 2.0 ± 0.5 |
| H-MOR-18.7 | 8.8 ± 0.1 | 3.0 ± 0.5 | 6.8 ± 0.1 | 16 ± 1 | 4.3 ± 0.1 | 78 ± 2 | 1.3 ± 0.2 | 3.0 ± 0.5 |
| H-MOR-19.8 | – | – | 6.8 ± 0.1 | 33 ± 2 | 4.0 ± 0.1 | 65 ± 2 | 1.3 ± 0.2 | 2.0 ± 0.5 |
| Partly dehydrated | ||||||||
| H-MOR-9.9 | 9.1 ± 0.1 | 7.0 ± 0.1 | 6.8 ± 0.1 | 36 ± 2 | 4.4 ± 0.1 | 46 ± 2 | 1.7 ± 0.2 | 11 ± 1 |
| H-MOR-15.0 | – | – | 6.9 ± 0.1 | 28 ± 2 | 4.1 ± 0.1 | 66 ± 2 | 1.3 ± 0.2 | 6.0 ± 0.5 |
| H-MOR-15.2 | 9.1 ± 0.1 | 9.0 ± 0.5 | 7.6 ± 0.1 | 8 ± 1 | 5.4 ± 0.1 | 79 ± 2 | 1.1 ± 0.2 | 4.0 ± 0.5 |
| H-MOR-15.7 | 9.1 ± 0.1 | 8.0 ± 0.5 | 7.6 ± 0.1 | 7 ± 1 | 5.4 ± 0.1 | 76 ± 2 | 1.1 ± 0.2 | 9 ± 1 |
| H-MOR-15.8 | 8.6 ± 0.1 | 12 ± 1 | 6.4 ± 0.1 | 19 ± 1 | 5.4 ± 0.1 | 67 ± 2 | 1.3 ± 0.2 | 2.0 ± 0.5 |
| H-MOR-18.7 | 8.4 ± 0.1 | 7.0 ± 0.5 | 6.7 ± 0.1 | 22 ± 2 | 4.4 ± 0.1 | 43 ± 2 | 1.8 ± 0.2 | 27 ± 2 |
| H-MOR-19.8 | 8.7 ± 0.1 | 1.0 ± 0.5 | 6.8 ± 0.1 | 37 ± 2 | 4.0 ± 0.1 | 40 ± 2 | 1.7 ± 0.2 | 22 ± 2 |
| Type of Sites | Lewis Acid Sites | Brønsted Acid Sites | Al Content | Silanol Hydroxyls | |||||
|---|---|---|---|---|---|---|---|---|---|
| ν (cm−1) | 2230–2200 | 2180–2170 | 3620 | 3747 | |||||
| ε (cm/μmol) | 0.2 1 | 2.1 1 | 3.06 2 | 1.5 2 | |||||
| Sample | ICO (cm/mg) | NL (μmol/g) | ICO (cm/mg) | NB (μmol/g) | IOH (cm/mg) | NB (μmol/g) | NAl (μmol/g) | IOH (cm/mg) | NS (μmol/g) |
| H-MOR-9.9 | 0.017 ± 0.002 4 | 85 ± 9 | 2.3 ± 0.1 | 1100 ± 60 | 9.4 ± 0.2 | 3100 ± 500 | 2860 ± 30 | 0.44 ± 0.09 | 300 ± 60 |
| H-MOR-15.0 | 0.008 ± 0.001 | 40 ± 6 | 2.1 ± 0.1 | 1000 ± 50 | 7,0 ± 0.1 | 2300 ± 100 | 2070 ± 20 | 0.19 ± 0.04 | 120 ± 20 |
| H-MOR-15.2 | 0.07 ± 0.01 | 350 ± 40 | 1.9 ± 0.1 | 900 ± 50 | off scale | 1670 ± 20 | 2.0 ± 0.8 | 1300 ± 500 | |
| H-MOR-15.7 | 0.087 ± 0.009 | 430 ± 40 | off scale | off scale | 1660 ± 20 | 1.5 ± 0.8 | 1000 ± 600 | ||
| H-MOR-15.8 | 0.085 ± 0.008 | 420 ± 40 | 2.4 ± 0.1 | 1140 ± 60 | 6.7 ± 0.9 | 2200 ± 300 | 1660 ± 20 | 2.1 ± 0.3 | 1460 ± 200 |
| H-MOR-18.7 | 0.027 ± 0.004 3 | 130 ± 20 | 2.3 ± 0.1 | 1100 ± 60 | 3.7 ± 0.6 | 1200 ± 200 | 1640 ± 20 | 0.28 ± 0.04 | 190 ± 30 |
| H-MOR-19.8 | 0.014 ± 0.003 | 70 ± 20 | 2.4 ± 0.1 | 1140 ± 60 | 8.3 ± 0.7 | 2700 ± 200 | 1660 ± 20 | 0.32 ± 0.05 | 210 ± 30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shelyapina, M.G.; Krylova, E.A.; Mazur, A.S.; Tsyganenko, A.A.; Shergin, Y.V.; Satikova, E.A.; Petranovskii, V. Active Sites in H-Mordenite Catalysts Probed by NMR and FTIR. Catalysts 2023, 13, 344. https://doi.org/10.3390/catal13020344
Shelyapina MG, Krylova EA, Mazur AS, Tsyganenko AA, Shergin YV, Satikova EA, Petranovskii V. Active Sites in H-Mordenite Catalysts Probed by NMR and FTIR. Catalysts. 2023; 13(2):344. https://doi.org/10.3390/catal13020344
Chicago/Turabian StyleShelyapina, Marina G., Ekaterina A. Krylova, Anton S. Mazur, Alexey A. Tsyganenko, Yaroslav V. Shergin, Elizaveta A. Satikova, and Vitalii Petranovskii. 2023. "Active Sites in H-Mordenite Catalysts Probed by NMR and FTIR" Catalysts 13, no. 2: 344. https://doi.org/10.3390/catal13020344
APA StyleShelyapina, M. G., Krylova, E. A., Mazur, A. S., Tsyganenko, A. A., Shergin, Y. V., Satikova, E. A., & Petranovskii, V. (2023). Active Sites in H-Mordenite Catalysts Probed by NMR and FTIR. Catalysts, 13(2), 344. https://doi.org/10.3390/catal13020344