

The Influence of Active Phase Content on Properties and Activity of Nd2O3-Supported Cobalt Catalysts for Ammonia Synthesis
 ,
,  ,
,  , , , , and
, , , , and
Abstract
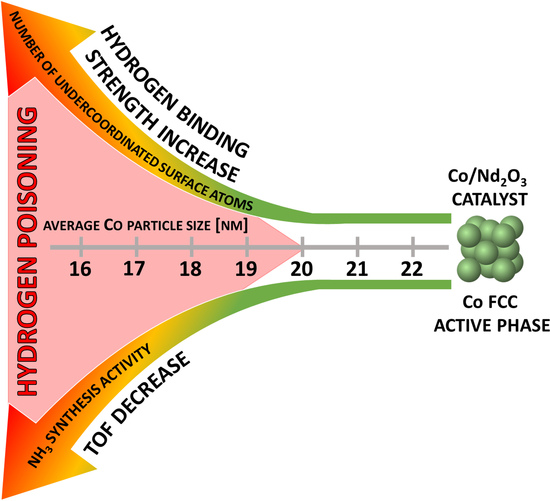
:
1. Introduction
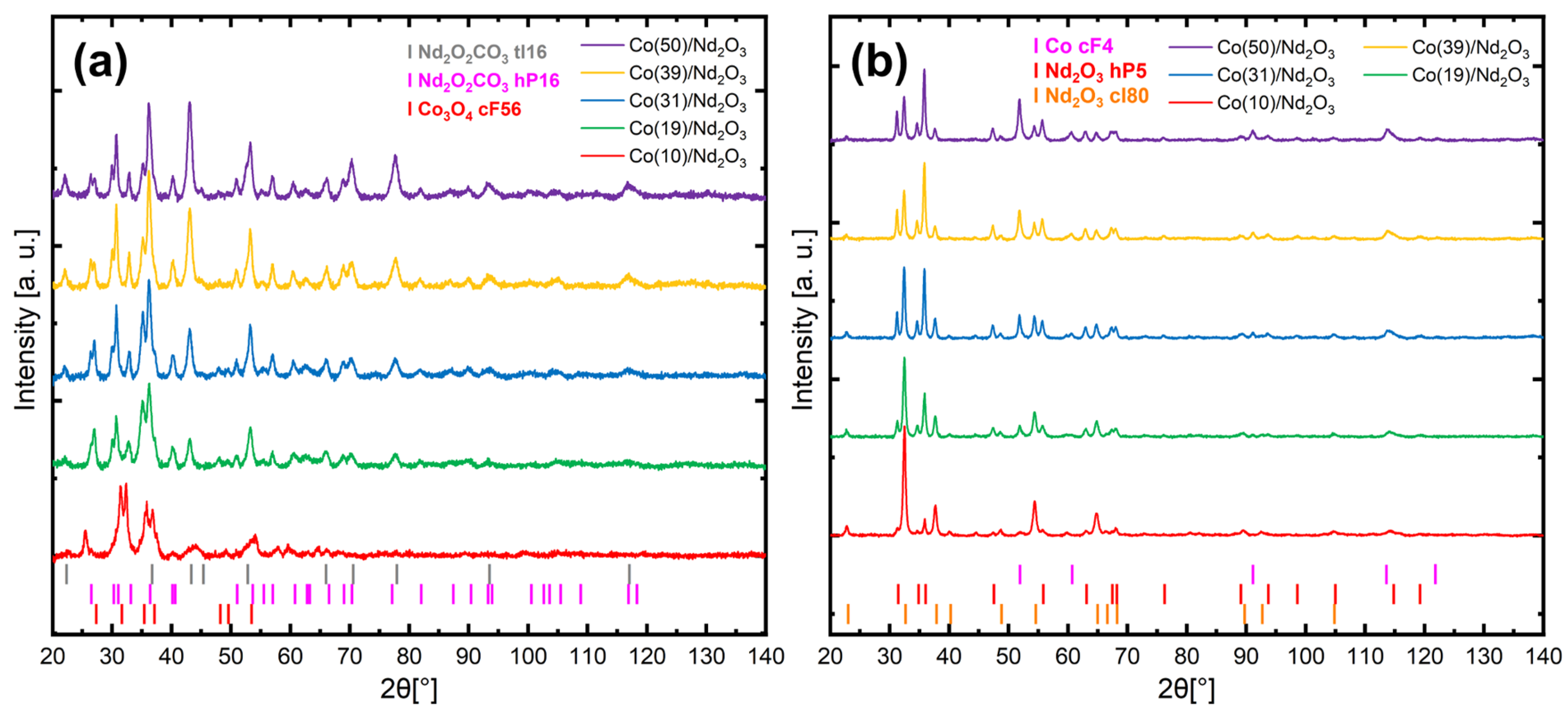
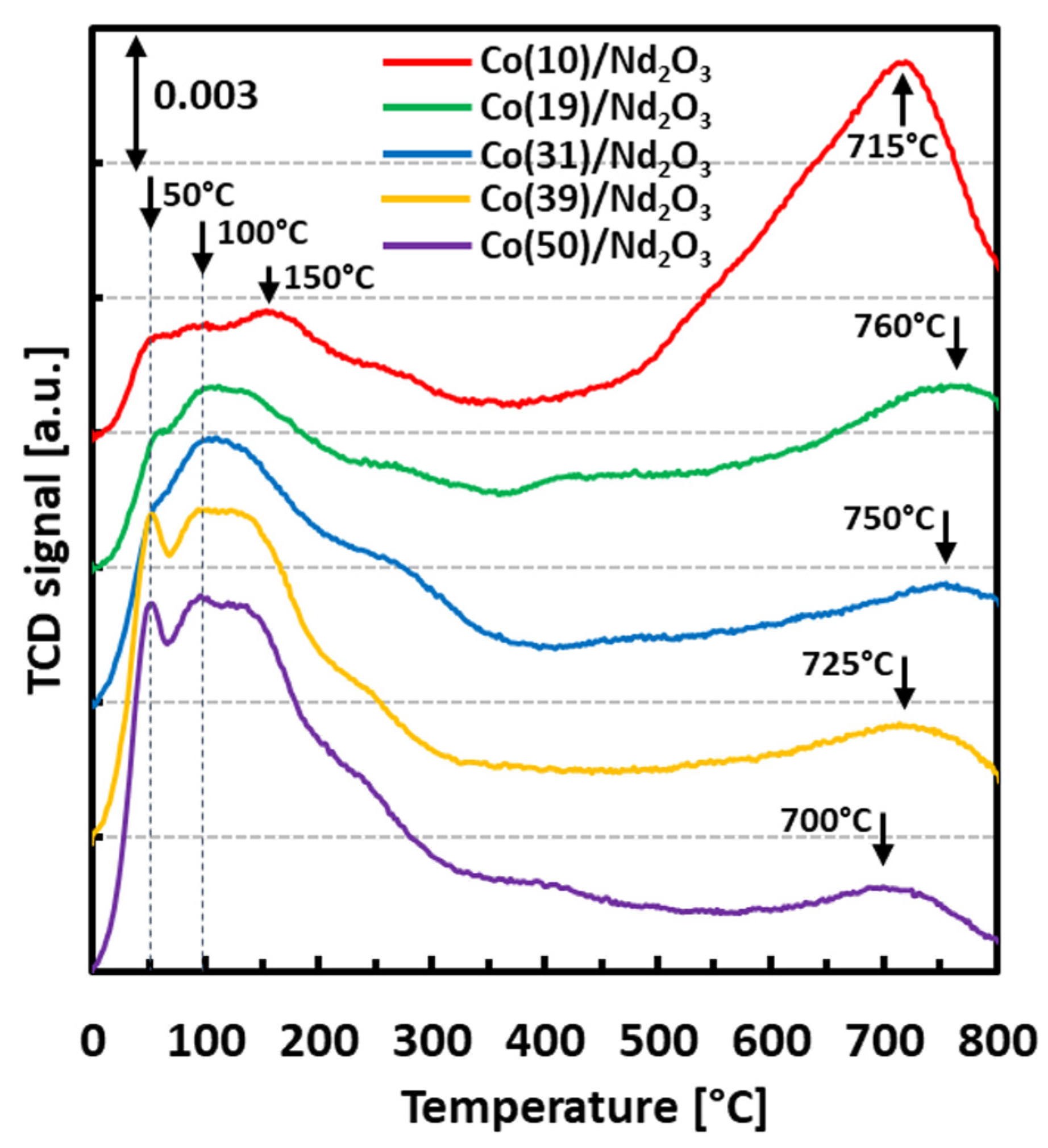
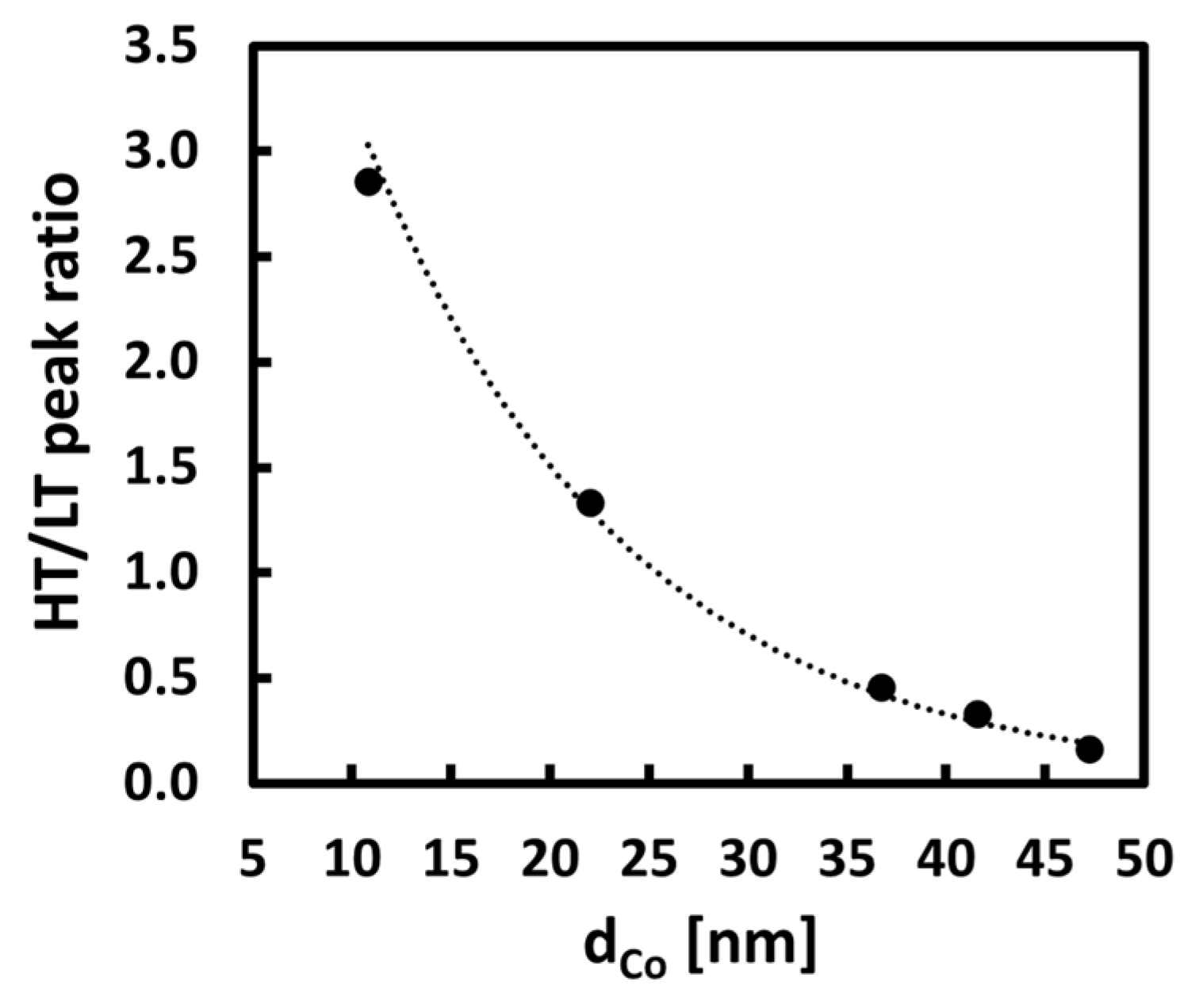
2. Results and Discussion
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Characterisation Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Smith, C.; Hill, A.K.; Torrente-Murciano, L. Current and future role of Haber–Bosch ammonia in a carbon-free energy landscape. Energy Environ. Sci. 2020, 13, 331–344. [Google Scholar] [CrossRef]
- Jennings, J.R. Catalytic Ammonia Synthesis—Fundamentals and Practice; Springer: New York, NY, USA, 2008. [Google Scholar]
- Liu, H. Ammonia Synthesis Catalysts; World Scientific: Singapore, 2013. [Google Scholar]
- Faria, J.A. Renaissance of ammonia synthesis for sustainable production of energy and fertilizers. Curr. Opin. Green Sustain. Chem. 2021, 29, 100466. [Google Scholar] [CrossRef]
- Nilsson, A.; Pettersson, L.G.M.; Hammer, B.; Bligaard, T.; Christensen, C.H.; Nørskov, J.K. The electronic structure effect in heterogeneous catalysis. Catal. Lett. 2005, 100, 111–114. [Google Scholar] [CrossRef]
- Hagen, S.; Barfod, R.; Fehrmann, R.; Jacobsen, C.J.H.H.; Teunissen, H.T.; Ståhl, K.; Chorkendorff, I. New efficient catalyst for ammonia synthesis: Barium-promoted cobalt on carbon. Chem. Commun. 2002, 11, 1206–1207. [Google Scholar] [CrossRef]
- Hagen, S.; Barfod, R.; Fehrmann, R.; Jacobsen, C.J.H.; Teunissen, H.T.; Chorkendorff, I. Ammonia synthesis with barium-promoted iron-cobalt alloys supported on carbon. J. Catal. 2003, 214, 327–335. [Google Scholar] [CrossRef]
- Raróg-Pilecka, W.; Miśkiewicz, E.; Kepiński, L.; Kaszkur, Z.; Kielar, K.; Kowalczyk, Z. Ammonia synthesis over barium-promoted cobalt catalysts supported on graphitised carbon. J. Catal. 2007, 249, 24–33. [Google Scholar] [CrossRef]
- Karolewska, M.; Truszkiewicz, E.; Wściseł, M.; Mierzwa, B.; Kȩpiński, L.; Raróg-Pilecka, W. Ammonia synthesis over a Ba and Ce-promoted carbon-supported cobalt catalyst. Effect of the cerium addition and preparation procedure. J. Catal. 2013, 303, 130–134. [Google Scholar] [CrossRef]
- Lin, B.; Qi, Y.; Wei, K.; Lin, J. Effect of pretreatment on ceria-supported cobalt catalyst for ammonia synthesis. RSC Adv. 2014, 4, 38093. [Google Scholar] [CrossRef]
- Lin, B.; Liu, Y.; Heng, L.; Ni, J.; Lin, J.; Jiang, L. Effect of ceria morphology on the catalytic activity of Co/CeO2 catalyst for ammonia synthesis. Catal. Commun. 2017, 101, 15–19. [Google Scholar] [CrossRef]
- Lin, B.; Liu, Y.; Heng, L.; Ni, J.; Lin, J.; Jiang, L. Effect of barium and potassium promoter on Co/CeO2 catalysts in ammonia synthesis. J. Rare Earths 2018, 36, 703–707. [Google Scholar] [CrossRef]
- Sato, K.; Miyahara, S.; Tsujimaru, K.; Wada, Y.; Toriyama, T.; Yamamoto, T.; Matsumura, S.; Inazu, K.; Mohri, H.; Iwasa, T.; et al. Barium Oxide Encapsulating Cobalt Nanoparticles Supported on Magnesium Oxide: Active Non-Noble Metal Catalysts for Ammonia Synthesis under Mild Reaction Conditions. ACS Catal. 2021, 11, 13050–13061. [Google Scholar] [CrossRef]
- Ronduda, H.; Zybert, M.; Patkowski, W.; Tarka, A.; Jodłowski, P.; Kępiński, L.; Sarnecki, A.; Moszyński, D.; Raróg-Pilecka, W. Tuning the catalytic performance of Co/Mg-La system for ammonia synthesis via the active phase precursor introduction method. Appl. Catal. A Gen. 2020, 598, 117553. [Google Scholar] [CrossRef]
- Ronduda, H.; Zybert, M.; Patkowski, W.; Tarka, A.; Ostrowski, A.; Raróg-Pilecka, W. Kinetic studies of ammonia synthesis over a barium-promoted cobalt catalyst supported on magnesium–lanthanum mixed oxide. J. Taiwan Inst. Chem. Eng. 2020, 114, 241–248. [Google Scholar] [CrossRef]
- Ronduda, H.; Zybert, M.; Patkowski, W.; Ostrowski, A.; Jodłowski, P.; Szymański, D.; Kępiński, L.; Raróg-Pilecka, W. Boosting the Catalytic Performance of Co/Mg/La Catalyst for Ammonia Synthesis by Selecting a Pre-Treatment Method. Catalysts 2021, 11, 941. [Google Scholar] [CrossRef]
- Ronduda, H.; Zybert, M.; Patkowski, W.; Ostrowski, A.; Jodłowski, P.; Szymański, D.; Kępiński, L.; Raróg-Pilecka, W. A high performance barium-promoted cobalt catalyst supported on magnesium–lanthanum mixed oxide for ammonia synthesis. RSC Adv. 2021, 11, 14218–14228. [Google Scholar] [CrossRef]
- Ronduda, H.; Zybert, M.; Patkowski, W.; Ostrowski, A.; Jodłowski, P.; Szymański, D.; Kępiński, L.; Raróg-Pilecka, W. Development of cobalt catalyst supported on MgO–Ln2O3 (Ln = La, Nd, Eu) mixed oxide systems for ammonia synthesis. Int. J. Hydrog. Energy 2022, 47, 6666–6678. [Google Scholar] [CrossRef]
- Inoue, Y.; Kitano, M.; Tokunari, M.; Taniguchi, T.; Ooya, K.; Abe, H.; Niwa, Y.; Sasase, M.; Hara, M.; Hosono, H. Direct Activation of Cobalt Catalyst by 12CaO7Al2O3 Electride for Ammonia Synthesis. ACS Catal. 2019, 9, 1670–1679. [Google Scholar] [CrossRef]
- Gao, W.; Wang, P.; Guo, J.; Chang, F.; He, T.; Wang, Q.; Wu, G.; Chen, P. Barium Hydride-Mediated Nitrogen Transfer and Hydrogenation for Ammonia Synthesis: A Case Study of Cobalt. ACS Catal. 2017, 7, 3654–3661. [Google Scholar] [CrossRef]
- Raróg-Pilecka, W.; Karolewska, M.; Truszkiewicz, E.E.; Iwanek, E.; Mierzwa, B. Cobalt Catalyst Doped with Cerium and Barium Obtained by Co-Precipitation Method for Ammonia Synthesis Process. Catal. Lett. 2011, 141, 678–684. [Google Scholar] [CrossRef]
- Tarka, A.; Patkowski, W.; Zybert, M.; Ronduda, H.; Wieciński, P.; Adamski, P.; Sarnecki, A.; Moszyński, D.; Raróg-Pilecka, W. Synergistic interaction of cerium and barium-new insight into the promotion effect in cobalt systems for ammonia synthesis. Catalysts 2020, 10, 658. [Google Scholar] [CrossRef]
- Zybert, M.; Wyszyńska, M.; Tarka, A.; Patkowski, W.; Ronduda, H.; Mierzwa, B.; Kępiński, L.; Sarnecki, A.; Moszyński, D.; Raróg-Pilecka, W. Surface enrichment phenomenon in the Ba-doped cobalt catalyst for ammonia synthesis. Vacuum 2019, 168, 108831. [Google Scholar] [CrossRef]
- Patkowski, W.; Kowalik, P.; Antoniak-Jurak, K.; Zybert, M.; Ronduda, H.; Mierzwa, B.; Próchniak, W.; Raróg-Pilecka, W. On the Effect of Flash Calcination Method on the Characteristics of Cobalt Catalysts for Ammonia Synthesis Process. Eur. J. Inorg. Chem. 2021, 2021, 1518–1529. [Google Scholar] [CrossRef]
- Karolewska, M.; Truszkiewicz, E.; Mierzwa, B.; Keopiński, L.; Raróg-Pilecka, W. Ammonia synthesis over cobalt catalysts doped with cerium and barium. Effect of the ceria loading. Appl. Catal. A Gen. 2012, 445–446, 280–286. [Google Scholar] [CrossRef]
- Zybert, M.; Tarka, A.; Mierzwa, B.; Kępiński, L.; Raróg-Pilecka, W. Promotion effect of lanthanum on the Co/La/Ba ammonia synthesis catalysts—The influence of lanthanum content. Appl. Catal. A Gen. 2016, 515, 16–24. [Google Scholar] [CrossRef]
- Zybert, M.; Tarka, A.; Patkowski, W.; Ronduda, H.; Mierzwa, B.; Kępiński, L.; Raróg-Pilecka, W. Structure Sensitivity of Ammonia Synthesis on Cobalt: Effect of the Cobalt Particle Size on the Activity of Promoted Cobalt Catalysts Supported on Carbon. Catalysts 2022, 12, 1285. [Google Scholar] [CrossRef]
- Ronduda, H.; Zybert, M.; Patkowski, W.; Sobczak, K.; Moszyński, D.; Albrecht, A.; Sarnecki, A.; Raróg-Pilecka, W. On the effect of metal loading on the performance of Co catalysts supported on mixed MgO–La2O3 oxides for ammonia synthesis. RSC Adv. 2022, 12, 33876–33888. [Google Scholar] [CrossRef]
- Niwa, Y.; Aika, K. The Effect of Lanthanide Oxides as a Support for Ruthenium Catalysts in Ammonia Synthesis. J. Catal. 1996, 162, 138–142. [Google Scholar] [CrossRef]
- Miyahara, S.I.; Sato, K.; Kawano, Y.; Imamura, K.; Ogura, Y.; Tsujimaru, K.; Nagaoka, K. Ammonia synthesis over lanthanoid oxide–supported ruthenium catalysts. Catal. Today 2020, 376, 2–5. [Google Scholar] [CrossRef]
- Sato, K.; Imamura, K.; Kawano, Y.; Miyahara, S.; Yamamoto, T.; Matsumura, S.; Nagaoka, K. A low-crystalline ruthenium nano-layer supported on praseodymium oxide as an active catalyst for ammonia synthesis. Chem. Sci. 2017, 8, 674–679. [Google Scholar] [CrossRef]
- Imamura, K.; Miyahara, S.I.; Kawano, Y.; Sato, K.; Nakasaka, Y.; Nagaoka, K. Kinetics of ammonia synthesis over Ru/Pr2O3. J. Taiwan Inst. Chem. Eng. 2019, 105, 50–56. [Google Scholar] [CrossRef]
- Bernal, S.; Botana, F.J.; García, R.; Rodríguez-Izquierdo, J.M. Behaviour of rare earth sesquioxides exposed to atmospheric carbon dioxide and water. React. Solids 1987, 4, 23–40. [Google Scholar] [CrossRef]
- Bernal, S.; Botana, F.J.; García, R.; Rodríguez-Izquierdo, J.M. Study of the interaction of two hexagonal neodymium oxides with atmospheric CO2 and H2O. J. Mater. Sci. 1988, 23, 1474–1480. [Google Scholar] [CrossRef]
- Turcotte, R.P.; Sawyer, J.O.; Eyring, L. Rare earth dioxymonocarbonates and their decomposition. Inorg. Chem. 1969, 8, 238–246. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Payne, B.P.; Grosvenor, A.P.; Lau, L.W.M.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. [Google Scholar] [CrossRef]
- Garces, L.J.; Hincapie, B.; Zerger, R.; Suib, S.L. The Effect of Temperature and Support on the Reduction of Cobalt Oxide: An in Situ X-ray Diffraction Study. J. Phys. Chem. C 2015, 119, 5484–5490. [Google Scholar] [CrossRef]
- Zybert, M.; Karasińska, M.; Truszkiewicz, E.; Mierzwa, B.; Raróg-Pilecka, W. Properties and activity of the cobalt catalysts for NH 3 synthesis obtained by co-precipitation—the effect of lanthanum addition. Pol. J. Chem. Technol. 2015, 17, 138–143. [Google Scholar] [CrossRef]
- Ciufo, R.A.; Han, S.; Floto, M.E.; Eichler, J.E.; Henkelman, G.; Mullins, C.B. Hydrogen desorption from the surface and subsurface of cobalt. Phys. Chem. Chem. Phys. 2020, 22, 15281–15287. [Google Scholar] [CrossRef]
- Huesges, Z.; Christmann, K. Interaction of Hydrogen with a Cobalt(0001) Surface. Z. Für Phys. Chem. 2013, 227, 881–899. [Google Scholar] [CrossRef]
- Kitakami, O.; Sato, H.; Shimada, Y.; Sato, F.; Tanaka, M. Size effect on the crystal phase of cobalt fine particles. Phys. Rev. B 1997, 56, 13849–13854. [Google Scholar] [CrossRef]
- Liu, J.X.; Li, W.X. Theoretical study of crystal phase effect in heterogeneous catalysis. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 571–583. [Google Scholar] [CrossRef]
- Freels, M.; Liaw, P.K.; Jiang, L.; Klarstrom, D.L. Advanced Structural Materials: Properties, Design Optimization, and Applications. In Advanced Structural Materials: Properties, Design Optimization, and Applications; Soboyejo, W.O., Srivatsan, T.S., Eds.; CRC Press: Boca Raton, FL, USA, 2007; pp. 187–224. [Google Scholar]
- Weststrate, C.J.; Mahmoodinia, M.; Farstad, M.H.; Svenum, I.-H.; Strømsheim, M.D.; Niemantsverdriet, J.W.; Venvik, H.J. Interaction of hydrogen with flat (0001) and corrugated (11–20) and (10–12) cobalt surfaces: Insights from experiment and theory. Catal. Today 2020, 342, 124–130. [Google Scholar] [CrossRef]
- Weststrate, C.J.; Garcia Rodriguez, D.; Sharma, D.; Niemantsverdriet, J.W. Structure-dependent adsorption and desorption of hydrogen on FCC and HCP cobalt surfaces. J. Catal. 2022, 405, 303–312. [Google Scholar] [CrossRef]
- Fernández, C.; Bion, N.; Gaigneaux, E.M.; Duprez, D.; Ruiz, P. Kinetics of hydrogen adsorption and mobility on Ru nanoparticles supported on alumina: Effects on the catalytic mechanism of ammonia synthesis. J. Catal. 2016, 344, 16–28. [Google Scholar] [CrossRef]
- Kojima, R.; Aika, K. Cobalt molybdenum bimetallic nitride catalysts for ammonia synthesis. Appl. Catal. A Gen. 2001, 218, 121–128. [Google Scholar] [CrossRef]
- Rosowski, F.; Hornung, A.; Hinrichsen, O.; Herein, D.; Muhler, M.; Ertl, G. Ruthenium catalysts for ammonia synthesis at high pressures: Preparation, characterization, and power-law kinetics. Appl. Catal. A Gen. 1997, 151, 443–460. [Google Scholar] [CrossRef]
- Rivera Rocabado, D.S.; Aizawa, M.; Noguchi, T.G.; Yamauchi, M.; Ishimoto, T. Uncovering the Mechanism of the Hydrogen Poisoning on Ru Nanoparticles via Density Functional Theory Calculations. Catalysts 2022, 12, 331. [Google Scholar] [CrossRef]
- Boudart, M. Kinetics and mechanism of ammonia synthesis. Catal. Rev. Eng. 1981, 23, 1–15. [Google Scholar] [CrossRef]
- Ertl, G.; Lee, S.B.; Weiss, M. Adsorption of nitrogen on potassium promoted Fe(111) and (100) surfaces. Surf. Sci. 1982, 114, 527–545. [Google Scholar] [CrossRef]
- Bozso, F.; Ertl, G.; Grunze, M.; Weiss, M. Chemisorption of hydrogen on iron surfaces. Appl. Surf. Sci. 1977, 1, 103–119. [Google Scholar] [CrossRef]
- Ertl, G. Surface Science and Catalysis—Studies on the Mechanism of Ammonia Synthesis: The P. H. Emmett Award Address. Catal. Rev. 1980, 21, 201–223. [Google Scholar] [CrossRef]
- Rosynek, M.P. Catalytic Properties of Rare Earth Oxides. Catal. Rev.—Sci. Eng. 1977, 16, 111–154. [Google Scholar] [CrossRef]
- Alvero, R.; Odriozola, J.A.; Trillo, J.M.; Bernal, S. Lanthanide oxides: Preparation and ageing. J. Chem. Soc. Dalt. Trans. 1984, 2, 87. [Google Scholar] [CrossRef]
- Bernal, S.; Blanco, G.; Calvino, J.J.; Omil, J.A.P.; Pintado, J.M. Some major aspects of the chemical behavior of rare earth oxides: An overview. J. Alloy. Compd. 2006, 408–412, 496–502. [Google Scholar] [CrossRef]
- Reuel, R.C.; Bartholomew, C.H. The stoichiometries of H2 and CO adsorptions on cobalt: Effects of support and preparation. J. Catal. 1984, 85, 63–77. [Google Scholar] [CrossRef]
- Borodziński, A.; Bonarowska, M. Relation between Crystallite Size and Dispersion on Supported Metal Catalysts. Langmuir 1997, 13, 5613–5620. [Google Scholar] [CrossRef]
- Kowalczyk, Z. Effect of potassium on the high pressure kinetics of ammonia synthesis over fused iron catalyst. Catal. Lett. 1996, 37, 173–179. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Co(10)/Nd2O3 | Co(19)/Nd2O3 | Co(31)/Nd2O3 | Co(39)/Nd2O3 | Co(50)/Nd2O3 |
|---|---|---|---|---|---|
| Specific surface area SBET 1 [m2 g−1] | 27.3 | 32.0 | 36.5 | 39.6 | 41.8 |
| Total pore volume Vpor 2 [cm3 g−1] | 0.104 | 0.125 | 0.132 | 0.139 | 0.142 |
| Parameter | Co(10)/Nd2O3 | Co(19)/Nd2O3 | Co(31)/Nd2O3 | Co(39)/Nd2O3 | Co(50)/Nd2O3 |
|---|---|---|---|---|---|
| Total hydrogen volume [cm3 g−1] | 1.99 | 1.82 | 1.68 | 1.82 | 2.02 |
| Low-temperature peak (LT) share [%] | 26 | 43 | 68 | 75 | 86 |
| High-temperature peak (HT) share [%] | 74 | 57 | 32 | 25 | 14 |
| Parameter | Co(10)/Nd2O3 | Co(19)/Nd2O3 | Co(31)/Nd2O3 | Co(39)/Nd2O3 | Co(50)/Nd2O3 |
|---|---|---|---|---|---|
| dH2-TPD [nm] | 11 | 22 | 37 | 42 | 47 |
| dSTEM [nm] | 27 | - | 36 | - | 51 |
| dXRPD [nm] | 17 | 21 | 21 | 20 | 19 |
| Parameter | Co(10)/Nd2O3 | Co(19)/Nd2O3 | Co(31)/Nd2O3 | Co(39)/Nd2O3 | Co(50)/Nd2O3 |
|---|---|---|---|---|---|
| ravg [gNH3 gcat−1 h−1] | 1.01 | 1.39 | 1.27 | 1.42 | 1.82 |
| Parameter | Co(10)/Nd2O3 | Co(19)/Nd2O3 | Co(31)/Nd2O3 | Co(39)/Nd2O3 | Co(50)/Nd2O3 |
|---|---|---|---|---|---|
| Co content [wt.%] | 9.9 | 19.4 | 30.8 | 38.6 | 49.6 |
| Nd content [wt.%] | 77.2 | 65.6 | 59.6 | 46.8 | 39.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patkowski, W.; Zybert, M.; Ronduda, H.; Gawrońska, G.; Albrecht, A.; Moszyński, D.; Fidler, A.; Dłużewski, P.; Raróg-Pilecka, W. The Influence of Active Phase Content on Properties and Activity of Nd2O3-Supported Cobalt Catalysts for Ammonia Synthesis. Catalysts 2023, 13, 405. https://doi.org/10.3390/catal13020405
Patkowski W, Zybert M, Ronduda H, Gawrońska G, Albrecht A, Moszyński D, Fidler A, Dłużewski P, Raróg-Pilecka W. The Influence of Active Phase Content on Properties and Activity of Nd2O3-Supported Cobalt Catalysts for Ammonia Synthesis. Catalysts. 2023; 13(2):405. https://doi.org/10.3390/catal13020405
Chicago/Turabian StylePatkowski, Wojciech, Magdalena Zybert, Hubert Ronduda, Gabriela Gawrońska, Aleksander Albrecht, Dariusz Moszyński, Aleksandra Fidler, Piotr Dłużewski, and Wioletta Raróg-Pilecka. 2023. "The Influence of Active Phase Content on Properties and Activity of Nd2O3-Supported Cobalt Catalysts for Ammonia Synthesis" Catalysts 13, no. 2: 405. https://doi.org/10.3390/catal13020405
APA StylePatkowski, W., Zybert, M., Ronduda, H., Gawrońska, G., Albrecht, A., Moszyński, D., Fidler, A., Dłużewski, P., & Raróg-Pilecka, W. (2023). The Influence of Active Phase Content on Properties and Activity of Nd2O3-Supported Cobalt Catalysts for Ammonia Synthesis. Catalysts, 13(2), 405. https://doi.org/10.3390/catal13020405