
Thermal and Plasma-Assisted CO2 Methanation over Ru/Zeolite: A Mechanistic Study Using In-Situ Operando FTIR


 ,
,  and
and
Abstract
:1. Introduction
2. Results
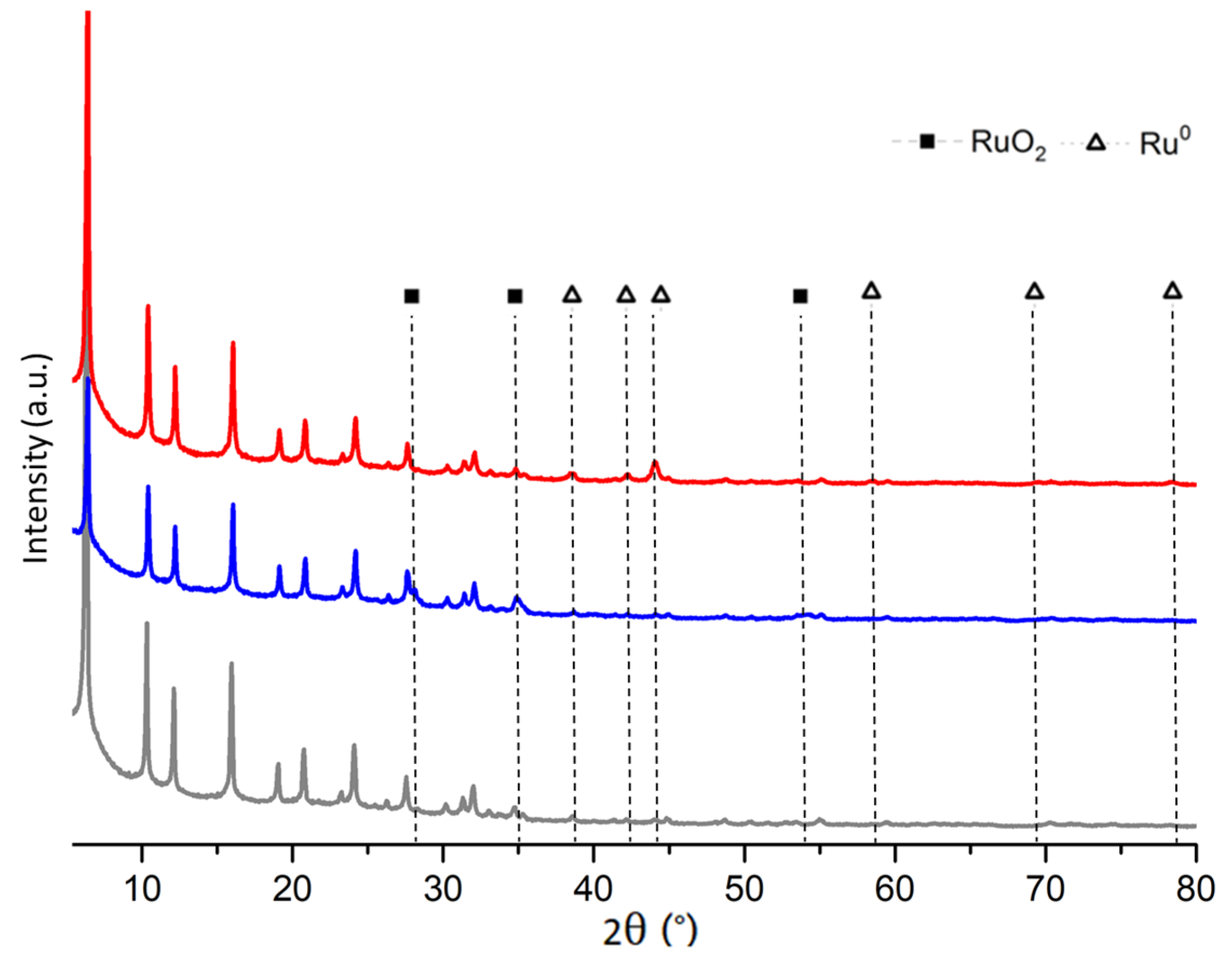
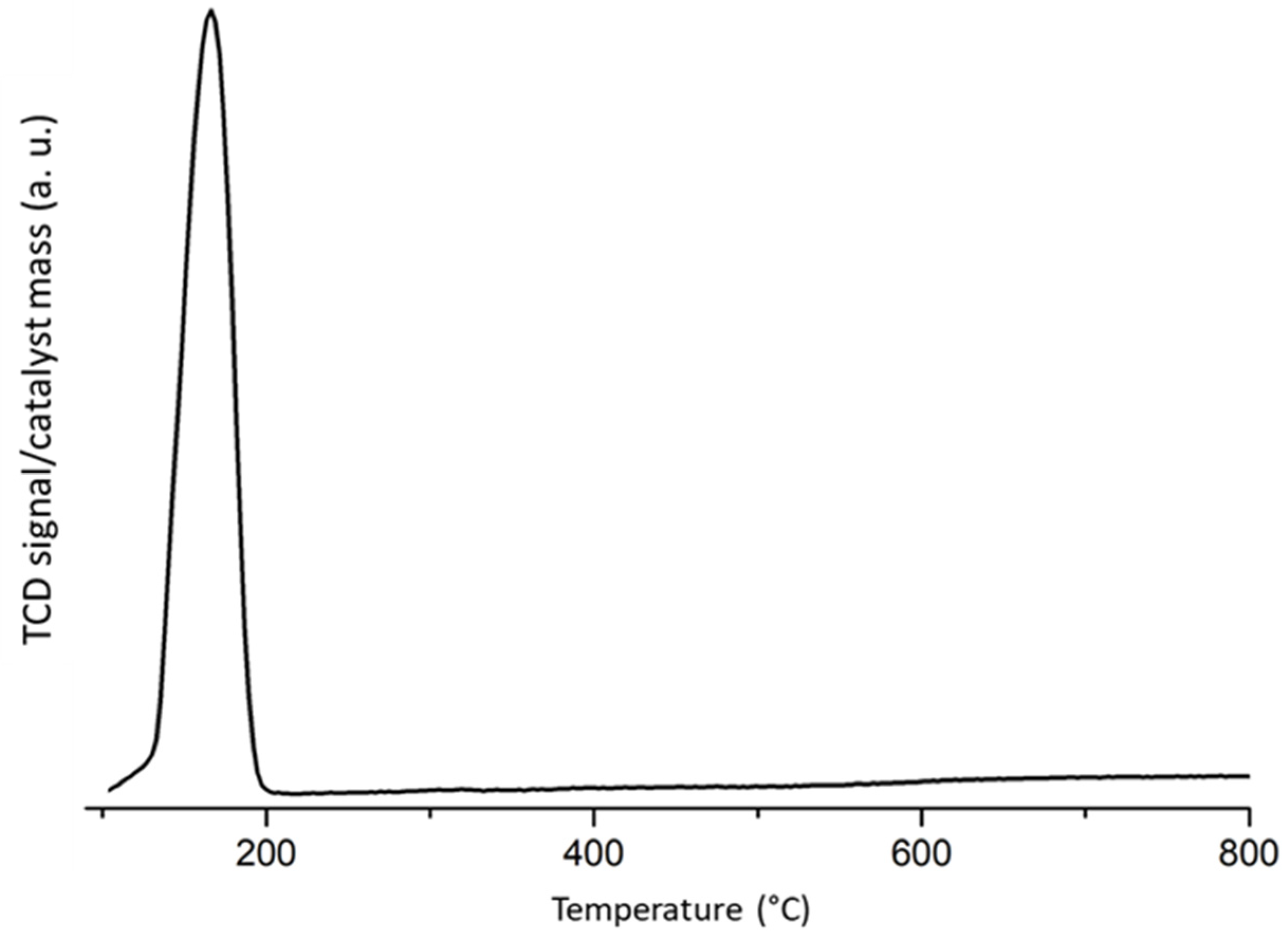
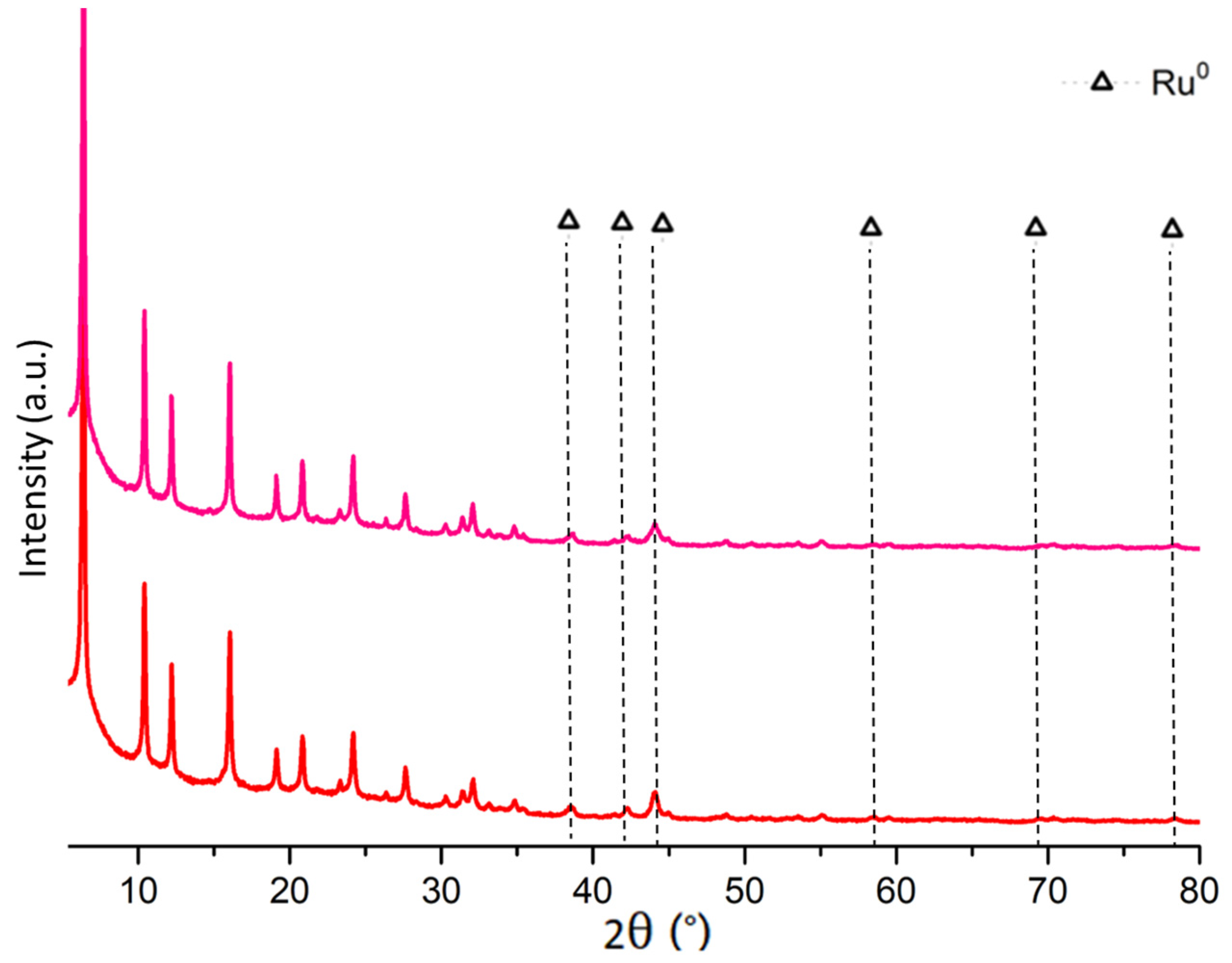
2.1. Fresh and Reduced Catalyst Characterization
2.2. In-Situ Operando FTIR
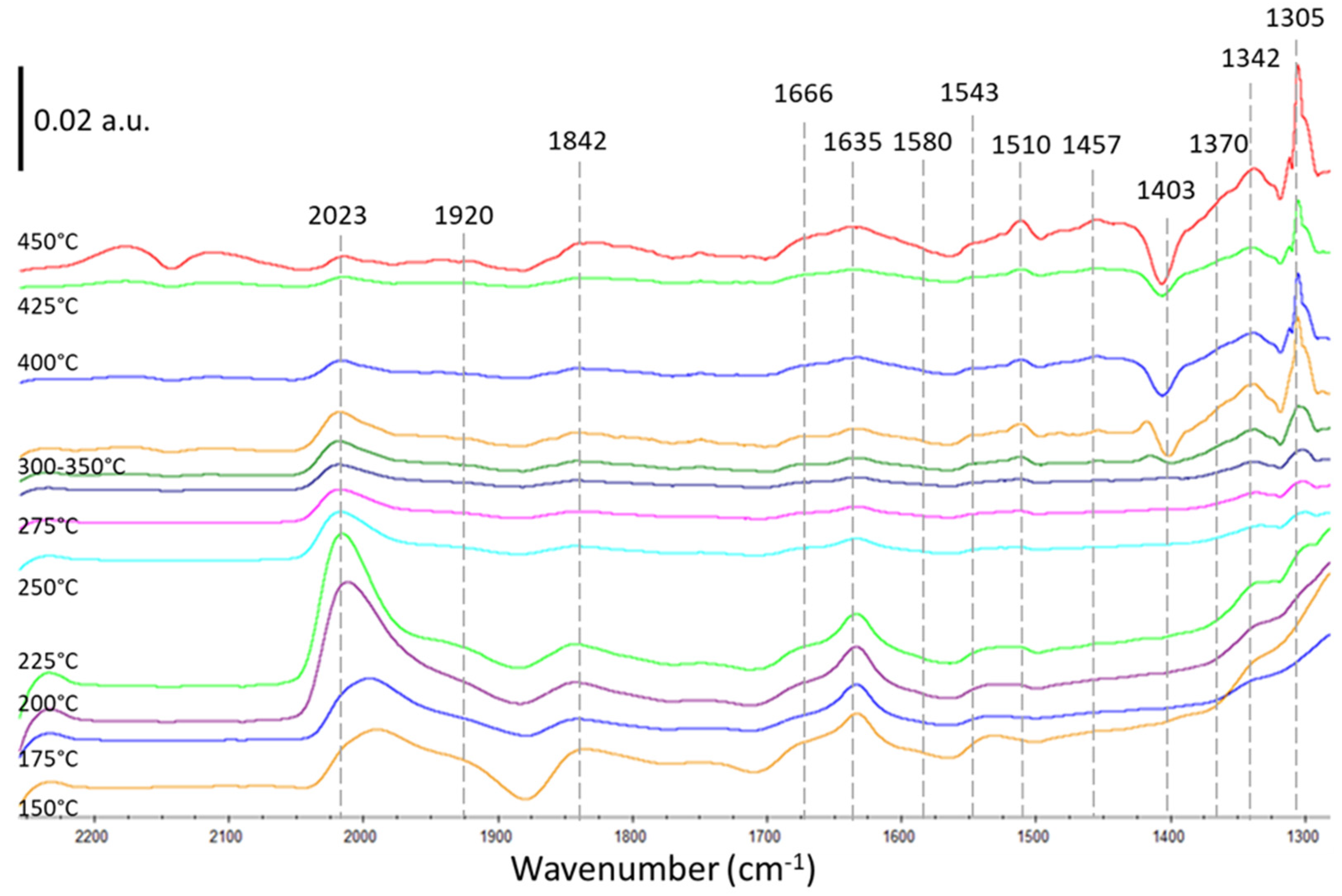
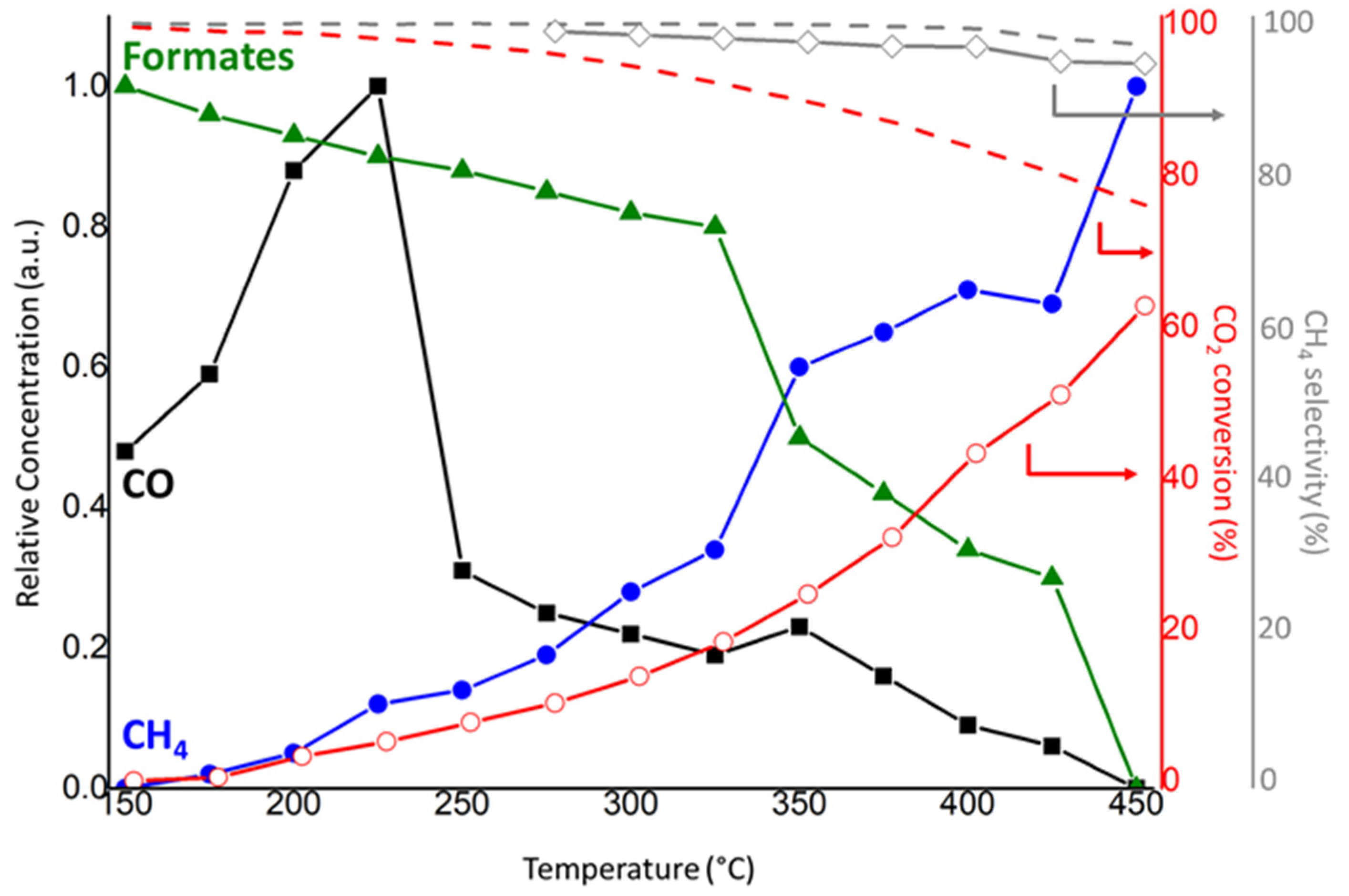
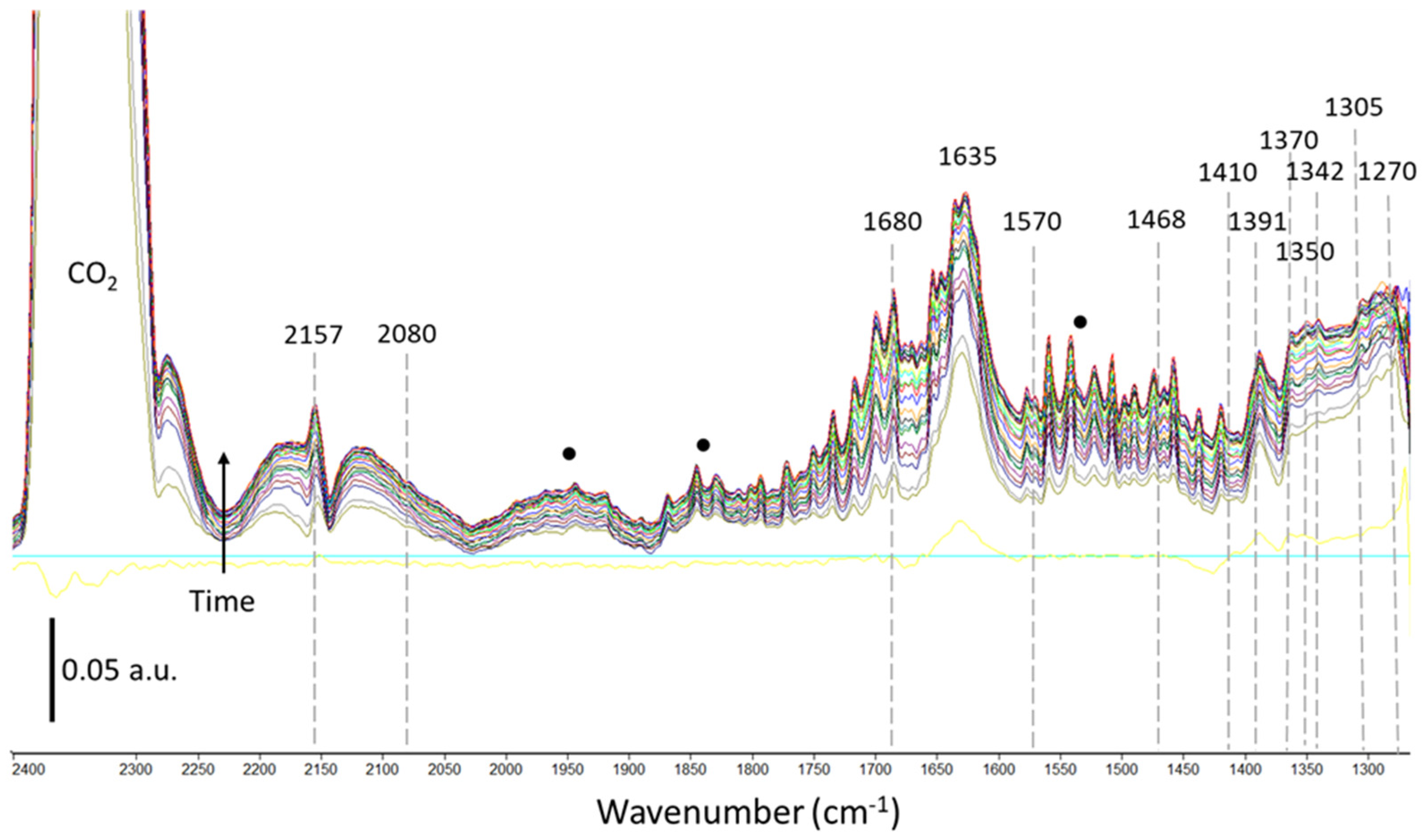
2.2.1. Thermal Catalysis
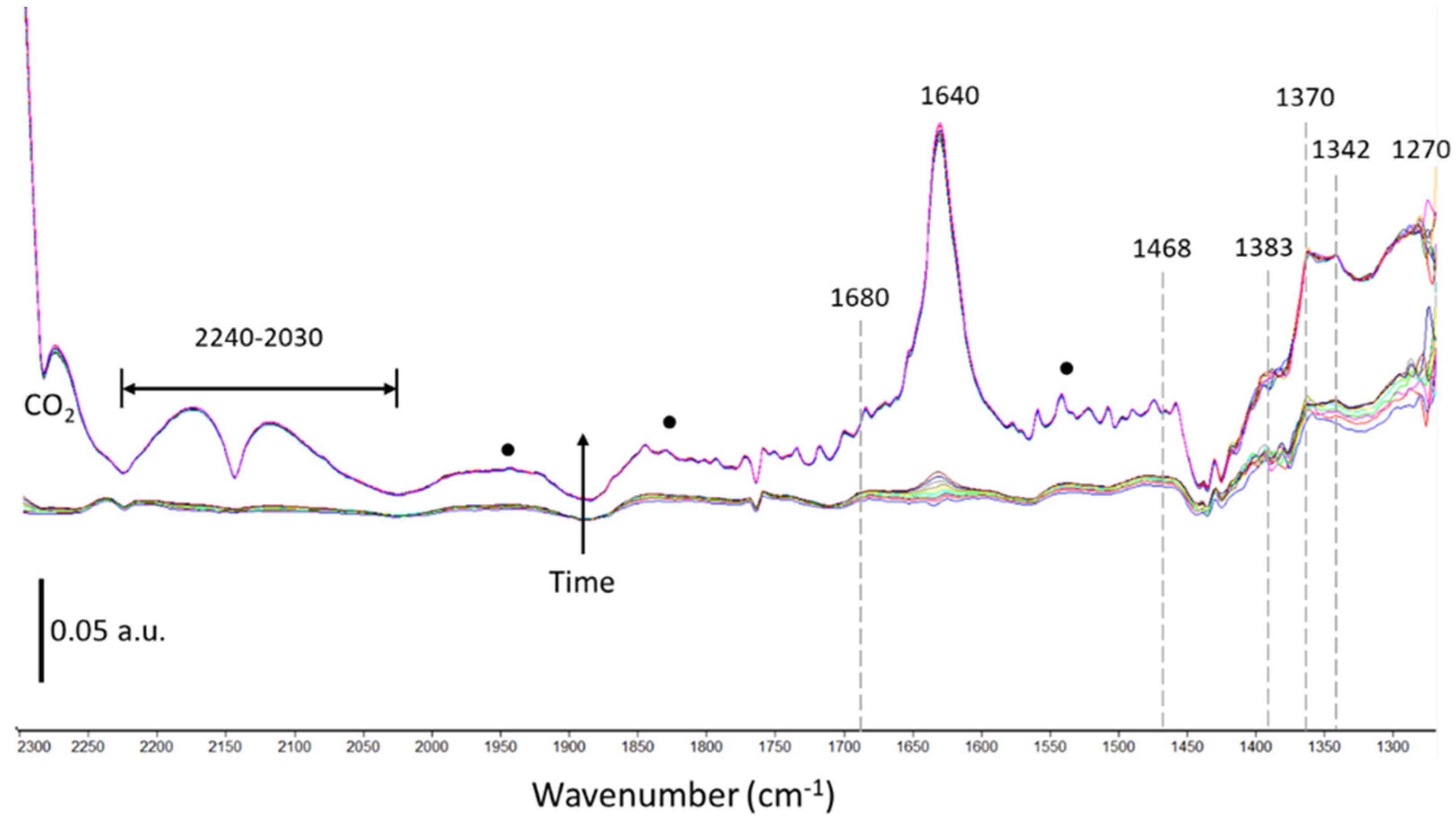
2.2.2. Plasma-Catalysis
2.3. Spent Catalyst Characterization
3. Discussion
4. Materials and Methods
4.1. Catalyst Synthesis
4.2. Catalyst Characterization
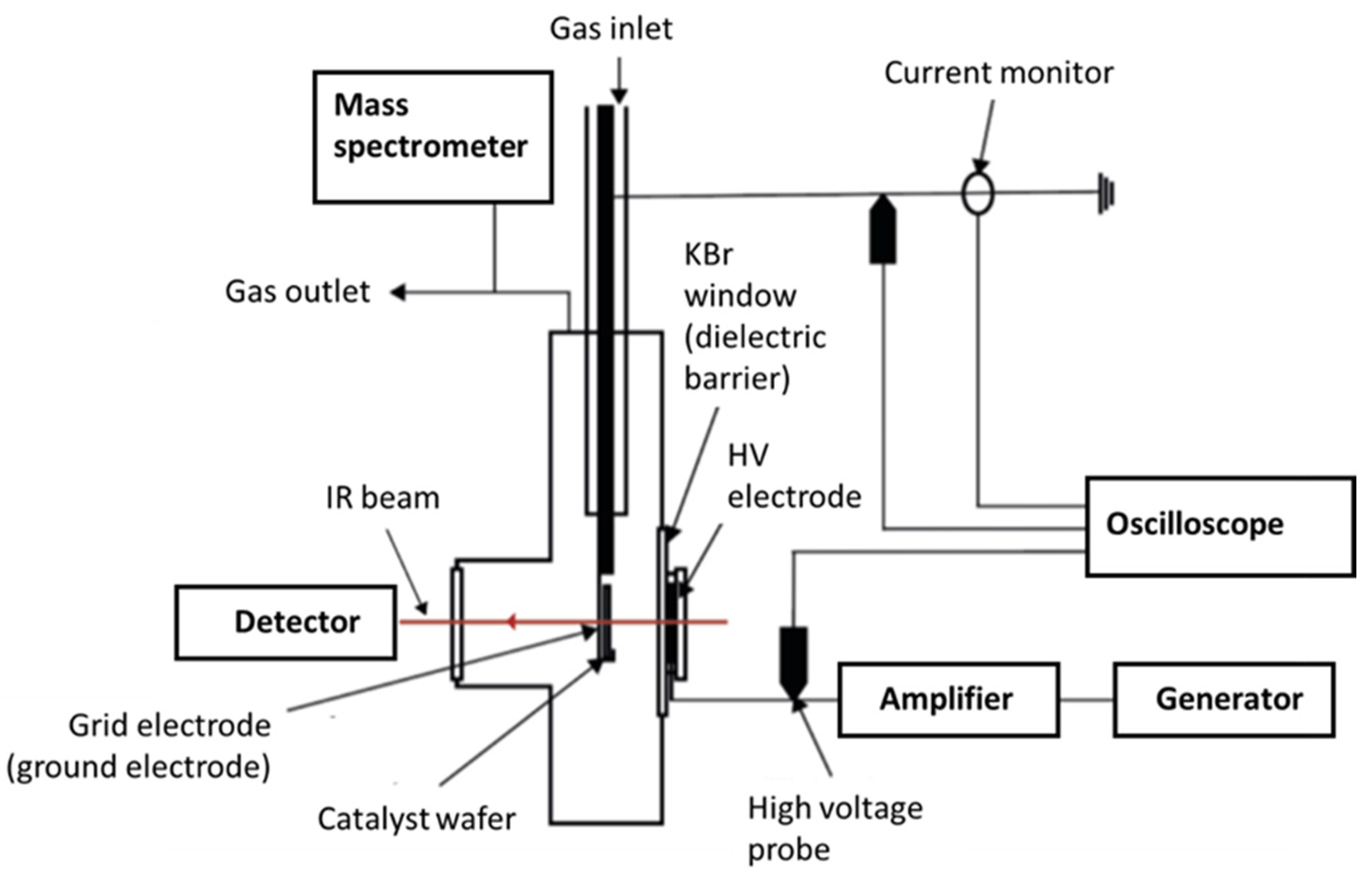
4.3. In-Situ Operando FTIR Experiments
4.3.1. Thermal Catalysis
4.3.2. Plasma-Catalysis
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- IPCC (Intergovernmental Panel on Climate Change). Climate Change 2022: The Physical Science Basis. Working Group I Contribution to the Sixth Assessment Report of the Intergovermental Panel on Climate Change, IPCC (Intergovernmental Panel on Climate Change). Available online: https://www.ipcc.ch/ar6-syr/ (accessed on 12 January 2023).
- Centi, G.; Perathoner, S. Opportunities and prospects in the chemical recycling of carbon dioxide to fuels. Catal. Today 2009, 148, 191–205. [Google Scholar] [CrossRef]
- Sreedhar, I.; Varun, Y.; Singh, S.A.; Venugopal, A.; Reddy, B.M. Developmental trends in CO2 methanation using various catalysts. Catal. Sci. Technol. 2019, 9, 4478–4504. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Garba, M.D.; Usman, M.; Khan, S.; Shehzad, F.; Galadima, A.; Ehsan, M.F.; Ghanem, A.S.; Humayun, M. CO2 towards fuels: A review of catalytic conversion of carbon dioxide to hydrocarbons. J. Environ. Chem. Eng. 2020, 9, 104756. [Google Scholar] [CrossRef]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported Catalysts for CO2 Methanation: A Review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Ridzuan, N.D.M.; Shaharun, M.S.; Anawar, M.A.; Ud-Din, I. Ni-Based Catalyst for Carbon Dioxide Methanation: A Review on Performance and Progress. Catalysts 2022, 12, 469. [Google Scholar] [CrossRef]
- Schubert, M.; Pokhrel, S.; Thomé, A.; Zielasek, V.; Gesing, T.M.; Roessner, F.; Mädler, L.; Bäumer, M. Highly active Co–Al2O3-based catalysts for CO2 methanation with very low platinum promotion prepared by double flame spray pyrolysis. Catal. Sci. Technol. 2016, 6, 7449–7460. [Google Scholar] [CrossRef] [Green Version]
- Kirchner, J.; Anolleck, J.K.; Lösch, H.; Kureti, S. Methanation of CO2 on iron based catalysts. Appl. Catal. B Environ. 2018, 223, 47–59. [Google Scholar] [CrossRef]
- Karelovic, A.; Ruiz, P. CO2 hydrogenation at low temperature over Rh/γ-Al2O3 catalysts: Effect of the metal particle size on catalytic performances and reaction mechanism. Appl. Catal. B Environ. 2012, 113–114, 237–249. [Google Scholar] [CrossRef]
- Quindimil, A.; De-La-Torre, U.; Pereda-Ayo, B.; Davó-Quiñonero, A.; Bailón-García, E.; Lozano-Castelló, D.; González-Marcos, J.A.; Bueno-López, A.; González-Velasco, J.R. Effect of metal loading on the CO2 methanation: A comparison between alumina supported Ni and Ru catalysts. Catal. Today 2019, 356, 419–432. [Google Scholar] [CrossRef]
- Zheng, J.; Wang, C.; Chu, W.; Zhou, Y.; Köhler, K. CO2Methanation over Supported Ru/Al2O3Catalysts: Mechanistic Studies by In situ Infrared Spectroscopy. Chemistryselect 2016, 1, 3197–3203. [Google Scholar] [CrossRef]
- Dębek, R.; Azzolina-Jury, F.; Travert, A.; Maugé, F. A review on plasma-catalytic methanation of carbon dioxide—Looking for an efficient catalyst. Renew. Sustain. Energy Rev. 2019, 116, 109427. [Google Scholar] [CrossRef]
- Bacariza, M.C.; Graça, I.; Lopes, J.M.; Henriques, C. Tuning Zeolite Properties towards CO2 Methanation: An Overview. Chemcatchem 2019, 11, 2388–2400. [Google Scholar] [CrossRef]
- Bogaerts, A.; Kozák, T.; van Laer, K.; Snoeckx, R. Plasma-based conversion of CO2: Current status and future challenges. Faraday Discuss. 2015, 183, 217–232. [Google Scholar] [CrossRef]
- Ashford, B.; Wang, Y.; Wang, L.; Tu, X. Plasma-Catalytic Conversion of Carbon Dioxide. In Plasma Catalysis: Fundamentals and Applications; Springer Series on Atomic, Optical, and Plasma Physics; Tu, X., Whitehead, J.C., Nozaki, T., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 271–307. ISBN 978-3-030-05189-1. [Google Scholar]
- Snoeckx, R.; Bogaerts, A. Plasma technology—A novel solution for CO2 conversion? Chem. Soc. Rev. 2017, 46, 5805–5863. [Google Scholar] [CrossRef] [Green Version]
- Nizio, M.; Albarazi, A.; Cavadias, S.; Amouroux, J.; Galvez, M.E.; Da Costa, P. Hybrid plasma-catalytic methanation of CO2 at low temperature over ceria zirconia supported Ni catalysts. Int. J. Hydrogen Energy 2016, 41, 11584–11592. [Google Scholar] [CrossRef] [Green Version]
- Bacariza, M.; Biset-Peiró, M.; Graça, I.; Guilera, J.; Morante, J.; Lopes, J.; Andreu, T.; Henriques, C. DBD plasma-assisted CO2 methanation using zeolite-based catalysts: Structure composition-reactivity approach and effect of Ce as promoter. J. CO2 Util. 2018, 26, 202–211. [Google Scholar] [CrossRef]
- Solis-Garcia, A.; Fierro-Gonzalez, J.C. Mechanistic Insights into the CO2 Methanation Catalyzed by Supported Metals: A Review. J. Nanosci. Nanotechnol. 2019, 19, 3110–3123. [Google Scholar] [CrossRef]
- Westermann, A.; Azambre, B.; Bacariza, M.C.; Graça, I.; Ribeiro, M.F.; Lopes, J.M.; Henriques, C. Insight into CO2 methanation mechanism over NiUSY zeolites: An operando IR study. Appl. Catal. B Environ. 2015, 174–175, 120–125. [Google Scholar] [CrossRef]
- Neyts, E.C.; Ostrikov, K.; Sunkara, M.K.; Bogaerts, A. Plasma Catalysis: Synergistic Effects at the Nanoscale. Chem. Rev. 2015, 115, 13408–13446. [Google Scholar] [CrossRef]
- Azzolina-Jury, F.; Thibault-Starzyk, F. Mechanism of Low Pressure Plasma-Assisted CO2 Hydrogenation Over Ni-USY by Microsecond Time-resolved FTIR Spectroscopy. Top. Catal. 2017, 60, 1709–1721. [Google Scholar] [CrossRef]
- Sun, Y.; Li, J.; Chen, P.; Wang, B.; Wu, J.; Fu, M.; Chen, L.; Ye, D. Reverse water-gas shift in a packed bed DBD reactor: Investigation of metal-support interface towards a better understanding of plasma catalysis. Appl. Catal. A Gen. 2020, 591, 117407. [Google Scholar] [CrossRef]
- Van Turnhout, J.; Aceto, D.; Travert, A.; Bazin, P.; Thibault-Starzyk, F.; Bogaerts, A.; Azzolina-Jury, F. Observation of surface species in plasma-catalytic dry reforming of methane in a novel atmospheric pressure dielectric barrier discharge in situ IR cell. Catal. Sci. Technol. 2022, 12, 6676–6686. [Google Scholar] [CrossRef]
- Bacariza, M.; Bértolo, R.; Graça, I.; Lopes, J.; Henriques, C. The effect of the compensating cation on the catalytic performances of Ni/USY zeolites towards CO2 methanation. J. CO2 Util. 2017, 21, 280–291. [Google Scholar] [CrossRef]
- Bacariza, M.; Graça, I.; Lopes, J.; Henriques, C. Enhanced activity of CO2 hydrogenation to CH4 over Ni based zeolites through the optimization of the Si/Al ratio. Microporous Mesoporous Mater. 2018, 267, 9–19. [Google Scholar] [CrossRef]
- Bacariza, M.; Maleval, M.; Graça, I.; Lopes, J.; Henriques, C. Power-to-methane over Ni/zeolites: Influence of the framework type. Microporous Mesoporous Mater. 2018, 274, 102–112. [Google Scholar] [CrossRef]
- Afzal, M.I.; Shahid, S.; Mansoor, S.; Javed, M.; Iqbal, S.; Hakami, O.; Yousef, E.S.; Al-Fawzan, F.F.; Elkaeed, E.B.; Pashameah, R.A.; et al. Fabrication of a Ternary Nanocomposite g-C3N4/Cu@CdS with Superior Charge Separation for Removal of Organic Pollutants and Bacterial Disinfection from Wastewater under Sunlight Illumination. Toxics 2022, 10, 657. [Google Scholar] [CrossRef]
- Fazal, T.; Iqbal, S.; Shah, M.; Ismail, B.; Shaheen, N.; Alharthi, A.I.; Awwad, N.S.; Ibrahium, H.A. Correlation between structural, morphological and optical properties of Bi2S3 thin films deposited by various aqueous and non-aqueous chemical bath deposition methods. Results Phys. 2022, 40, 105817. [Google Scholar] [CrossRef]
- Huang, W.-S.; Humphrey, B.D.; MacDiarmid, A.G. Polyaniline, a novel conducting polymer. Morphology and chemistry of its oxidation and reduction in aqueous electrolytes. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1986, 82, 2385–2400. [Google Scholar] [CrossRef]
- Treacy, M.M.J. Collection of simulated XRD powder patterns for zeolites. Appl. Catal. 1986, 21, 388–389. [Google Scholar]
- Gamliel, D.P.; Karakalos, S.; Valla, J.A. Liquid phase hydrodeoxygenation of anisole, 4-ethylphenol and benzofuran using Ni, Ru and Pd supported on USY zeolite. Appl. Catal. A Gen. 2018, 559, 20–29. [Google Scholar] [CrossRef]
- Shen, X.; Garces, L.-J.; Ding, Y.; Laubernds, K.; Zerger, R.P.; Aindow, M.; Neth, E.J.; Suib, S.L. Behavior of H2 chemisorption on Ru/TiO2 surface and its application in evaluation of Ru particle sizes compared with TEM and XRD analyses. Appl. Catal. A Gen. 2008, 335, 187–195. [Google Scholar] [CrossRef]
- Biswas, A.; Paul, S.; Banerjee, A. Carbon nanodots, Ru nanodots and hybrid nanodots: Preparation and catalytic properties. J. Mater. Chem. A 2015, 3, 15074–15081. [Google Scholar] [CrossRef]
- Qu, L.; Li, J.; Hao, Z.; Li, L. Catalytic Oxidation of Nitric Oxide to Nitrogen Dioxide on Ru-FAU. Catal. Lett. 2009, 131, 656–662. [Google Scholar] [CrossRef] [Green Version]
- Juurlink, L.; Killelea, D.; Utz, A. State-resolved probes of methane dissociation dynamics. Prog. Surf. Sci. 2009, 84, 69–134. [Google Scholar] [CrossRef]
- Wierzbicki, D.; Debek, R.; Motak, M.; Grzybek, T.; Gálvez, M.E.; Da Costa, P. Novel Ni-La-hydrotalcite derived catalysts for CO2 methanation. Catal. Commun. 2016, 83, 5–8. [Google Scholar] [CrossRef] [Green Version]
- Boukha, Z.; Jiménez-González, C.; Gil-Calvo, M.; de Rivas, B.; González-Velasco, J.R.; Gutiérrez-Ortiz, J.I.; López-Fonseca, R. MgO/NiAl2O4 as a new formulation of reforming catalysts: Tuning the surface properties for the enhanced partial oxidation of methane. Appl. Catal. B Environ. 2016, 199, 372–383. [Google Scholar] [CrossRef]
- Meldrum, B.J.; Rochester, C.H. Fourier-transform infrared study of surface species on carbon mixed with KBr and reacted with CO2, O2 and CO. J. Chem. Soc. Faraday Trans. 1990, 86, 3647–3652. [Google Scholar] [CrossRef]
- Goodwin, J.G., Jr.; Naccache, C. CO adsorption on ion-exchanged Ru zeolite catalyst. J. Catal. 1980, 64, 482–486. [Google Scholar] [CrossRef]
- Hadjiivanov, K.; Lavalley, J.-C.; Lamotte, J.; Maugé, F.; Saint-Just, J.; Che, M. FTIR Study of CO Interaction with Ru/TiO2Catalysts. J. Catal. 1998, 176, 415–425. [Google Scholar] [CrossRef]
- Haq, S.; Love, J.; Sanders, H.; King, D. Adsorption and decomposition of formic acid on Ni{110}. Surf. Sci. 1995, 325, 230–242. [Google Scholar] [CrossRef]
- Wierzbicki, D.; Moreno, M.V.; Ognier, S.; Motak, M.; Grzybek, T.; Da Costa, P.; Gálvez, M.E. Ni-Fe layered double hydroxide derived catalysts for non-plasma and DBD plasma-assisted CO2 methanation. Int. J. Hydrogen Energy 2019, 45, 10423–10432. [Google Scholar] [CrossRef]
- Mikhail, M.; Da Costa, P.; Amouroux, J.; Cavadias, S.; Tatoulian, M.; Ognier, S.; Gálvez, M.E. Effect of Na and K impurities on the performance of Ni/CeZrOx catalysts in DBD plasma-catalytic CO2 methanation. Fuel 2021, 306, 121639. [Google Scholar] [CrossRef]
- Parastaev, A.; Kosinov, N.; Hensen, E.J.M. Mechanistic study of catalytic CO2 hydrogenation in a plasma by operando DRIFT spectroscopy. J. Phys. D Appl. Phys. 2021, 54, 264004. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Vmicro (cm3 g−1) | Vmeso (cm3 g−1) | Sext (m2 g−1) |
|---|---|---|---|
| CsUSY | 0.18 | 0.32 | 315 |
| 3Ru/CsUSY | 0.14 | 0.22 | 170 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aceto, D.; Bacariza, M.C.; Travert, A.; Henriques, C.; Azzolina-Jury, F. Thermal and Plasma-Assisted CO2 Methanation over Ru/Zeolite: A Mechanistic Study Using In-Situ Operando FTIR. Catalysts 2023, 13, 481. https://doi.org/10.3390/catal13030481
Aceto D, Bacariza MC, Travert A, Henriques C, Azzolina-Jury F. Thermal and Plasma-Assisted CO2 Methanation over Ru/Zeolite: A Mechanistic Study Using In-Situ Operando FTIR. Catalysts. 2023; 13(3):481. https://doi.org/10.3390/catal13030481
Chicago/Turabian StyleAceto, Domenico, Maria Carmen Bacariza, Arnaud Travert, Carlos Henriques, and Federico Azzolina-Jury. 2023. "Thermal and Plasma-Assisted CO2 Methanation over Ru/Zeolite: A Mechanistic Study Using In-Situ Operando FTIR" Catalysts 13, no. 3: 481. https://doi.org/10.3390/catal13030481