


Methane Activation and Coupling Pathways on Ni2P Catalyst

 ,
, 

Abstract
:1. Introduction
2. Results and Discussion
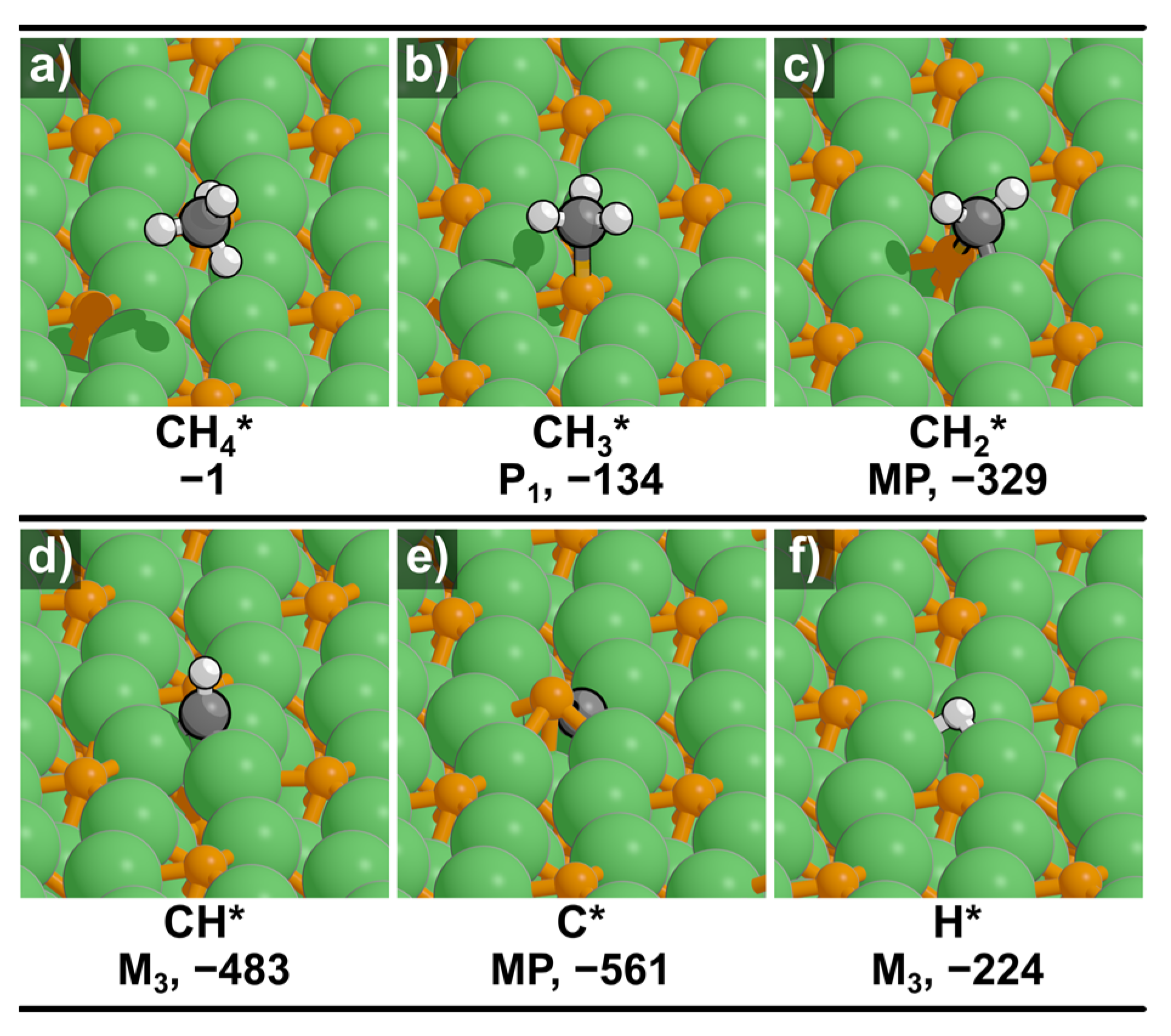
2.1. Optimized Structures and Binding Energies on Ni2P(001) Surface
2.2. Methane Dehydrogenation Reactions
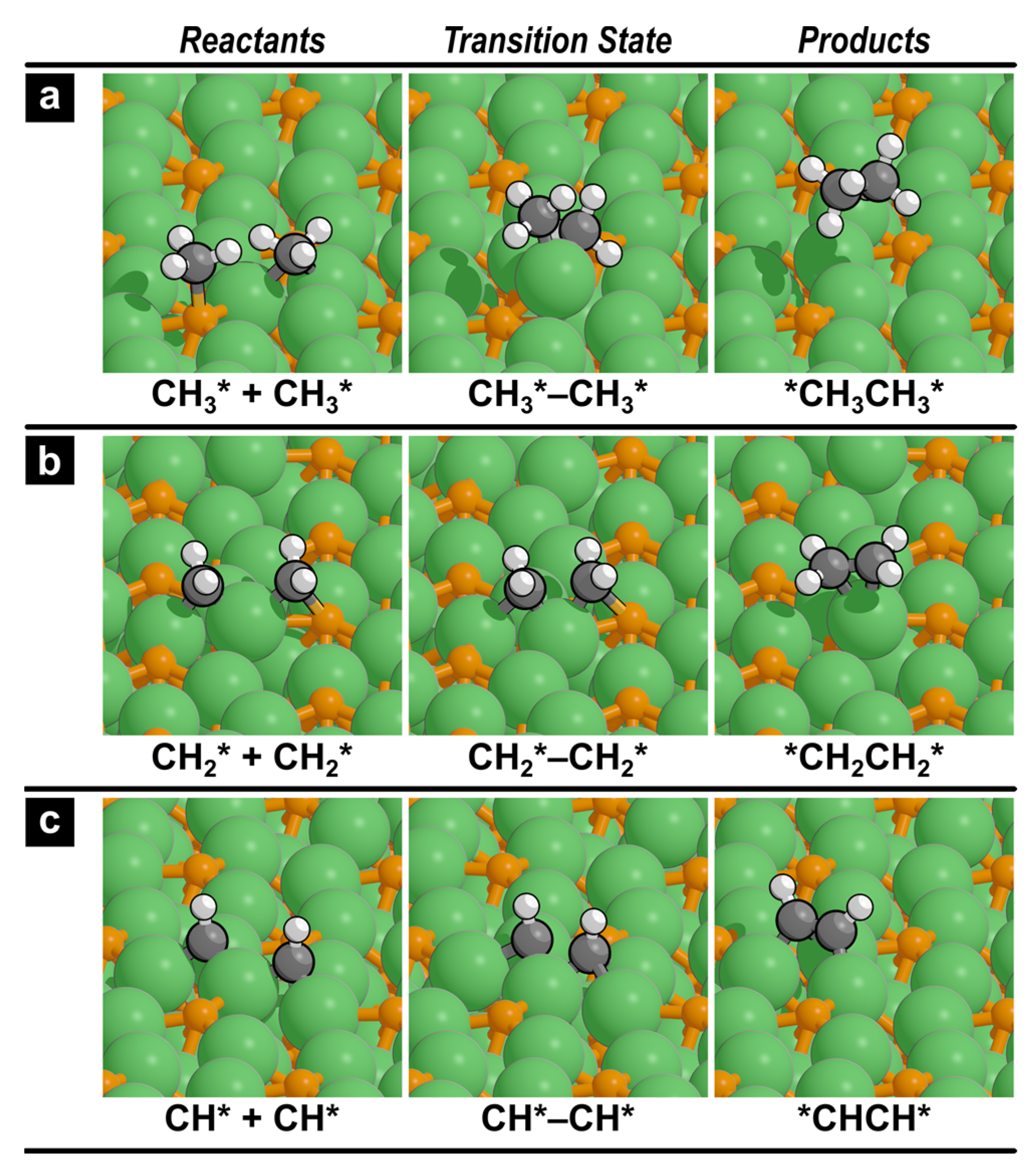
2.3. C–C Coupling Reactions
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liang, Z.; Li, T.; Kim, M.; Asthagiri, A.; Weaver, J.F. Low-Temperature Activation of Methane on the IrO2(110) Surface. Science 2017, 356, 299–303. [Google Scholar] [CrossRef]
- Kim, D.; Ju, Y.; Kang, D.; Kang, S.B.; Kim, M. Potential of Intrinsic Reactivity toward Value Added Products from Methane Oxidation on RhO2(1 1 0) Surface. Appl. Surf. Sci. 2022, 596, 153499. [Google Scholar] [CrossRef]
- Zhang, T.; Holiharimanana, D.; Yang, X.; Ge, Q. DFT Study of Methane Activation and Coupling on the (0001) and (112¯ 0) Surfaces of α-WC. J. Phys. Chem. C 2020, 124, 26722–26729. [Google Scholar] [CrossRef]
- Boekfa, B.; Treesukol, P.; Injongkol, Y.; Maihom, T.; Maitarad, P.; Limtrakul, J. The Activation of Methane on Ru, Rh, and Pd Decorated Carbon Nanotube and Boron Nitride Nanotube: A DFT Study. Catalysts 2018, 8, 190. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Li, X.; Liu, Y.; Yu, Z.; Li, B.; Zhao, Z. The C-H Bond Activation Triggered by Subsurface Mo Dopant on MgO Catalyst in Oxidative Coupling of Methane. Catalysts 2022, 12, 1083. [Google Scholar] [CrossRef]
- Zhang, T.; Yang, X.; Ge, Q. A DFT Study of Methane Conversion on Mo-Terminated Mo2C Carbides: Carburization vs C–C Coupling. Catal. Today 2021, 368, 140–147. [Google Scholar] [CrossRef]
- Rosen, A.S.; Notestein, J.M.; Snurr, R.Q. Structure-Activity Relationships That Identify Metal-Organic Framework Catalysts for Methane Activation. ACS Catal. 2019, 9, 3576–3587. [Google Scholar] [CrossRef]
- Mehmood, A.; Chae, S.Y.; Park, E.D. Photoelectrochemical Conversion of Methane into Value-Added Products. Catalysts 2021, 11, 1387. [Google Scholar] [CrossRef]
- Latimer, A.A.; Kakekhani, A.; Kulkarni, A.R.; Nørskov, J.K. Direct Methane to Methanol: The Selectivity–Conversion Limit and Design Strategies. ACS Catal. 2018, 8, 6894–6907. [Google Scholar] [CrossRef]
- West, N.M.; Miller, A.J.M.; Labinger, J.A.; Bercaw, J.E. Homogeneous Syngas Conversion. Coord. Chem. Rev. 2011, 255, 881–898. [Google Scholar] [CrossRef]
- Blanksby, S.J.; Ellison, G.B. Bond Dissociation Energies of Organic Molecules. Acc Chem. Res. 2003, 36, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Keller, G. Synthesis of Ethylene via Oxidative Coupling of Methane I. Determination of Active Catalysts. J. Catal. 1982, 73, 9–19. [Google Scholar] [CrossRef]
- Kim, M.; Franklin, A.; Martin, R.; Feng, F.; Li, T.; Liang, Z.; Asthagiri, A.; Weaver, J.F. Adsorption and Oxidation of CH4 on Oxygen-Rich IrO2 (110). J. Phys. Chem. C 2019, 123, 27603–27614. [Google Scholar] [CrossRef]
- Kim, M.; Franklin, A.D.; Martin, R.; Bian, Y.; Weaver, J.F.; Asthagiri, A. Kinetics of Low-Temperature Methane Activation on IrO2(1 1 0): Role of Local Surface Hydroxide Species. J. Catal. 2020, 383, 181–192. [Google Scholar] [CrossRef]
- Wang, C.-C.; Siao, S.S.; Jiang, J.-C. C–H Bond Activation of Methane via σ–d Interaction on the IrO2(110) Surface: Density Functional Theory Study. J. Phys. Chem. C 2012, 116, 6367–6370. [Google Scholar] [CrossRef]
- Erlekam, U.; Paulus, U.A.; Wang, Y.; Bonzel, H.P.; Jacobi, K.; Ertl, G. Adsorption of Methane and Ethane on RuO2(110) Surfaces. Z. Für Phys. Chem. 2005, 219, 891–903. [Google Scholar] [CrossRef] [Green Version]
- Weaver, J.F.; Hakanoglu, C.; Hawkins, J.M.; Asthagiri, A. Molecular Adsorption of Small Alkanes on a PdO(101) Thin Film: Evidence of σ-Complex Formation. J. Chem. Phys. 2010, 132, 024709. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Wang, J.; Wu, X.; Liu, H.; Cai, J.; Xu, G.Q.; Wang, S.L. Room-Temperature Coupling of Methane with Singlet Oxygen. Environ. Sci. Adv. 2022, 1, 438–442. [Google Scholar] [CrossRef]
- Wu, X.Y.; Tang, Z.; Zhao, X.; Luo, X.; John Pennycook, S.; Wang, S.L. Visible-Light Driven Room-Temperature Coupling of Methane to Ethane by Atomically Dispersed Au on WO3. J. Energy Chem. 2021, 61, 195–202. [Google Scholar] [CrossRef]
- Zeng, Y.; Luo, X.; Li, F.; Huang, A.; Wu, H.; Xu, G.Q.; Wang, S.L. Noble Metal-Free FeOOH/Li0.1 WO3 Core–Shell Nanorods for Selective Oxidation of Methane to Methanol with Visible–NIR Light. Environ. Sci. Technol. 2021, 55, 7711–7720. [Google Scholar] [CrossRef]
- Li, W.; Dhandapani, B.; Oyama, S.T. Molybdenum Phosphide: A Novel Catalyst for Hydrodenitrogenation. Chem. Lett. 1998, 27, 207–208. [Google Scholar] [CrossRef]
- Bui, P.; Cecilia, J.A.; Oyama, S.T.; Takagaki, A.; Infantes-Molina, A.; Zhao, H.; Li, D.; Rodríguez-Castellón, E.; Jiménez López, A. Studies of the Synthesis of Transition Metal Phosphides and Their Activity in the Hydrodeoxygenation of a Biofuel Model Compound. J. Catal. 2012, 294, 184–198. [Google Scholar] [CrossRef]
- Almithn, A.; Alhulaybi, Z. A Mechanistic Study of Methanol Steam Reforming on Ni2P Catalyst. Catalysts 2022, 12, 1174. [Google Scholar] [CrossRef]
- Witzke, M.E.; Almithn, A.; Conrad, C.L.; Hibbitts, D.D.; Flaherty, D.W. Mechanisms and Active Sites for C–O Bond Rupture within 2-Methyltetrahydrofuran over Ni, Ni12P5, and Ni2P Catalysts. ACS Catal. 2018, 8, 7141–7157. [Google Scholar] [CrossRef]
- Witzke, M.E.; Almithn, A.; Conrad, C.L.; Triezenberg, M.D.; Hibbitts, D.D.; Flaherty, D.W. In Situ Methods for Identifying Reactive Surface Intermediates during Hydrogenolysis Reactions: C–O Bond Cleavage on Nanoparticles of Nickel and Nickel Phosphides. J. Am. Chem. Soc. 2019, 141, 16671–16684. [Google Scholar] [CrossRef]
- Dipu, A.L.; Nishikawa, Y.; Inami, Y.; Iguchi, S.; Yamanaka, I. Development of Highly Active Silica-Supported Nickel Phosphide Catalysts for Direct Dehydrogenative Conversion of Methane to Higher Hydrocarbons. Catal. Lett. 2022, 152, 199–212. [Google Scholar] [CrossRef]
- Dipu, A.L.; Ohbuchi, S.; Nishikawa, Y.; Iguchi, S.; Ogihara, H.; Yamanaka, I. Direct Nonoxidative Conversion of Methane to Higher Hydrocarbons over Silica-Supported Nickel Phosphide Catalyst. ACS Catal. 2020, 10, 375–379. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Han, Z.; Yang, Z.; Han, M. Comprehensive Investigation of Methane Conversion over Ni(111) Surface under a Consistent DFT Framework: Implications for Anti-Coking of SOFC Anodes. Appl. Surf. Sci. 2019, 480, 243–255. [Google Scholar] [CrossRef]
- Pham, T.L.M.; Leggesse, E.G.; Jiang, J.C. Ethylene Formation by Methane Dehydrogenation and C-C Coupling Reaction on a Stoichiometric IrO2(110) Surface—A Density Functional Theory Investigation. Catal. Sci. Technol. 2015, 5, 4064–4071. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal–Amorphous-Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kravchenko, P.; Plaisance, C.; Hibbitts, D. A New Computational Interface for Catalysis. ChemRxiv 2019, preprint. [Google Scholar]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 1999, 59, 7413–7421. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, W. Comment on “Generalized Gradient Approximation Made Simple”. Phys. Rev. Lett. 1998, 80, 890. [Google Scholar] [CrossRef]
- Larsson, E. An X-Ray Investigation of Ni-P System and Crystal Structures of NiP and NiP2. Arkiv. Kemi. 1965, 23, 335. [Google Scholar]
- Rem, J.; Wang, J.; Li, J.; Li, Y. Density Functional Theory Study on Crystal Nickel Phosphides. J. Fuel Chem. Technol. 2007, 35, 458–464. [Google Scholar] [CrossRef]
- Rundqvist, S. X-Ray Investigations of Mn3P, Mn2P, and Ni2P. Acta Chem. Scand 1962, 16, 992–998. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. “Special Points for Brillouin-Zone Integrations”—A Reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Jónsson, H.; Mills, G.; Jacobsen, K.W. Nudged Elastic Band Method for Finding Minimum Energy Paths of Transitions. In Proceedings of the Classical and Quantum Dynamics in Condensed Phase Simulations, Lerici, Italy, 7—18 July 1997; World Scientific: Singapore, 1998; pp. 385–404. [Google Scholar]
- Henkelman, G.; Jónsson, H. A Dimer Method for Finding Saddle Points on High Dimensional Potential Surfaces Using Only First Derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Adsorption Mode | ΔEads | d(C–Ni) | d(C–P) |
|---|---|---|---|---|
| kJ mol−1 | (Å) | (Å) | ||
| CH4* | Physisorbed | −1 | – | – |
| CH3* | P1 | −134 | – | 1.90 |
| CH2* | MP | −329 | 2.06 | 1.78 |
| CH* | M3 | −483 | 1.89 | – |
| C* | MP | −561 | 1.92 | 1.77 |
| H* | M3 | −224 | 1.80 | – |
| No. | Reaction | ΔHact | ΔHrxn |
|---|---|---|---|
| kJ mol−1 | kJ mol−1 | ||
| 1 | CH4*→CH3* + ½H2(g) | 246 | 99 |
| 2 | CH3*→CH2* + ½H2(g) | 65 | 44 |
| 3 | CH2*→CH* + ½H2(g) | 84 | 74 |
| 4 | CH*→C* + ½H2(g) | 102 | 41 |
| No. | Reaction | ΔHact | ΔHrxn |
|---|---|---|---|
| kJ mol−1 | kJ mol−1 | ||
| 1 | CH3* + CH3*→*CH3CH3* | 98 | −96 |
| 2 | CH2* + CH2*→*CH2CH2* | 56 | −79 |
| 3 | CH* + CH*→*CHCH* | 56 | −133 |
| 4 | CH3* + CH2*→*CH3CH2* | 191 | −31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almithn, A.; Alghanim, S.N.; Mohammed, A.A.; Alghawinim, A.K.; Alomaireen, M.A.; Alhulaybi, Z.; Hossain, S.S. Methane Activation and Coupling Pathways on Ni2P Catalyst. Catalysts 2023, 13, 531. https://doi.org/10.3390/catal13030531
Almithn A, Alghanim SN, Mohammed AA, Alghawinim AK, Alomaireen MA, Alhulaybi Z, Hossain SS. Methane Activation and Coupling Pathways on Ni2P Catalyst. Catalysts. 2023; 13(3):531. https://doi.org/10.3390/catal13030531
Chicago/Turabian StyleAlmithn, Abdulrahman, Salem N. Alghanim, Abdullah A. Mohammed, Abdullah K. Alghawinim, Mazen A. Alomaireen, Zaid Alhulaybi, and SK Safdar Hossain. 2023. "Methane Activation and Coupling Pathways on Ni2P Catalyst" Catalysts 13, no. 3: 531. https://doi.org/10.3390/catal13030531
APA StyleAlmithn, A., Alghanim, S. N., Mohammed, A. A., Alghawinim, A. K., Alomaireen, M. A., Alhulaybi, Z., & Hossain, S. S. (2023). Methane Activation and Coupling Pathways on Ni2P Catalyst. Catalysts, 13(3), 531. https://doi.org/10.3390/catal13030531