Abstract
Despite their potential as a protein source for human consumption, the health benefits of black soldier fly larvae (BSFL) proteins following human gastrointestinal (GI) digestion are poorly understood. This computational study explored the potential of BSFL proteins to release health-promoting peptides after human GI digestion. Twenty-six proteins were virtually proteolyzed with GI proteases. The resultant peptides were screened for high GI absorption and non-toxicity. Shortlisted peptides were searched against the BIOPEP-UWM and Scopus databases to identify their bioactivities. The potential of the peptides as inhibitors of myeloperoxidase (MPO), NADPH oxidase (NOX), and xanthine oxidase (XO), as well as a disruptor of Keap1–Nrf2 protein–protein interaction, were predicted using molecular docking and dynamics simulation. Our results revealed that about 95% of the 5218 fragments generated from the proteolysis of BSFL proteins came from muscle proteins. Dipeptides comprised the largest group (about 25%) of fragments arising from each muscular protein. Screening of 1994 di- and tripeptides using SwissADME and STopTox tools revealed 65 unique sequences with high GI absorption and non-toxicity. A search of the databases identified 16 antioxidant peptides, 14 anti-angiotensin-converting enzyme peptides, and 17 anti-dipeptidyl peptidase IV peptides among these sequences. Results from molecular docking and dynamic simulation suggest that the dipeptide DF has the potential to inhibit Keap1–Nrf2 interaction and interact with MPO within a short time frame, whereas the dipeptide TF shows promise as an XO inhibitor. BSFL peptides were likely weak NOX inhibitors. Our in silico results suggest that upon GI digestion, BSFL proteins may yield high-GI-absorbed and non-toxic peptides with potential health benefits. This study is the first to investigate the bioactivity of peptides liberated from BSFL proteins following human GI digestion. Our findings provide a basis for further investigations into the potential use of BSFL proteins as a functional food ingredient with significant health benefits.
1. Introduction
Black soldier fly larvae (BSFL) (Hermetia illucens L.) are largely regarded as a sustainable, cost-effective source of proteins for animal feed [1]. Additionally supporting this is the evidence of the nutritional benefits of BSFL-based animal feed in the literature [2]. Notably, recent studies reported that BSFL proteins could serve as sources of bioactive peptides following treatment with proteases [3,4,5,6]. It was proposed that BSFL-derived bioactive peptides may have potential applications in the development of functional food ingredients, cosmetics, and pharmaceuticals [4,6].
Bioactive peptides are short peptides, often comprising 2-20 residues, and can exert beneficial physiological functions in the human body. The peptides are initially encrypted in an inactive state within the precursor proteins. They can be liberated from the proteins through enzymatic hydrolysis, microbial fermentation, and, naturally, during the gastrointestinal (GI) digestion of dietary proteins in vivo [7,8,9]. In fact, enzymatic hydrolysis of proteins is the key strategy adopted by many studies to generate bioactive peptides from diverse bioresources. Enzymatic proteolysis of animal and plant proteins, both in vitro and in silico, has led to the discovery of peptides exhibiting diverse health-promoting activities, such as antioxidant [10,11,12], antihypertensive [13,14], antidiabetic [13], and anticancer [15,16,17] activities. Notably, many bioactive peptides were identified from the protein hydrolysates of edible insects, such as crickets, locusts, mealworms, and silkworm pupae [18]. By contrast, the discovery of bioactive peptides from BSFL is a relatively new development when compared with that from other bioresources. Zhu and co-workers [6] compared protein hydrolysates prepared from BSFL that were proteolyzed with alcalase, neutrase, trypsin, and papain, revealing the strongest free radical scavenging activity in the alcalase hydrolysate. Seventeen peptides with features typical of other known antioxidant peptides were identified from the hydrolysate [6]. Lu and co-workers [4] also reported the discovery of putative antioxidant peptides from the alcalase hydrolysate of BSFL fed with food waste. Several other studies reported the production and analysis of BSFL protein hydrolysates, although without identifying any bioactive peptides [3,5,19,20,21]. In short, BSFL-derived bioactive peptides remain a gap of knowledge; available findings are confined to free radical scavenging peptides.
The potential of BSFL as a viable protein-rich food source for human consumption has been highlighted recently [1], yet little is known about the health benefits of BSFL proteins. In this study, we addressed a novel research question that has not been explored in the existing literature: whether BSFL proteins can release health-promoting peptides upon consumption and digestion by humans. We adopted an in silico approach to discover peptides that could be released from BSFL proteins after virtual digestion by using three GI proteases: chymotrypsin (EC 3.4.21.1), trypsin (EC 3.4.21.4), and pepsin (EC 3.4.23.1). The three GI proteases were regularly employed through the BIOPEP-UWM server [22] in previous studies to virtually simulate human GI digestion of proteins [13,23]. BIOPEP-UWM is a free, curated database now housing more than 4000 peptides annotated with 62 types of bioactivities, with antioxidant, anti-angiotensin-converting enzyme (anti-ACE), and anti-dipeptidyl peptidase IV (anti-DPP-IV) peptides being the three major categories [22]. The applications of antioxidant peptides as food preservatives, functional food ingredients, and therapeutic agents were previously highlighted [9]. Anti-ACE and anti-DPP-IV peptides are valuable antihypertensive [24,25] and antidiabetic [26] agents, respectively. Through a search of the BIOPEP-UWM database, it would be possible to reveal whether peptides generated by in silico GI digestion of BSFL proteins comprise those with previously reported antioxidant, anti-ACE, anti-DPP-IV, and other bioactivities.
Mouithys-Mickalad and co-workers [5] reported that a commercial BSFL protein hydrolysate mitigated cellular oxidative damage by inhibiting myeloperoxidase (MPO) activity. Although the anti-MPO peptides were not identified [5], the finding implies that BSFL-derived peptides may also exert their antioxidant actions through mechanisms other than free radical scavenging, such as by modulating enzymatic and non-enzymatic regulators of cellular redox status. To explore this possibility, we also aimed to investigate the ability of BSFL peptides to interact with or inhibit four protein targets known to be regulators of cellular redox status: MPO, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX), xanthine oxidase (XO), and Kelch-like ECH-associated protein 1 (Keap1). MPO, NOX, and XO are prooxidant enzymes. MPO is an abundant heme-containing enzyme in human neutrophils. MPO can catalyze the reaction between hydrogen peroxide and chloride, leading to the formation of hypochlorous acid, which is a strong oxidant [27,28]. NOX is a membrane-bound enzyme complex whose main function is the generation of superoxide radicals. The assembly of NOX requires the participation of the cytosolic factor p47phox and transmembrane component p22phox [29,30]. Thus, the p47phox–p22phox interaction in NOX activation can be targeted in virtual screening for NOX inhibitors [31]. XO catalyzes the oxidation of hypoxanthine and xanthine to uric acid, producing reactive oxygen species (ROS) in the reaction [32,33]. On the other hand, the Keap1-nuclear factor erythroid 2-related factor 2 (Nrf2) pathway is a fundamental signaling cascade responsible for cellular protection against oxidative injury. Nrf2 is the principal regulator of antioxidant response element-containing cytoprotective genes whose expression is induced in response to increased oxidative stress. Nevertheless, Keap1–Nrf2 interaction leads to the ubiquitylation and proteasomal degradation of Nrf2. By contrast, inhibitors of Keap1–Nrf2 protein–protein interaction can preserve the transcription-activating role of Nrf2, hence alleviating ROS-mediated cellular injury [34,35].
In this study, the ability of BSFL peptides to inhibit MPO, NOX, XO, and Keap1 will be computationally evaluated by using molecular docking and molecular dynamic (MD) simulation. Both biomolecular simulation techniques are regularly used to cost-effectively and rapidly perform structure-based virtual screening for drug and bioactive compound discovery [10,13,17,36]. Molecular docking is a computational technique that can predict the preferred orientation and binding affinity of a peptide in the binding site of a target protein besides elucidating their intermolecular interactions [36]. Often, an effective peptide inhibitor would be anticipated to bind to the active site or key catalytic residues of a targeted enzyme. This would preclude access to those sites by the natural substrate of the enzyme. Meanwhile, a peptide targeting Keap1–Nrf2 protein–protein interaction would be expected to bind stably to key residues in the binding interface between the two proteins [10,17]. Further validation of a docked peptide–target complex by MD simulation would allow the behavior of the peptide and target protein to be captured in full atomic detail and at nanosecond resolution. In other words, the details of the dynamic behavior or time evolution of the intermolecular interactions between the peptide and the target protein can be unraveled [36]. In this study, the CHARMM27 force field was adopted in MD simulations. The force field is considered comprehensive and accurate due to its ability to analyze diverse types of atoms and its inclusion of many parameters that can be optimized by users [37,38]. Furthermore, the parameters in CHARMM27 were derived from a wide range of experimental and theoretical data sources, which included hydrogen bonding interactions that are important for accurately describing the behavior of biological systems [39].
In this in silico study, our objectives were three-fold: (a) to compile peptides generated by virtual GI digestion of BSFL proteins, followed by screening for high-GI-absorption, non-toxic peptides; (b) to identify high-GI-absorption and non-toxic peptide sequences previously reported to have bioactivities; (c) to perform structure-based screening of the peptides for the ability to target MPO, NOX, XO, and Keap1 by molecular docking analyses, followed by MD validation of peptide–target interactions.
2. Results and Discussion
2.1. In Silico GI Digestion
To discover whether BSFL proteins could release potentially bioavailable, health-promoting peptides upon human consumption and GI digestion, we performed virtual enzymatic digestion of the proteins using GI proteases. A total pool of 5218 fragments, 3229 of being peptides, were liberated from the 19 muscular and seven cuticular BSFL proteins (Table 1) that were virtually proteolyzed. About 95% of the liberated fragments (4934 fragments) originated from the 19 muscular proteins, whereas 5% (284) came from the seven cuticular proteins. This could be attributed not only to 2.7-fold more muscular proteins being proteolyzed than cuticular proteins. Moreover, the muscular proteins are overall larger (157–1949 residues; 17.9–222.8 kDa) compared with the cuticular proteins (76–271 residues; 8.5–28.8 kDa) (Table 1). Hence, there is greater availability of GI protease cleavage sites in muscular proteins in general.

Table 1.
Muscular and cuticular proteins of black soldier fly larvae 1.
Figure 1 summarizes the outcome of in silico GI digestion of 26 BSFL proteins. The number of peptide fragments released from the muscular proteins ranged between 465 fragments (A0A0R1DVF3_DROYA, Mhc, isoform D) and 38 fragments (A0A0L0BTD6_LUCCU, troponin C, isoform 3). The number of peptide fragments liberated from the cuticular proteins ranged between 50 fragments (A0A026VY81_OOCBI, cuticle protein) and 17 fragments (A0A1A9Z940_GLOPL, uncharacterized protein) (Figure 1A). Notably, the number of peptide fragments liberated from muscular protein (A0A0R1DVF3_DROYA, Mhc, isoform D) is about 27-fold higher than that from cuticular protein (A0A1A9Z940_GLOPL, uncharacterized protein). The average number of fragments released by each muscular protein is 260 fragments, whereas the average of that released by each cuticular protein is only 41 fragments. Taken together, our results suggest that in future studies aimed at investigating peptides following GI proteolysis of BSFL proteins, focusing on muscular proteins may be a promising approach to enhance peptide generation.
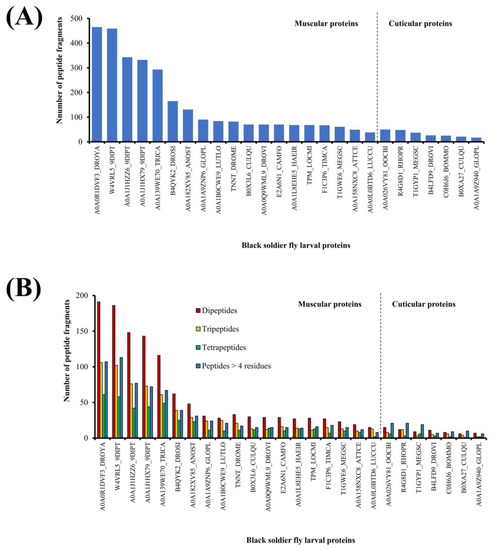
Figure 1.
Number (A) and length distribution (B) of peptide fragments liberated by in silico GI digestion of black soldier fly larval proteins.
The 26 BSFL proteins analyzed in this study collectively released 1994 dipeptide and tripeptide fragments, comprising 1281 dipeptides and 713 tripeptides. Overall, dipeptides comprised the largest portion (25% on average) of fragments released from each muscular protein when virtually hydrolyzed by GI proteases (Figure 1B). By contrast, peptides consisting of five or more residues comprised the largest group (36% on average) of fragments released by five of the cuticular proteins (A0A026VY81_OOCBI, R4G8D1_RHOPR, T1GYP1_MEGSC, C0H6J6_BOMMO, and B0XA27_CULQU). This implies that the occurrence of GI protease-specific cleavage sites may be less frequent in the sequences of those cuticular proteins, hence more frequent liberation of longer fragments (>four residues) following virtual proteolysis. Protein conformation is unlikely to be a deterring factor causing fewer dipeptides to be released from the cuticular proteins. This is because in silico proteolysis assumes the polypeptide chain of a protein to be fully extended, hence full accessibility to the cleavage sites.
Among the muscular proteins, the top five producers of dipeptides and tripeptides are those from the myosin (A0A0R1DVF3_DROYA, W4VRL5_9DIPT, and A0A139WE70_TRICA) and tropomyosin (A0A1J1HZZ6_9DIPT and A0A1J1HX79_9DIPT) groups (Figure 1B). These five proteins also have the largest molecular sizes (1225–1949 residues; 141.1–222.8 kDa) among the 26 BSFL proteins analyzed in this study (Table 1). In fact, the 1654 dipeptide and tripeptide fragments released by the myosin and tropomyosin groups collectively made up 83% of all 1994 dipeptide and tripeptide fragments liberated by virtual GI proteolysis of the 26 BSFL proteins. Thus, where a more targeted strategy is desired in the future to enzymatically generate dipeptides and tripeptides from BSFL proteins by using GI proteases, the myosin and tropomyosin isolates of BSFL are favorable substrates.
2.2. Screening for High GI Absorption and Non-Acute Oral Toxicity
Dipeptides and tripeptides are potentially bioavailable. This is because digested proteins are absorbed primarily in the form of dipeptides and tripeptides by the body, not as longer peptides or as free amino acids [40,41,42,43]. Thus, in this study, we focused on the 1994 di- and tripeptides released from virtually GI-proteolyzed BSFL proteins. It is worth noting that not all those di- and tripeptides will be absorbed by the GI tract, although they, being a product of in silico GI digestion, can be assumed to be GI-protease-resistant. The screening of the 1994 peptide fragments by using SwissADME [44] revealed 657 high-GI-absorption peptides. Interestingly, all those 657 peptides were predicted to have no acute oral toxicity by STopTox [45]. This in silico finding implies that the consumption of BSFL proteins is not associated with the potential uptake of toxic di- and tripeptides by the body. Meanwhile, the 1337 peptides, not predicted to be highly absorbed by the GI tract, were not subjected to further screening for toxicity or any other bioactivity in this study. Although we cannot confirm whether any of these 1337 peptides are toxic or bioactive, we believe their likelihood of exerting any significant activity in the body is minimal due to their predicted low GI absorption.
Upon eliminating repetitive sequences, the 657 peptide fragments can be consolidated into 65 unique sequences; the frequency of occurrence of each sequence is depicted in Figure 2. Collectively, the first 25 sequences in Figure 2 represented 548 fragments, i.e., 83.4% of the 657 high-GI-absorbable and non-toxic peptide fragments. All those abundant 25 sequences (EL, AL, SL, DL, AK, TL, VK, GK, IK, AF, VL, IL, GF, PK, IM, PL, TF, GL, SF, AM, AY, TY, DF, GY, and GM) are dipeptides. They ranged between 188 and 282 Da in mass. The 25 dipeptide sequences each represent more than 1% of the 657 fragments. Peptide fragments liberated from five muscular proteins, three myosins (A0A0R1DVF3_DROYA, W4VRL5_9DIPT, and A0A139WE70_TRICA), and two tropomyosins (A0A1J1HZZ6_9DIPT and A0A1J1HX79_9DIPT), contributed to 60% of peptide fragments having the 25 aforementioned sequences. This suggests that the five proteins are the most promising BSFL sources of potentially bioavailable peptides upon GI digestion.
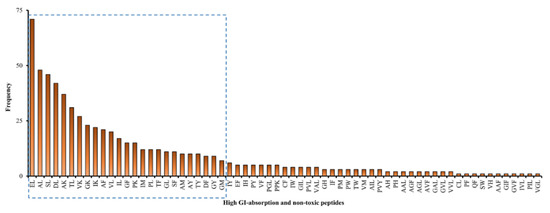
Figure 2.
Frequency of each of the 65 unique peptide sequences comprising the 657 high-GI-absorbable and non-toxic peptide fragments liberated from black soldier fly larval proteins. The 25 peptide sequences in the blue dashed box contribute between 10.8% (EL) and 1.1% (GM) to the total of 657 fragments.
2.3. Searching Databases for Bioactivity
To predict the potential health benefits of BSFL proteins for human consumption, we searched the BIOPEP-UWM database and publications in the Scopus database for bioactivities reported for the 25 most abundant, high-GI-absorbable, and non-toxic peptide sequences (Figure 2). The peptide DL is the only one having no previously reported bioactivity. The remaining 24 dipeptides were previously reported to have at least one reported bioactivity. Overall, the three major bioactivities for the peptide sequences are as follows: antioxidant; anti-ACE; and anti-DPP-IV activities (Figure 3).
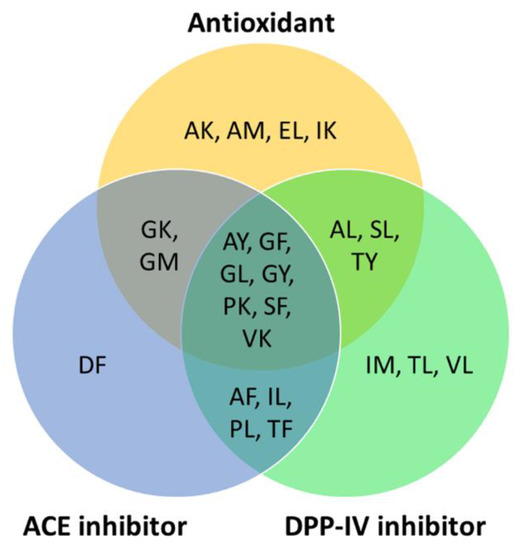
Figure 3.
High-GI-absorbable and non-toxic peptide sequences reported to have antioxidant, anti-ACE, and anti-DPP-IV activities based on records in the BIOPEP-UWM databases and the published literature in the Scopus database (accessed on 30 June 2022).
Sixteen of the 25 peptide sequences, including the most abundant EL, were previously reported to be antioxidant peptides (Figure 3). Five sequences, AK, GK, IK, PK, and VK, were demonstrated to have antioxidant activity as determined by using the ferric thiocyanate method [46]. Five dipeptides, AL, GL, SF, SL [47], and EL [48], were shown to have superoxide anion radical scavenging activity. Two dipeptides, AM and GM, were reported to have peroxyl radical scavenging activity [49]. Two other dipeptides, GF and GY, were found to have 2,2-diphenyl-1-picrylhydrazyl (DPPH) and hydroxyl radical scavenging activities [50]. AY exhibited DPPH scavenging activity and ferric-reducing potential [51]. TY exhibited 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) radical scavenging activity and inhibited peroxyl radical-induced oxidation, as revealed by the oxygen radical absorbance capacity assay [52]. The aforementioned evidence, although primarily derived from in vitro assays, implies that the 16 high-GI-absorption peptides may be capable of playing an antioxidant role, protecting against oxidative stress in the body.
Fourteen of the 25 peptide sequences (AF, AY, DF, GF, GK, GL, GM, GY, IL, PK, PL, SF, TF, and VK) were previously shown to exhibit anti-ACE activity [53,54,55,56,57,58,59,60]. Four of the anti-ACE peptides were previously identified in edible plant sources, corn gluten meal (AY) [56], garlic (SF) [55], wheat milling byproducts and rapeseed (TF) [14,53], and Tartary buckwheat (VK) [57], respectively. On the other hand, 17 peptide sequences (AF, AL, AY, GF, GL, GY, IL, IM, PK, PL, SF, SL, TF, TL, TY, VK, and VL) were previously demonstrated to have anti-DPP-IV activity [61,62,63] (Figure 3). Many of the 25 abundant, high-GI-absorption, non-toxic dipeptides have multi-functionality. Nine of the dipeptides are bifunctional, having both antioxidant and anti-ACE activities. Ten of the dipeptides have both antioxidant and anti-DPP-IV activities. Eleven of the dipeptides have both anti-ACE and anti-DPP-IV activities. Notably, seven of the peptides are trifunctional, having antioxidant, anti-ACE, and anti-DPP-IV activities (Figure 3). Besides the aforementioned major bioactivities, four other bioactivities were found in a small number of the 25 peptides, DPP-III inhibitor (GF and TF) [64,65], renin inhibitor (SF and TF) [14,66], stimulator of glucose uptake (IL and VL) [67], and regulator of phosphoglycerate kinase activity (SL) [68].
The most abundant dipeptide EL (71 fragments) (Figure 2) was released by 17 BSFL proteins. EL has only been reported to have a single bioactivity (antioxidant) [48]. By contrast, TF is a more well-characterized antihypertensive peptide among the 25 sequences. Previously, TF was identified from alcalase-hydrolyzed rapeseed proteins [14]. TF not only exhibited both anti-ACE and anti-renin activities but also in vivo antihypertensive effects in spontaneously hypertensive rats following oral administration of the dipeptide [14]. Nonetheless, in this study, TF (12 fragments) was less abundant than EL, being released by only nine BSFL proteins following in silico GI proteolysis.
Antioxidant peptides can dampen ROS-mediated disease development and diseases-related complications [8,9]. Meanwhile, ACE and DPP-IV are therapeutic targets for the management of the hypertension [24,25] and type 2 diabetes [26], respectively. Thus, the release of highly-GI-absorbable antioxidant, anti-ACE, and anti-DPP-IV peptides from BSFL proteins (Figure 3) implies that consumption of BSFL proteins may benefit the control of blood pressure and glucose levels in the body. The BSFL proteins can also be explored for the formulation of functional foods or nutraceuticals. Taken together, the 25 most abundant dipeptide sequences depicted in Figure 2, except for DL, possess health-promoting bioactivities. Despite their predicted high-GI-absorption tendency, whether GI digestion of BSFL proteins could release such peptides quantitatively sufficient to exert in vivo effects is a question that requires verification by using animal studies in the future.
2.4. Molecular Docking Analysis
To further explore the potential of the 25 most abundant, high-GI-absorption dipeptide sequences as inhibitors of MPO, NOX, and XO activities, as well as Keap1–Nrf2 protein–protein interactions, molecular docking analysis was conducted, followed by MD simulation analysis. Molecular docking analysis can be used to screen for the most stable peptide–target complexes rapidly, where the most negative binding affinity or docking score is taken as an indicator of the greatest binding stability between a peptide and its target. Two tools, Webina and HPEPDOCK, were used in this study to generate the docking results. Webina provided binding affinity data, while HPEPDOCK generated docking score data. The docking score from HPEPDOCK is a numerical value that predicts the binding affinity and stability of a peptide–protein complex. A more negative docking score suggests a stronger affinity between the peptide and protein, which indicates a more stable predicted complex. However, the docking score is a relative measure that only provides a ranking of different binding modes of a peptide–protein complex. While the docking score can be a useful predictor of binding affinity, it is not equivalent to the actual binding affinity of a peptide–protein complex [69].
In this study, we have selected the crystal structure of the bovine XO–quercetin complex (3NVY) for our study for several reasons. Firstly, no crystal structure of the human XO protein–ligand complex is currently available. Moreover, the resolution of 3NVY (2.00 Å) [33] is higher than those of other crystal structures of human XO without bound ligands, such as 2CKJ (3.59 Å) [70] and 2E1Q (2.60 Å) [71]). Furthermore, the 3NVY crystal has the greatest resolution (2.0 Å) among the bovine XO proteins deposited in the Protein Data Bank [72]. Secondly, XO is highly conserved across different species, including humans and bovines. Notably, the bovine milk XO (1332 residues) shares 90% sequence identity with the human liver XO (1333 residues) [73]. Lastly, the quercetin binding site in the bovine XO crystal structure (3NVY) is both structurally and sequentially conserved between bovine and human enzymes [33]. Therefore, the quercetin binding mode in the bovine XO can be used as a reasonable approximation for the human XO–quercetin complex. Taken together, these factors indicate that 3NVY is a suitable model for investigating the human XO–peptide interaction in our study.
Prior to analyzing the docking of the BSFL-derived peptides and reference peptides, we performed a redocking step as described in the Materials and Methods. Root Mean Square Deviation (RMSD) values computed by comparing the poses of the redocked ligands with their original crystallographic poses were 0.891, 0.689, 1.820, and 1.458, respectively (Figure 4). The four values are considered satisfactory because RMSD values less than 2.0 Å indicate that the poses are considered “near-native” [74]. In light of the satisfactory RMSD values, we proceeded to conduct molecular docking analyses on the BSFL-derived peptides and reference peptides. Generally, the ranges of binding affinity were similar for the three targets, MPO, XO, and Keap1 (Figure 5A–C). Among the 25 dipeptide sequences, DF had the most negative binding affinity when docked against MPO and Keap1, suggesting that DF could bind most stably with the two targets. The most negative binding affinity computed for TF when complexed with XO indicates the strongest binding between TF and XO. Meanwhile, the most negative docking score computed for TY (Figure 5D) indicates that TY-p47phox could be the most stable when compared with complexes between p47phox and the other 24 peptides. Nevertheless, none of the 25 peptides surpassed the co-crystallized ligands in terms of binding affinities and docking scores. ETGE and p22phox likely had a higher affinity to Keap1 and p47phox, respectively, than the 25 BSFL peptides due to their longer length, providing more sites for interaction with the two proteins. In this study, the binding modes between the 25 peptides and p47phox were not analyzed further. This is because their docking scores were all drastically less negative than those of the co-crystallized ligand of p47phox, a proline-rich peptide derived from p22phox (Figure 5D). The p22phox-derived peptide is beyond the scope of Webina. Redocking of p22phox to p47phox with Webina failed to work, making it impossible for us to validate the docking procedure. We, therefore, used HPEPDOCK, a well-established peptide docking tool in the case of p22phox–p47phox interaction. Based on the binding affinities presented in Figure 5A–C, we narrowed down the 25 dipeptide sequences to several promising sequences and investigated their binding modes with MPO, XO, and Keap1, as discussed further below.
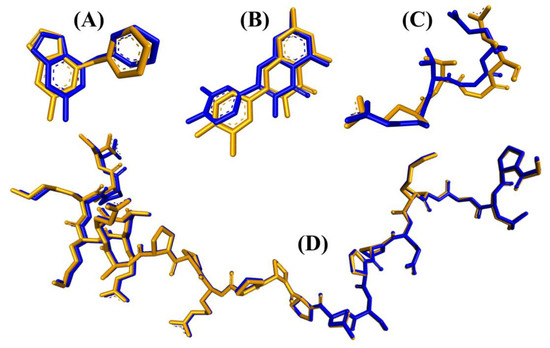
Figure 4.
Superimposition of the co-crystallized (orange) and the redocked (blue) poses of the same ligands for the four target proteins: (A) MPO, (B) XO, (C) Keap1, and (D) p47phox.
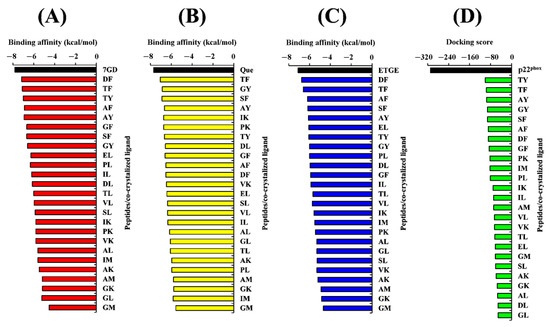
Figure 5.
Binding affinities of 25 peptides complexed with (A) MPO, (B) XO, and (C) Keap1, in comparison with the co-crystallized ligand of each target protein. Docking scores of 25 peptides complexed with p47phox in comparison with the co-crystallized ligand (D). For (A), 7GD is 7-benzyl-1H-[1,2,3]triazolo [4,5-b]pyridin-5-amine, the co-crystallized inhibitor of MPO. For (B), Que is quercetin, the co-crystallized inhibitor of XO. For (C), ETGE is the key motif of the Nrf2 peptide co-crystallized with Keap1. For (D), p22phox represents the co-crystalized p22phox-derived proline-rich peptide (GPLGSKQPPSNPPPRPPAEARKKPS).
Table 2 shows the intermolecular interactions between three selected BSFL peptides and MPO. Because the co-crystallized ligand in the MPO crystal is not a peptide, we compared the three BSFL peptides with N-acetyl lysyltyrosylcysteine amide (KYC), an experimentally-validated anti-MPO peptide [75]. The binding affinities of the three dipeptides, DF, TF, and TY, are comparable to that of KYC. Furthermore, similar to KYC, the three dipeptides could each form hydrophobic interaction with Arg239 and Phe407, key residues in the active site of MPO, as well as with the heme group (Hec606) of MPO (Table 2). The 7-benzyl-1H-[1,2,3]triazolo [4,5-b]pyridin-5-amine, the co-crystallized inhibitor of the MPO crystal used in this study, were reported to bind to the two residues Arg239 and Phe407 [76]. Moreover, Arg239 is a catalytic residue of MPO [77]. Our data show that DF, TF, and TY could potentially bind to the active site of MPO as do other MPO inhibitors [75,77], thus implying the anti-MPO potential of the three dipeptides. Hydrophobic interactions with MPO are critical to the potency of MPO inhibitors [76]. In line with this, we found that hydrophobic interactions accounted for 77-90% of MPO–peptide interactions involving DF, TF, and TY (Table 2). Among the three BSFL dipeptides, DF stood out as the most promising candidate as an MPO inhibitor, considering its strongest binding affinity to MPO and its binding to the key catalytic/active-site residues. A graphical illustration of the DF-MPO docked model and its intermolecular interactions in the 2D structure are shown in Figure 6.
Table 2.
Intermolecular interactions between BSFL peptides and MPO.
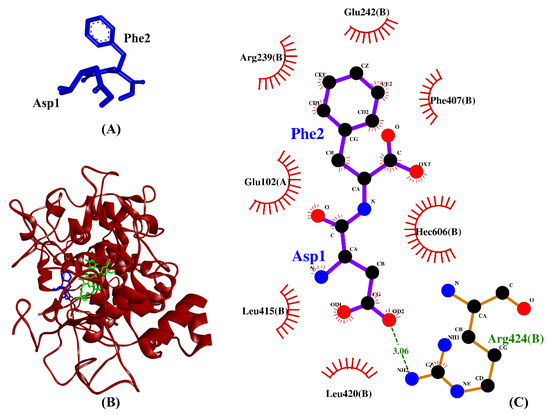
Figure 6.
(A) 3D diagram of DF; (B) 3D diagram of DF-MPO docked model; (C) 2D diagram of DF–MPO interactions. MPO is shown in red ribbon, whereas DF and the heme group are displayed in blue- and green-stick styles, respectively, in (B). In the 2D diagram (C), bonds of MPO are in orange, whereas those of DF are in purple. Hydrophobic interactions and hydrogen bonds are shown in red spoked arcs and green dashed lines, respectively.
Table 3 shows the intermolecular interactions between three selected BSFL peptides and XO. Three experimentally-validated anti-XO peptides, namely, IW [63], GPY [78], and ACECD [79], were taken as positive controls. In this study, the binding affinities of TF, GY, and SF were 9–28% more negative than those of GPY and ACECD (Table 3). This implies that the three dipeptides bound to XO more stably than did two anti-XO peptides. Similar to the three validated anti-XO peptides, the three BSFL dipeptides could form hydrophobic interactions with the catalytic residue Glu802 and with at least one other inhibitor-binding residue (Phe914). Interestingly, our observation on ACECD suggests that the ability to bind to a greater number of the inhibitor-binding residues in XO does not necessarily lead to a more stable binding between a peptide and XO. GY is the only BSFL dipeptide that could make hydrogen bond with catalytic residue Glu802. However, the results in Table 3 show that the formation of a hydrogen bond to Glu802 is not crucial to the stable binding of a peptide to XO; neither is it associated with the ability to inhibit XO experimentally. The majority of the intermolecular interactions shown in Table 3 are hydrophobic interactions, in line with the previous finding that the main interaction forces between XO and XO-inhibitory peptides were hydrophobic interaction [80]. Taken together, the resemblance between the three BSFL dipeptides and the three known anti-XO peptides implies that the BSFL peptides are potential XO inhibitors. Among them, TF is the most promising considering its strongest binding affinity to XO. A graphical illustration of the TF-XO docked model and its intermolecular interactions in the 2D structure are shown in Figure 7.
Table 3.
Intermolecular interactions between BSFL peptides and XO.
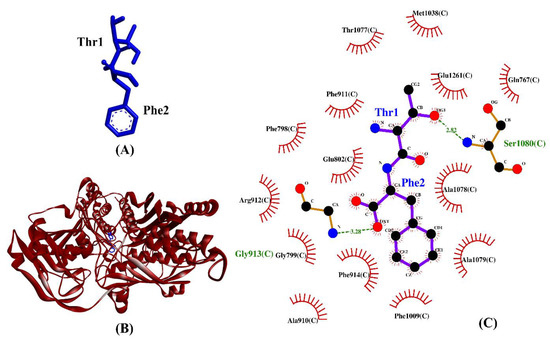
Figure 7.
(A) 3D diagram of TF; (B) 3D diagram of TF-XO docked model; (C) 2D diagram of TF–XO interactions. TF is displayed in a blue-stick style, whereas XO is in red ribbon in (B). In the 2D diagram (C), bonds of XO are in orange, whereas those of TF are in purple. Hydrophobic interactions and hydrogen bonds are shown in red spoked arcs and green dashed lines, respectively.
Our molecular docking simulation suggests DF and TF be potential inhibitors of Keap1–Nrf2 protein–protein interaction. As shown in Table 4, the hydrogen bonds and hydrophobic interactions formed by DF, TF, DDW, and DKK with Keap1 are similarly dominated by key Keap1 residues known for binding to Nrf2. The two reference peptides, DDW and DKK, were experimentally-verified inhibitors of the Keap1–Nrf2 interaction [81]. The binding affinities of DF and TF toward Keap1 were superior or comparable to that of DKK. This implies that the two dipeptides bound to Keap1 more stably than did DKK. Meanwhile, DDW and DF could both form hydrogen bonds with Arg415 and Arg483, hydrophobic interactions with Tyr334, Arg415, Arg483, Ser508, Tyr525, Ser555, Tyr572, Phe577, and Ser602, as well as a salt bridge with Arg415. All the aforementioned Keap1 residues are key to the formation of a stable Keap1–Nrf2 complex [82]. Among the 25 BSFL peptides, DF is likely to be the best candidate for inhibitor of Keap1–Nrf2 interaction based on its binding affinity, as well as the resemblance of its binding mode to those of the co-crystalized ETGE motif and DDW. A graphical illustration of the DF-Keap1 docked model and its intermolecular interactions in the 2D structure are shown in Figure 8.
Table 4.
Intermolecular interactions between BSFL peptides and Keap1.
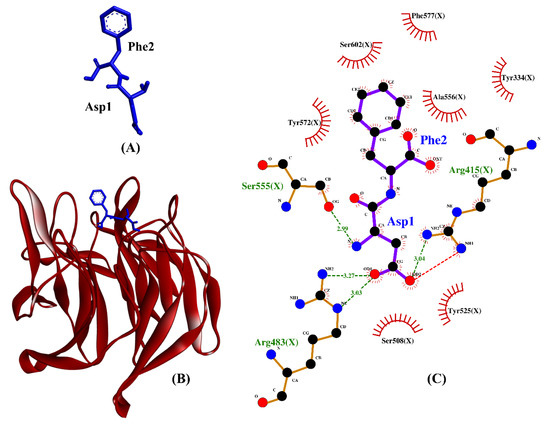
Figure 8.
(A) 3D diagram of DF; (B) 3D diagram of DF-Keap1 docked model; (C) 2D diagram of DF-Keap1 interactions. DF is displayed in a blue-stick style, whereas Keap1 is displayed as red ribbon in (B). In the 2D diagram (C), bonds of Keap1 are in orange, whereas those of DF are in purple. Hydrophobic interactions, hydrogen bonds, and salt bridges are represented in red spoked arcs and green and red dashed lines, respectively.
2.5. Molecular Dynamics Simulation
The DF-MPO, TF-XO, and DF-Keap1 docked models, which exhibited the most favorable binding affinities, were subjected to 100 ns and independently repeated MD simulation analysis. Four parameters were evaluated in the following analyses: RMSD; the radius of gyration (Rg); intermolecular hydrogen bond formation; and intermolecular protein–peptide distance. RMSD is a common metric used to quantify the difference between the positions of atoms in two structures in MD [83].
Figure 9A shows that the RMSD values of the DF-docked Keap1 were mostly stable during the 100 ns simulation. By contrast, the RMSD values of DF-docked MPO and TF-docked XO showed a more obvious increase during the 100 ns simulation. A slight peak can be observed at around 75–80 ns for MPO, implying a minor movement of the protein, before stabilizing again up to 100 ns. The RMSD values of the three proteins, when averaged over the 100 ns duration and presented as mean ± standard deviation (SD), were 1.4051 ± 0.0926 Å (Keap1), 2.3343 ± 0.2130 Å (XO), and 2.4127 ± 0.2901 Å (MPO). These low mean values of approximately 2.000 Å and below, as well as the low SD values, indicate that the three proteins did not undergo much conformational change [84]. On the other hand, the averaged RMSD values of the three docked dipeptides, presented as mean ± SD, were 1.414 ± 0.2290 Å (DF, docked to MPO), 1.5290 ± 0.2198 Å (TF, docked to XO), and 1.7928 ± 0.1917 Å (DF, docked to Keap1). As observed in Figure 9B, the docked peptides displayed greater fluctuations in their docking positions compared with the three target proteins. Generally, several factors contribute to higher RMSD fluctuations in a ligand compared to its receptor, including the size of the ligand [85], its flexibility [86], and the nature of protein–ligand interactions [87]. The ligand is usually smaller in size compared to the receptor, and hence, its atoms are more susceptible to fluctuations and movements [88]. In addition, the ligand has more flexible and dynamic regions compared to the receptor, resulting in higher RMSD fluctuations, which captured its full range of motions [89]. The ligand is also involved in specific interactions with the receptor, and any changes in these interactions can result in changes in the ligand’s conformation and higher RMSD fluctuations [90]. The plot depicted in Figure 9B indicates that the dipeptide DF, upon docking to MPO, exhibited substantial fluctuations, with the residues’ backbone rotations likely occurring more aggressively over time, particularly beyond the 50 ns timepoint. On the other hand, the dipeptide TF bound to XO demonstrated stability in its docked position against the protein. The differential behaviors of dipeptides DF and TF could be attributed, at least in part, to the presence of aspartate (D) or threonine (T) residues. The side chain of aspartate is more hydrophilic than threonine, which affects the hydropathy of the dipeptides. This may have facilitated the interactions between DF and water molecules in the MPO’s active site microenvironment. In contrast, threonine’s less hydrophilic side chain may result in TF interacting less with water in the XO binding site. Consequently, DF showed an increased tendency to leave the MPO active site after a brief period of time [91,92,93,94].
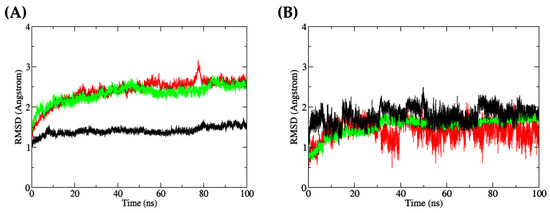
Figure 9.
(A) all-atom RMSD of receptor proteins (red: MPO; green: XO; black: Keap1); (B) all-atom RMSD of dipeptide ligands bound to the receptor proteins (red: DF (bound to MPO); green: TF (bound to XO); black: DF (bound to Keap1)).
Rg is defined as the distance between the center of mass of a molecule (protein or ligand) and the average position of its constituent atoms [95]. Thus, Rg is an important parameter for assessing protein folding and the flexibility of a ligand in a simulated environment over time [96]. As shown in Figure 10A, the overall stability of the Rg values of peptide-docked MPO, XO, and Keap1 throughout the 100 ns simulation time suggests negligible levels of protein unfolding. The Rg values of the three proteins, when averaged over the 100 ns duration and presented as mean ± SD, were 17.7963 ± 0.0343 Å (Keap1), 23.9047 ± 0.0679 Å (MPO), and 28.1789 ± 0.0700 Å (XO). The observation implies limited protein structure fluctuations [97]. On the other hand, the averaged Rg values of the three docked dipeptides, presented as mean ± SD, were 3.2508 ± 0.0572 Å (TF, docked to XO), 3.3069 ± 0.0861 Å (DF, docked to MPO), and 3.3889 ± 0.1072 Å (DF, docked to Keap1). As shown in Figure 10B, among the three dipeptides analyzed, the Rg values for MPO-bound dipeptide DF fluctuated the most, followed by Keap1-bound DF, especially after the 30 ns timepoint. In contrast, it is likely that the TF bound to XO underwent comparatively minimal unfolding at the docking site, thereby resulting in a more stable conformation.
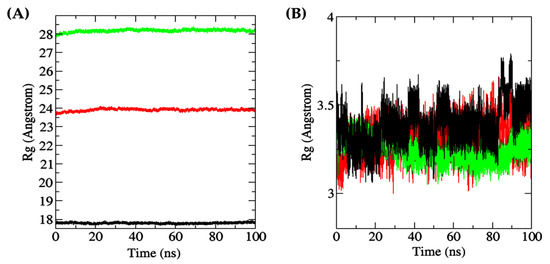
Figure 10.
(A) all-atom Rg of receptor proteins (red: MPO; green: XO; black: Keap1); (B) all-atom Rg of dipeptide ligands bound to receptor proteins (red: DF (bound to MPO); green: TF (bound to XO); black: DF (bound to Keap1)).
Intermolecular hydrogen bonds play a crucial role in demonstrating the interaction between proteins and ligands, as they substantially contribute to the stability and specificity of the ligand–protein complex formation [98]. In this context, Figure 11A depicts that during the 100 ns simulation, the number of intermolecular hydrogen bonds was the most stable in the DF–Keap1 complex, followed by the TF–XO complex. When compared with the other two complexes, the TF–XO complex had the highest number of hydrogen bonds initially before gradually decreasing after 25 ns of simulation. The present study observed an increase in the number of intermolecular hydrogen bonds between DF and Keap1 after the 45 ns timepoint, which subsequently reached equilibrium by the 100 ns timepoint. In contrast, the number of hydrogen bonds between the DF–MPO complex demonstrated significant fluctuation throughout the simulation, with frequent losses of hydrogen bonds occurring at various time points, particularly after the 20 ns time point. The number of intermolecular hydrogen bonds formed in the DF–Keap1 complex, when averaged over the 100 ns duration and presented as mean ± SD, was 3.0355 ± 0.7736, which was the highest among the three complexes studied. In contrast, the DF–MPO complex exhibited the least hydrogen bond formation, with a mean of 1.4520 ± 0.8746 bonds. The TF–XO complex showed a moderate level of intermolecular hydrogen bond formation, with a mean of 2.0076 ± 0.9574 bonds. This phenomenon was also translated in the protein–ligand distance of the complex. In this study, the DF peptide, upon binding to Keap1, and the TF peptide, upon binding to XO, were found to be in close proximity, i.e., within a distance of 3 Å, to their respective receptors, with intermolecular distances of 1.7190 ± 0.0798 Å and 1.7840 ± 0.0966 Å, respectively (Figure 11B). In contrast, the DF–MPO complex displayed a greater average intermolecular distance of 4.1404 ± 4.2743 Å. Figure 11B also shows that the intermolecular distance of the DF–MPO complex increased over the course of the simulation, indicating that DF may detach from the protein after 40 ns. Our findings suggest that the DF–MPO complex exhibited the least stability among the three peptide–protein complexes that were examined, likely attributable to the loss of hydrogen bonds and an increase in overall protein–ligand distance. Although a low minimum distance with a mean value of less than 10.0 Å between a ligand and its receptor protein may still suggest strong affinity and stable binding of the ligand to its receptor [99], protein–ligand clustering and mutagenesis targeting active binding residues are warranted in the future to evaluate the stability of the DF–MPO complex.
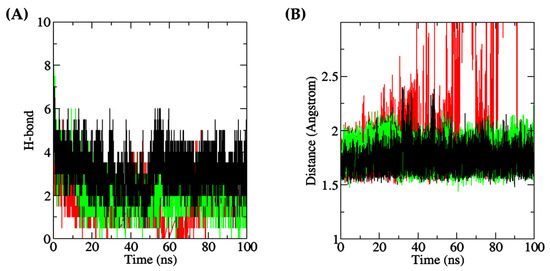
Figure 11.
(A) Number of intermolecular (protein–peptide) hydrogen bonds (red: MPO; green: XO; black: Keap1); (B) Minimum distance between each protein and its dipeptide ligand (red: DF, bound to MPO; green: TF, bound to XO; black: DF, bound to Keap1).
3. Materials and Methods
3.1. BSFL Protein Sequences
Seven cuticular and 19 muscular proteins previously identified as the major proteins in BSFL [100] were studied. The sequences of the 26 BSFL proteins were retrieved from the UniProt Knowledgebase (https://www.uniprot.org/) [101] (accessed on 25 April 2022). The number of residues and molecular mass of each BSFL protein were recorded.
3.2. In Silico GI Digestion
The 26 protein sequences were submitted to the BIOPEP-UWM: Bioactive peptides server (https://biochemia.uwm.edu.pl/en/biopep-uwm-2/) [22] (accessed on 3 May 2022) for simulating in silico GI digestion by using the “ENZYME(S) ACTION” tool. Three GI proteases were used, namely, pepsin (pH 1.3) (EC 3.4.23.1), trypsin (EC 3.4.21.4), and chymotrypsin A (EC 3.4.21.1), as previously described [13,23,102]. The fragments released by each protein were recorded and categorized by length: one residue (free amino acids), two residues (dipeptides), three residues (tripeptides), four residues (tetrapeptides), and peptides > four residues.
3.3. Screening for High GI Absorption and Oral Toxicity
All peptide fragments liberated by in silico GI digestion were screened for GI absorption by using the SwissADME server (http://www.swissadme.ch/) [44] (accessed on 11 May 2022). Peptides predicted to be high-GI-absorption were subsequently analyzed for acute oral toxicity via the STopTox app (https://stoptox.mml.unc.edu/) [45] (accessed on 11 May 2022). Peptide sequences in the Simplified Molecular-Input Line-Entry System (SMILES) format, which were required as input for analysis on SwissADME and STopTox, were generated by using the “SMILES” tool on BIOPEP-UWM (accessed on 11 May 2022). The molecular masses of selected peptide sequences were predicted to have high GI absorption, and no oral toxicity was calculated by using PepDraw (https://www2.tulane.edu/~biochem/WW/PepDraw/) (accessed on 11 May 2022).
3.4. Database Searching for Peptide Bioactivity
Peptide sequences were searched against the BIOPEP-UWM [22] and Scopus databases for previously reported bioactivities (accessed on 30 June 2022).
3.5. Molecular Docking Analysis
The crystal structures of the four target proteins were downloaded from RCSB Protein Data Bank (https://www.rcsb.org/) [103] on 15 October 2022. The four crystal structures were (i) human MPO in complex with 7-benzyl triazolopyridines (PDB ID: 6WYD) [76], (ii) bovine XO in complex with quercetin (PDB ID: 3NVY) [33], (iii) human Keap1 Kelch domain in complex with a 16-mer peptide of Nrf2 (PDB ID: 2FLU) [82], and (iv) human p47phox in complex with a p22phox-derived proline-rich peptide (GPLGSKQPPSNPPPRPPAEARKKPS)(PDB ID: 1WLP) [30].
The three-dimensional (3D) structures of the peptides used in molecular docking were obtained from the dataset of 3D structures of dipeptides and tripeptides provided by Prasasty and Istyastono [104] (accessed on 15 October 2022). The structure files were converted into the PDB format via the BIOVIA Discovery Studio Visualizer (BIOVIA, Dassault Systèmes, BIOVIA Discovery Studio Visualizer, Version 20.1.0.192, San Diego: Dassault Systèmes, CA, USA, 2020) before use. The 3D structure of the peptide ACECD, which was used as a positive control for docking to XO, was constructed by using PEP-FOLD 3.5 (https://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::PEP-FOLD3) [105,106,107] (accessed on 21 October 2022). One hundred simulations were run. The best output model sorted by sOPEP energy was downloaded and used for molecular docking analysis. KYC is a known anti-MPO peptide that was used as a positive control in molecular docking to MPO. KYC was downloaded as a two-dimensional structure (PubChem CID 51352312) in the SDF format from PubChem (https://pubchem.ncbi.nlm.nih.gov/) [108] (accessed on 20 October 2022). The 3D structure of KYC that was required for molecular docking was constructed on Frog2 (https://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::Frog2) [109] (accessed on 20 October 2022).
To prepare for molecular docking, the proteins and co-crystalized ligands were first separated from the crystals of MPO, XO, and Keap1 via the BIOVIA Discovery Studio Visualizer. This was followed by the removal of water and the addition of polar hydrogens and Kollman charges via the AutoDock Tools 1.5.7 [110]. The proteins were energy-minimized with the Swiss-PdbViewer 4.1 software [111]. Via AutoDock Tools 1.5.7, the three proteins were prepared as receptors, while the co-crystalized ligands and the peptides to be docked were prepared as ligands; all of them were saved in the PDBQT format. Molecular docking on MPO, XO, and Keap1 was performed on Webina 1.0.3 (https://durrantlab.pitt.edu/webina/) [112] (accessed on 15 October 2022) in triplicates. The coordinates of the box center and box sizes used for molecular docking are presented in Table S1. Following docking analyses, the top (most negative) binding affinity computed for each peptide was recorded. The analysis of the intermolecular interactions of selected docked models was performed with LigPlot+ v.2.2.4 [113,114].
To validate the docking procedures, we redocked the co-crystallized ligands separated from the original crystal structures to the respective proteins. The pose of the resultant best-score (most negative) docked ligand model was compared with the pose of the original co-crystallized ligand to evaluate conformational differences. The redocking of the co-crystalized ligands to MPO and XO was performed on Webina 1.0.3 using the same box center and box size settings as in Table S1. RMSD values were calculated on DockRMSD (https://zhanggroup.org/DockRMSD/) [74] (accessed on 22 October 2022). Redocking to Keap1 and RMSD determination were performed in a similar manner, except that a tetrapeptide ETGE, the key motif of the 16-mer peptide of Nrf2, was redocked to Keap1 as recommended [81].
For the docking of peptides to human p47phox, the protein and its co-crystalized ligand were separated from the crystal (PDB ID: 1WLP), and each was added with polar hydrogen via BIOVIA Discovery Studio Visualizer. Energy minimization of the protein was then performed via the Swiss-PdbViewer 4.1. Molecular docking between p47phox and peptides was performed on the HPEPDOCK 2.0 server (http://huanglab.phys.hust.edu.cn/hpepdock/) [69,115,116,117,118,119] (accessed on 13 October 2022). Following docking analyses, the top (most negative) docking score predicted for each peptide was recorded. Redocking of the co-crystalized peptide GPLGSKQPPSNPPPRPPAEARKKPS to p47phox was also performed on HPEPDOCK 2.0; RMSD value was computed on DockRMSD (accessed on 13 October 2022).
3.6. Molecular Dynamics Simulation
MD simulations were performed by using the GROMACS 2020 simulation package. Each system was modeled as a solvated protein–peptide complex in the Simple-Point-Charge water model. The CHARMM27 force field was selected, and sodium chloride was included to neutralize the total charge of the system [120]. The starting structures of three systems, namely, the complexes of DF docked to Keap1, DF docked to MPO, and TF docked to XO, were obtained from the aforementioned docking simulation steps. Each system was subjected to energy minimization to remove residual forces or strains, as described by Bibi and co-workers [121]. The simulation was performed in the isothermal-isobaric ensemble at a temperature of 310 K and a pressure of 1 bar with the Berendsen barostat. The particle-mesh Ewald method was used to handle long-range electrostatic interactions. A time step of 2 fs was used for the integration of the equations of motion, and the system was simulated for 100 ns. Two replicates of MD simulation were performed, and all MD data reported in this paper were mean values of the two replicates. The stability of the complex was analyzed by monitoring the RMSD and Rg of the protein structure and the peptide over time, as described in Surti and co-workers [122]. The number of intermolecular hydrogen bonds in each protein–peptide complex and the protein–peptide minimum distance was determined by using analysis tools in GROMACS following the methods described in Shamsi and co-workers [123]. Each graph was plotted by Xmgrace and averaged, and the mean values for 100 ns with standard deviations were recorded.
4. Conclusions
In this in silico study, the virtual screening of 5218 fragments released from 26 muscular and cuticular proteins of BSFL revealed the presence of dipeptide sequences with previously reported antioxidant, anti-ACE, and anti-DPP-IV activities. Molecular docking and dynamics simulations indicate that selected BSFL dipeptides could serve as potential antagonists of MPO, XO, and Keap1. Our findings suggest the potential of BSFL proteins as sources of health-promoting peptides that are non-toxic and easily absorbed by the GI tract. In vitro and in vivo studies are necessary to validate the identity and quantity of peptides released from BSFL proteins after GI digestion, besides characterizing their bioavailability. Moving forward, future research should prioritize evaluating the anti-ACE and anti-DPP-IV activity and, by extension, anti-hypertension and anti-hyperglycemia effects of BSFL proteins following oral consumption and GI digestion in animal models. Additionally, the antioxidant activity of BSFL peptides, including their role as antagonists or inhibitors of cellular modulators of oxidative stress (such as MPO, XO, and Keap1–Nrf2 protein–protein interaction), is worth exploring. Our study provides a basis for further research on the use of BSFL protein in human nutrition and highlights the potential of using computational approaches to explore the health benefits of novel protein sources.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal13030605/s1, Table S1: Coordinates of box centers and box sizes used in the molecular docking of MPO, XO, and Keap1.
Author Contributions
Conceptualization, F.-C.W., F.A.M. and T.-T.C.; methodology, T.-T.C., J.-H.O. and M.Z.S.; validation, J.-H.O., Y.-H.L. and M.Z.S.; formal analysis, J.-H.O., Y.-H.L. and M.Z.S.; investigation, F.-C.W., J.-H.O. and Y.-H.L.; data curation, F.-C.W. and T.-T.C.; writing—original draft preparation, J.-H.O., Y.-H.L. and M.Z.S.; writing—review and editing, F.-C.W. and T.-T.C.; visualization, J.-H.O., Y.-H.L. and M.Z.S.; supervision, F.A.M. and T.-T.C.; project administration, F.A.M., F.-C.W. and T.-T.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bessa, L.W.; Pieterse, E.; Marais, J.; Hoffman, L.C. Why for feed and not for human consumption? The black soldier fly larvae. Compr. Rev. Food Sci. Food Saf. 2020, 19, 2747–2763. [Google Scholar] [CrossRef]
- Lu, S.; Taethaisong, N.; Meethip, W.; Surakhunthod, J.; Sinpru, B.; Sroichak, T.; Archa, P.; Thongpea, S.; Paengkoum, S.; Purba, R.A.P.; et al. Nutritional composition of black soldier fly larvae (Hermetia illucens L.) and its potential uses as alternative protein sources in animal diets: A review. Insects 2022, 13, 831. [Google Scholar] [CrossRef] [PubMed]
- Firmansyah, M.; Abduh, M.Y. Production of protein hydrolysate containing antioxidant activity from Hermetia illucens. Heliyon 2019, 5, e02005. [Google Scholar] [CrossRef]
- Lu, J.; Guo, Y.; Muhmood, A.; Zeng, B.; Qiu, Y.; Wang, P.; Ren, L. Probing the antioxidant activity of functional proteins and bioactive peptides in Hermetia illucens larvae fed with food wastes. Sci. Rep. 2022, 12, 2799. [Google Scholar] [CrossRef]
- Mouithys-Mickalad, A.; Schmitt, E.; Dalim, M.; Franck, T.; Tome, N.M.; van Spankeren, M.; Serteyn, D.; Paul, A. Black soldier fly (Hermetia illucens) larvae protein derivatives: Potential to promote animal health. Animals 2020, 10, 941. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Huang, X.; Tu, F.; Wang, C.; Yang, F. Preparation, antioxidant activity evaluation, and identification of antioxidant peptide from black soldier fly (Hermetia illucens L.) larvae. J. Food Biochem. 2020, 44, e13186. [Google Scholar] [CrossRef]
- Apostolopoulos, V.; Bojarska, J.; Chai, T.-T.; Elnagdy, S.; Kaczmarek, K.; Matsoukas, J.; New, R.; Parang, K.; Lopez, O.P.; Parhiz, H.; et al. A global review on short peptides: Frontiers and perspectives. Molecules 2021, 26, 430. [Google Scholar] [CrossRef]
- Chai, T.-T.; Ee, K.-Y.; Kumar, D.T.; Manan, F.A.; Wong, F.-C. Plant bioactive peptides: Current status and prospects towards use on human health. Protein Pept. Lett. 2021, 28, 623–642. [Google Scholar] [PubMed]
- Wong, F.-C.; Xiao, J.; Wang, S.; Ee, K.-Y.; Chai, T.-T. Advances on the antioxidant peptides from edible plant sources. Trends Food Sci. Technol. 2020, 99, 44–57. [Google Scholar] [CrossRef]
- Ong, J.-H.; Koh, J.-A.; Cao, H.; Tan, S.-A.; Manan, F.A.; Wong, F.-C.; Chai, T.-T. Purification, identification and characterization of antioxidant peptides from corn silk tryptic hydrolysate: An integrated in vitro-in silico approach. Antioxidants 2021, 10, 1822. [Google Scholar] [CrossRef]
- Wong, F.-C.; Xiao, J.; Ong, M.G.L.; Pang, M.-J.; Wong, S.-J.; Teh, L.-K.; Chai, T.-T. Identification and characterization of antioxidant peptides from hydrolysate of blue-spotted stingray and their stability against thermal, pH and simulated gastrointestinal digestion treatments. Food Chem. 2019, 271, 614–622. [Google Scholar] [CrossRef]
- Chai, T.-T.; Xiao, J.; Mohana Dass, S.; Teoh, J.-Y.; Ee, K.-Y.; Ng, W.-J.; Wong, F.-C. Identification of antioxidant peptides derived from tropical jackfruit seed and investigation of the stability profiles. Food Chem. 2021, 340, 127876. [Google Scholar] [CrossRef]
- Chai, T.-T.; Wong, C.C.-C.; Sabri, M.Z.; Wong, F.-C. Seafood paramyosins as sources of anti-angiotensin-converting-enzyme and anti-dipeptidyl-peptidase peptides after gastrointestinal digestion: A cheminformatic investigation. Molecules 2022, 27, 3864. [Google Scholar] [CrossRef]
- He, R.; Malomo, S.A.; Alashi, A.; Girgih, A.T.; Ju, X.; Aluko, R.E. Purification and hypotensive activity of rapeseed protein-derived renin and angiotensin converting enzyme inhibitory peptides. J. Funct. Foods 2013, 5, 781–789. [Google Scholar] [CrossRef]
- Quah, Y.; Mohd Ismail, N.I.; Ooi, J.L.S.; Affendi, Y.A.; Abd Manan, F.; Teh, L.K.; Wong, F.-C.; Chai, T.-T. Purification and identification of novel cytotoxic oligopeptides from soft coral Sarcophyton glaucum. J. Zhejiang Univ. Sci. B 2019, 20, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Quah, Y.; Mohd Ismail, N.I.; Ooi, J.L.S.; Affendi, Y.A.; Abd Manan, F.; Wong, F.-C.; Chai, T.-T. Identification of novel cytotoxic peptide KENPVLSLVNGMF from marine sponge Xestospongia testudinaria, with characterization of stability in human serum. Int. J. Pept. Res. Ther. 2018, 24, 189–199. [Google Scholar] [CrossRef]
- Chai, T.-T.; Koh, J.-A.; Wong, C.C.-C.; Sabri, M.Z.; Wong, F.-C. Computational screening for the anticancer potential of seed-derived antioxidant peptides: A cheminformatic approach. Molecules 2021, 26, 7396. [Google Scholar] [CrossRef]
- Quah, Y.; Tong, S.-R.; Bojarska, J.; Giller, K.; Tan, S.-A.; Ziora, Z.M.; Esatbeyoglu, T.; Chai, T.-T. Bioactive peptide discovery from edible insects for potential applications in human health and agriculture. Molecules 2023, 28, 1233. [Google Scholar] [CrossRef] [PubMed]
- Batish, I.; Brits, D.; Valencia, P.; Miyai, C.; Rafeeq, S.; Xu, Y.; Galanopoulos, M.; Sismour, E.; Ovissipour, R. Effects of enzymatic hydrolysis on the functional properties, antioxidant activity and protein structure of black soldier fly (Hermetia illucens) protein. Insects 2020, 11, 876. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wierenga, P.A.; Hendriks, W.H.; Jansman, A.J.M. In vitro protein digestion kinetics of protein sources for pigs. Animal 2019, 13, 1154–1164. [Google Scholar] [CrossRef]
- Traksele, L.; Speiciene, V.; Smicius, R.; Alencikiene, G.; Salaseviciene, A.; Garmiene, G.; Zigmantaite, V.; Grigaleviciute, R.; Kucinskas, A. Investigation of in vitro and in vivo digestibility of black soldier fly (Hermetia illucens L.) larvae protein. J. Funct. Foods 2021, 79, 104402. [Google Scholar] [CrossRef]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM database of bioactive peptides: Current opportunities. Int. J. Mol. Sci. 2019, 20, 5978. [Google Scholar] [CrossRef]
- Koh, J.-A.; Ong, J.-H.; Abd Manan, F.; Ee, K.-Y.; Wong, F.-C.; Chai, T.-T. Discovery of bifunctional anti-DPP-IV and anti-ACE peptides from housefly larval proteins after in silico gastrointestinal digestion. Biointerface Res. Appl. Chem. 2022, 12, 4929–4944. [Google Scholar]
- Kaya, A.; Tatlisu, M.A.; Kaplan Kaya, T.; Yildirimturk, O.; Gungor, B.; Karatas, B.; Yazici, S.; Keskin, M.; Avsar, S.; Murat, A. Sublingual vs. oral captopril in hypertensive crisis. J. Emerg. Med. 2016, 50, 108–115. [Google Scholar] [CrossRef]
- Majumder, K.; Wu, J. Molecular targets of antihypertensive peptides: Understanding the mechanisms of action based on the pathophysiology of hypertension. Int. J. Mol. Sci. 2014, 16, 256–283. [Google Scholar] [CrossRef] [PubMed]
- Nishio, S.; Abe, M.; Ito, H. Anagliptin in the treatment of type 2 diabetes: Safety, efficacy, and patient acceptability. Diabetes Metab Syndr Obes 2015, 8, 163–171. [Google Scholar] [PubMed]
- Mika, D.; Guruvayoorappan, C. Myeloperoxidase: The yin and yang in tumour progression. J. Exp. Ther. Oncol. 2011, 9, 93–100. [Google Scholar]
- Kargapolova, Y.; Geißen, S.; Zheng, R.; Baldus, S.; Winkels, H.; Adam, M. The enzymatic and non-enzymatic function of myeloperoxidase (MPO) in inflammatory communication. Antioxidants 2021, 10, 562. [Google Scholar] [CrossRef]
- Vermot, A.; Petit-Härtlein, I.; Smith, S.M.E.; Fieschi, F. NADPH oxidases (NOX): An overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants 2021, 10, 890. [Google Scholar] [CrossRef]
- Ogura, K.; Nobuhisa, I.; Yuzawa, S.; Takeya, R.; Torikai, S.; Saikawa, K.; Sumimoto, H.; Inagaki, F. NMR solution structure of the tandem Src homology 3 domains of p47phox complexed with a p22phox-derived proline-rich peptide. J. Biol. Chem. 2006, 281, 3660–3668. [Google Scholar] [CrossRef] [PubMed]
- Macías Pérez, M.E.; Hernández Rodríguez, M.; Cabrera Pérez, L.C.; Fragoso-Vázquez, M.J.; Correa-Basurto, J.; Padilla-Martínez, I.I.; Méndez Luna, D.; Mera Jiménez, E.; Flores Sandoval, C.; Tamay Cach, F.; et al. Aromatic regions govern the recognition of NADPH oxidase inhibitors as diapocynin and its analogues. Arch. Der Pharm. 2017, 350, 1700041. [Google Scholar] [CrossRef]
- Oh, S.-H.; Choi, S.-Y.; Choi, H.-J.; Ryu, H.-M.; Kim, Y.-J.; Jung, H.-Y.; Cho, J.-H.; Kim, C.-D.; Park, S.-H.; Kwon, T.-H.; et al. The emerging role of xanthine oxidase inhibition for suppression of breast cancer cell migration and metastasis associated with hypercholesterolemia. FASEB J. 2019, 33, 7301–7314. [Google Scholar] [CrossRef]
- Cao, H.; Pauff, J.M.; Hille, R. X-ray crystal structure of a xanthine oxidase complex with the flavonoid inhibitor quercetin. J. Nat. Prod. 2014, 77, 1693–1699. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1–Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Tascioglu Aliyev, A.; Panieri, E.; Stepanić, V.; Gurer-Orhan, H.; Saso, L. Involvement of NRF2 in breast cancer and possible therapeutical role of polyphenols and melatonin. Molecules 2021, 26, 1853. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Limon, A.; Aguilar-Toalá, J.E.; Liceaga, A.M. Integration of molecular docking analysis and molecular dynamics simulations for studying food proteins and bioactive peptides. J. Agric. Food Chem. 2022, 70, 934–943. [Google Scholar] [CrossRef]
- Riniker, S. Fixed-charge atomistic force fields for molecular dynamics simulations in the condensed phase: An overview. J. Chem. Inf. Model. 2018, 58, 565–578. [Google Scholar] [CrossRef] [PubMed]
- Polêto, M.D.; Lemkul, J.A. Integration of experimental data and use of automated fitting methods in developing protein force fields. Commun. Chem. 2022, 5, 38. [Google Scholar] [CrossRef]
- Kührová, P.; Mlýnský, V.; Zgarbová, M.; Krepl, M.; Bussi, G.; Best, R.B.; Otyepka, M.; Šponer, J.; Banáš, P. Improving the performance of the Amber RNA force field by tuning the hydrogen-bonding interactions. J. Chem. Theory Comput. 2019, 15, 3288–3305. [Google Scholar] [CrossRef]
- Adibi, S.A. The oligopeptide transporter (Pept-1) in human intestine: Biology and function. Gastroenterology 1997, 113, 332–340. [Google Scholar] [CrossRef]
- Cheng, H.M.; Mah, K.K.; Seluakumaran, K. Protein absorption. In Defining Physiology: Principles, Themes, Concepts. Volume 2: Neurophysiology and Gastrointestinal Systems; Cheng, H.M., Mah, K.K., Seluakumaran, K., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 71–73. [Google Scholar]
- Leibach, F.H.; Ganapathy, V. Peptide transporters in the intestine and the kidney. Annu. Rev. Nutr. 1996, 16, 99–119. [Google Scholar] [CrossRef]
- Mathews, D.M.; Adibi, S.A. Peptide absorption. Gastroenterology 1976, 71, 151–161. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Borba, J.V.B.; Alves, V.M.; Braga, R.C.; Korn, D.R.; Overdahl, K.; Silva, A.C.; Hall, S.U.S.; Overdahl, E.; Kleinstreuer, N.; Strickland, J.; et al. STopTox: An in silico alternative to animal testing for acute systemic and topical toxicity. Environ. Health Perspect. 2022, 130, 027012. [Google Scholar] [CrossRef] [PubMed]
- Suetsuna, K.; Chen, J.-R. Studies on biologically active peptide derived from fish and shellfish—V. Antioxidant activities of Undaria pinnatifida dipeptide derivatives. J. Natl. Fish. Univ. 2002, 51, 1–5. [Google Scholar]
- Nongonierma, A.B.; FitzGerald, R.J. Dipeptidyl peptidase IV inhibitory and antioxidative properties of milk protein-derived dipeptides and hydrolysates. Peptides 2013, 39, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Suetsuna, K.; Ukeda, H.; Ochi, H. Isolation and characterization of free radical scavenging activities peptides derived from casein. J. Nutr. Biochem. 2000, 11, 128–131. [Google Scholar] [CrossRef]
- Torkova, A.; Koroleva, O.; Khrameeva, E.; Fedorova, T.; Tsentalovich, M. Structure-functional study of tyrosine and methionine dipeptides: An approach to antioxidant activity prediction. Int. J. Mol. Sci. 2015, 16, 25353–25376. [Google Scholar] [CrossRef]
- Weng, W.; Tang, L.; Wang, B.; Chen, J.; Su, W.; Osako, K.; Tanaka, M. Antioxidant properties of fractions isolated from blue shark (Prionace glauca) skin gelatin hydrolysates. J. Funct. Foods 2014, 11, 342–351. [Google Scholar] [CrossRef]
- Tkaczewska, J.; Bukowski, M.; Mak, P. Identification of antioxidant peptides in enzymatic hydrolysates of carp (Cyprinus carpio) skin gelatin. Molecules 2019, 24, 97. [Google Scholar] [CrossRef]
- Feng, L.; Peng, F.; Wang, X.; Li, M.; Lei, H.; Xu, H. Identification and characterization of antioxidative peptides derived from simulated in vitro gastrointestinal digestion of walnut meal proteins. Food Res. Int. 2019, 116, 518–526. [Google Scholar] [CrossRef]
- Nogata, Y.; Nagamine, T.; Yanaka, M.; Ohta, H. Angiotensin I converting enzyme inhibitory peptides produced by autolysis reactions from wheat bran. J. Agric. Food Chem. 2009, 57, 6618–6622. [Google Scholar] [CrossRef]
- Cheung, H.S.; Wang, F.L.; Ondetti, M.A.; Sabo, E.F.; Cushman, D.W. Binding of peptide substrates and inhibitors of angiotensin-converting enzyme. Importance of the COOH-terminal dipeptide sequence. J. Biol. Chem. 1980, 255, 401–407. [Google Scholar] [CrossRef]
- Suetsuna, K. Isolation and characterization of angiotensin I-converting enzyme inhibitor dipeptides derived from Allium sativum L (garlic). J. Nutr. Biochem. 1998, 9, 415–419. [Google Scholar] [CrossRef]
- Yano, S.; Suzuki, K.; Funatsu, G. Isolation from alpha-zein of thermolysin peptides with angiotensin I-converting enzyme inhibitory activity. Biosci Biotechnol Biochem 1996, 60, 661–663. [Google Scholar] [CrossRef]
- Li, C.H.; Matsui, T.; Matsumoto, K.; Yamasaki, R.; Kawasaki, T. Latent production of angiotensin I-converting enzyme inhibitors from buckwheat protein. J. Pept. Sci. 2002, 8, 267–274. [Google Scholar] [CrossRef]
- Wu, J.; Aluko, R.E.; Nakai, S. Structural requirements of angiotensin i-converting enzyme inhibitory peptides: Quantitative structure−activity relationship study of di- and tripeptides. J. Agric. Food Chem. 2006, 54, 732–738. [Google Scholar] [CrossRef]
- Byun, H.G.; Kim, S.K. Structure and activity of angiotensin I converting enzyme inhibitory peptides derived from Alaskan pollack skin. J. Biochem. Mol. Biol. 2002, 35, 239–243. [Google Scholar] [CrossRef]
- Kim, H.-J.; Kang, S.-G.; Jaiswal, L.; Li, J.; Choi, J.-H.; Moon, S.-M.; Cho, J.-Y.; Ham, K.-S. Identification of four new angiotensin I-converting enzyme inhibitory peptides from fermented anchovy sauce. Appl. Biol. Chem. 2016, 59, 25–31. [Google Scholar] [CrossRef]
- Lan, V.T.T.; Ito, K.; Ohno, M.; Motoyama, T.; Ito, S.; Kawarasaki, Y. Analyzing a dipeptide library to identify human dipeptidyl peptidase IV inhibitor. Food Chem. 2015, 175, 66–73. [Google Scholar] [CrossRef]
- Gallego, M.; Aristoy, M.-C.; Toldrá, F. Dipeptidyl peptidase IV inhibitory peptides generated in Spanish dry-cured ham. Meat Sci. 2014, 96, 757–761. [Google Scholar] [CrossRef]
- Nongonierma, A.B.; Mooney, C.; Shields, D.C.; FitzGerald, R.J. Inhibition of dipeptidyl peptidase IV and xanthine oxidase by amino acids and dipeptides. Food Chem. 2013, 141, 644–653. [Google Scholar] [CrossRef]
- Dhanda, S.; Singh, J.; Singh, H. Hydrolysis of various bioactive peptides by goat brain dipeptidylpeptidase-III homologue. Cell Biochem Funct 2008, 26, 339–345. [Google Scholar] [CrossRef]
- Lee, C.M.; Snyder, S.H. Dipeptidyl-aminopeptidase III of rat brain. Selective affinity for enkephalin and angiotensin. J. Biol. Chem. 1982, 257, 12043–12050. [Google Scholar] [CrossRef]
- Udenigwe, C.C.; Li, H.; Aluko, R.E. Quantitative structure-activity relationship modeling of renin-inhibiting dipeptides. Amino Acids 2012, 42, 1379–1386. [Google Scholar] [CrossRef]
- Morifuji, M.; Koga, J.; Kawanaka, K.; Higuchi, M. Branched-chain amino acid-containing dipeptides, identified from whey protein hydrolysates, stimulate glucose uptake rate in L6 myotubes and isolated skeletal muscles. J. Nutr. Sci. Vitaminol. 2009, 55, 81–86. [Google Scholar] [CrossRef]
- Luzarowski, M.; Vicente, R.; Kiselev, A.; Wagner, M.; Schlossarek, D.; Erban, A.; de Souza, L.P.; Childs, D.; Wojciechowska, I.; Luzarowska, U.; et al. Global mapping of protein–metabolite interactions in Saccharomyces cerevisiae reveals that Ser-Leu dipeptide regulates phosphoglycerate kinase activity. Commun. Biol. 2021, 4, 181. [Google Scholar] [CrossRef]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.Y. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef]
- Pearson, A.R.; Godber, B.; Eisenthal, R.; Taylor, G.; Harrison, R. Human milk xanthine oxidoreductase. [CrossRef]
- Yamaguchi, Y.; Matsumura, T.; Ichida, K.; Okamoto, K.; Nishino, T. Human xanthine oxidase changes its substrate specificity to aldehyde oxidase type upon mutation of amino acid residues in the active site: Roles of active site residues in binding and activation of purine substrate. J. Biochem. 2007, 141, 513–524. [Google Scholar] [CrossRef]
- Ghallab, D.S.; Shawky, E.; Metwally, A.M.; Celik, I.; Ibrahim, R.S.; Mohyeldin, M.M. Integrated in silico—In vitro strategy for the discovery of potential xanthine oxidase inhibitors from Egyptian propolis and their synergistic effect with allopurinol and febuxostat. RSC Adv. 2022, 12, 2843–2872. [Google Scholar] [CrossRef]
- Enroth, C.; Eger, B.T.; Okamoto, K.; Nishino, T.; Nishino, T.; Pai, E.F. Crystal structures of bovine milk xanthine dehydrogenase and xanthine oxidase: Structure-based mechanism of conversion. Proc. Natl. Acad. Sci. USA 2000, 97, 10723–10728. [Google Scholar] [CrossRef]
- Bell, E.W.; Zhang, Y. DockRMSD: An open-source tool for atom mapping and RMSD calculation of symmetric molecules through graph isomorphism. J. Cheminformat. 2019, 11, 40. [Google Scholar] [CrossRef]
- Zhang, H.; Jing, X.; Shi, Y.; Xu, H.; Du, J.; Guan, T.; Weihrauch, D.; Jones, D.W.; Wang, W.; Gourlay, D.; et al. N-acetyl lysyltyrosylcysteine amide inhibits myeloperoxidase, a novel tripeptide inhibitor. J. Lipid Res. 2013, 54, 3016–3029. [Google Scholar] [CrossRef]
- Shaw, S.A.; Vokits, B.P.; Dilger, A.K.; Viet, A.; Clark, C.G.; Abell, L.M.; Locke, G.A.; Duke, G.; Kopcho, L.M.; Dongre, A.; et al. Discovery and structure activity relationships of 7-benzyl triazolopyridines as stable, selective, and reversible inhibitors of myeloperoxidase. Bioorganic Med. Chem. 2020, 28, 115723. [Google Scholar] [CrossRef]
- Davey, C.A.; Fenna, R.E. 2.3 Å resolution x-ray crystal structure of the bisubstrate analogue inhibitor salicylhydroxamic acid bound to human myeloperoxidase: A model for a prereaction complex with hydrogen peroxide. Biochemistry 1996, 35, 10967–10973. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Ai, X.; Pan, F.; Zhou, N.; Zhao, L.; Cai, S.; Tang, X. Novel peptides with xanthine oxidase inhibitory activity identified from macadamia nuts: Integrated in silico and in vitro analysis. Eur. Food Res. Technol. 2022, 248, 2031–2042. [Google Scholar] [CrossRef]
- Zhong, H.; Zhang, Y.; Deng, L.; Zhao, M.; Tang, J.; Zhang, H.; Feng, F.; Wang, J. Exploring the potential of novel xanthine oxidase inhibitory peptide (ACECD) derived from Skipjack tuna hydrolysates using affinity-ultrafiltration coupled with HPLC–MALDI-TOF/TOF-MS. Food Chem. 2021, 347, 129068. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Chen, H.; Guan, K.; Sun, Y.; Wang, R.; Li, Q.; Ma, Y. Novel xanthine oxidase inhibitory peptides derived from whey protein: Identification, in vitro inhibition mechanism and in vivo activity validation. Bioorganic Chem. 2022, 128, 106097. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, J.; Nie, S.; Ding, L.; Wang, L.; Liu, J.; Liu, W.; Zhang, T. Direct inhibition of Keap1–Nrf2 interaction by egg-derived peptides DKK and DDW revealed by molecular docking and fluorescence polarization. RSC Adv. 2017, 7, 34963–34971. [Google Scholar] [CrossRef]
- Lo, S.-C.; Li, X.; Henzl, M.T.; Beamer, L.J.; Hannink, M. Structure of the Keap1:Nrf2 interface provides mechanistic insight into Nrf2 signaling. EMBO J. 2006, 25, 3605–3617. [Google Scholar] [CrossRef]
- Lazar, T.; Guharoy, M.; Vranken, W.; Rauscher, S.; Wodak, S.J.; Tompa, P. Distance-based metrics for comparing conformational ensembles of intrinsically disordered proteins. Biophys. J. 2020, 118, 2952–2965. [Google Scholar] [CrossRef]
- Liu, K.; Watanabe, E.; Kokubo, H. Exploring the stability of ligand binding modes to proteins by molecular dynamics simulations. J. Comput. -Aided Mol. Des. 2017, 31, 201–211. [Google Scholar] [CrossRef]
- Dey, D.; Paul, P.K.; Al Azad, S.; Al Mazid, M.F.; Khan, A.M.; Sharif, M.A.; Rahman, M.H. Molecular optimization, docking, and dynamic simulation profiling of selective aromatic phytochemical ligands in blocking the SARS-CoV-2 S protein attachment to ACE2 receptor: An in silico approach of targeted drug designing. J. Adv. Vet. Anim. Res. 2021, 8, 24–35. [Google Scholar] [CrossRef]
- Ramírez, D.; Caballero, J. Is it reliable to take the molecular docking top scoring position as the best solution without considering available structural data? Molecules 2018, 23, 1038. [Google Scholar] [CrossRef]
- Arefin, A.; Ismail Ema, T.; Islam, T.; Hossen, S.; Islam, T.; Al Azad, S.; Uddin Badal, N.; Islam, A.; Biswas, P.; Alam, N.U.; et al. Target specificity of selective bioactive compounds in blocking α-dystroglycan receptor to suppress Lassa virus infection: An in silico approach. J. Biomed. Res. 2021, 35, 459–473. [Google Scholar] [CrossRef]
- Girdhar, K.; Dehury, B.; Kumar Singh, M.; Daniel, V.P.; Choubey, A.; Dogra, S.; Kumar, S.; Mondal, P. Novel insights into the dynamics behavior of glucagon-like peptide-1 receptor with its small molecule agonists. J. Biomol. Struct. Dyn. 2019, 37, 3976–3986. [Google Scholar] [CrossRef]
- Sahoo, C.R.; Paidesetty, S.K.; Dehury, B.; Padhy, R.N. Molecular dynamics and computational study of Mannich-based coumarin derivatives: Potent tyrosine kinase inhibitor. J. Biomol. Struct. Dyn. 2020, 38, 5419–5428. [Google Scholar] [CrossRef]
- Arshia, A.H.; Shadravan, S.; Solhjoo, A.; Sakhteman, A.; Sami, A. De novo design of novel protease inhibitor candidates in the treatment of SARS-CoV-2 using deep learning, docking, and molecular dynamic simulations. Comput. Biol. Med. 2021, 139, 104967. [Google Scholar] [CrossRef]
- Di Rienzo, L.; Miotto, M.; Bò, L.; Ruocco, G.; Raimondo, D.; Milanetti, E. Characterizing hydropathy of amino acid side chain in a protein environment by investigating the structural changes of water molecules network. Front. Mol. Biosci. 2021, 8, 626837. [Google Scholar] [CrossRef]
- Mogany, T.; Kumari, S.; Swalaha, F.M.; Bux, F. In silico analysis of enzymes involved in mycosporine-like amino acids biosynthesis in Euhalothece sp.: Structural and functional characterization. Algal Res. 2022, 66, 102806. [Google Scholar] [CrossRef]
- Harris, T.K.; Turner, G.J. Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life 2002, 53, 85–98. [Google Scholar] [CrossRef]
- Yuce, M.; Cicek, E.; Inan, T.; Dag, A.B.; Kurkcuoglu, O.; Sungur, F.A. Repurposing of FDA-approved drugs against active site and potential allosteric drug-binding sites of COVID-19 main protease. Proteins 2021, 89, 1425–1441. [Google Scholar] [CrossRef]
- Khezri, A.; Karimi, A.; Yazdian, F.; Jokar, M.; Mofradnia, S.R.; Rashedi, H.; Tavakoli, Z. Molecular dynamic of curcumin/chitosan interaction using a computational molecular approach: Emphasis on biofilm reduction. Int. J. Biol. Macromol. 2018, 114, 972–978. [Google Scholar] [CrossRef]
- Islam, R.; Parves, M.R.; Paul, A.S.; Uddin, N.; Rahman, M.S.; Mamun, A.A.; Hossain, M.N.; Ali, M.A.; Halim, M.A. A molecular modeling approach to identify effective antiviral phytochemicals against the main protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 39, 3213–3224. [Google Scholar] [CrossRef]
- Ul Haq, F.; Abro, A.; Raza, S.; Liedl, K.R.; Azam, S.S. Molecular dynamics simulation studies of novel β-lactamase inhibitor. J. Mol. Graph. Model. 2017, 74, 143–152. [Google Scholar] [CrossRef]
- Lokhande, K.B.; Doiphode, S.; Vyas, R.; Swamy, K.V. Molecular docking and simulation studies on SARS-CoV-2 M(pro) reveals Mitoxantrone, Leucovorin, Birinapant, and Dynasore as potent drugs against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 7294–7305. [Google Scholar] [CrossRef]
- Zaki, A.A.; Ashour, A.; Elhady, S.S.; Darwish, K.M.; Al-Karmalawy, A.A. Calendulaglycoside A showing potential activity against SARS-CoV-2 main protease: Molecular docking, molecular dynamics, and SAR studies. J. Tradit. Complement. Med. 2022, 12, 16–34. [Google Scholar] [CrossRef]
- Leni, G.; Tedeschi, T.; Faccini, A.; Pratesi, F.; Folli, C.; Puxeddu, I.; Migliorini, P.; Gianotten, N.; Jacobs, J.; Depraetere, S.; et al. Shotgun proteomics, in-silico evaluation and immunoblotting assays for allergenicity assessment of lesser mealworm, black soldier fly and their protein hydrolysates. Sci. Rep. 2020, 10, 1228. [Google Scholar] [CrossRef]
- Consortium, T.U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2020, 49, D480–D489. [Google Scholar] [CrossRef]
- Wong, F.-C.; Ong, J.-H.; Chai, T.-T. Identification of putative cell-entry-inhibitory peptides against SARS-CoV-2 from edible insects: An in silico study. eFood 2020, 1, 357–368. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021, 49, D437–D451. [Google Scholar] [CrossRef]
- Prasasty, V.D.; Istyastono, E.P. Data of small peptides in SMILES and three-dimensional formats for virtual screening campaigns. Data Brief 2019, 27, 104607. [Google Scholar] [CrossRef]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef]
- Shen, Y.; Maupetit, J.; Derreumaux, P.; Tufféry, P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J. Chem. Theory Comput. 2014, 10, 4745–4758. [Google Scholar] [CrossRef]
- Thévenet, P.; Shen, Y.; Maupetit, J.; Guyon, F.; Derreumaux, P.; Tufféry, P. PEP-FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012, 40, W288–W293. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2020, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Miteva, M.A.; Guyon, F.; Tufféry, P. Frog2: Efficient 3D conformation ensemble generator for small compounds. Nucleic Acids Res. 2010, 38, W622–W627. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An Environment for Comparative Protein Modeling. Electrophoresis 1997, 18, 2714. [Google Scholar] [CrossRef]
- Kochnev, Y.; Hellemann, E.; Cassidy, K.C.; Durrant, J.D. Webina: An open-source library and web app that runs AutoDock Vina entirely in the web browser. Bioinformatics 2020, 36, 4513–4515. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef]
- Pearson, W.R.; Lipman, D.J. Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. USA 1988, 85, 2444–2448. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Martí-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sánchez, R.; Melo, F.; Šali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sabri, M.Z.; Hamid, A.A.A.; Hitam, S.M.S.; Rahim, M.Z.A. The assessment of three dimensional modelling design for single strand DNA aptamers for computational chemistry application. Biophys. Chem. 2020, 267, 106492. [Google Scholar] [CrossRef]
- Bibi, S.; Khan, M.S.; El-Kafrawy, S.A.; Alandijany, T.A.; El-Daly, M.M.; Yousafi, Q.; Fatima, D.; Faizo, A.A.; Bajrai, L.H.; Azhar, E.I. Virtual screening and molecular dynamics simulation analysis of Forsythoside A as a plant-derived inhibitor of SARS-CoV-2 3CLpro. Saudi Pharm. J. 2022, 30, 979–1002. [Google Scholar] [CrossRef]
- Surti, M.; Patel, M.; Adnan, M.; Moin, A.; Ashraf, S.A.; Siddiqui, A.J.; Snoussi, M.; Deshpande, S.; Reddy, M.N. Ilimaquinone (marine sponge metabolite) as a novel inhibitor of SARS-CoV-2 key target proteins in comparison with suggested COVID-19 drugs: Designing, docking and molecular dynamics simulation study. RSC Adv. 2020, 10, 37707–37720. [Google Scholar] [CrossRef] [PubMed]
- Shamsi, A.; Shahwan, M.; Khan, M.S.; Alhumaydhi, F.A.; Alsagaby, S.A.; Al Abdulmonem, W.; Abdullaev, B.; Yadav, D.K. Mechanistic insight into binding of huperzine a with human serum albumin: Computational and spectroscopic approaches. Molecules 2022, 27, 797. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).