

Density Functional Theory Study of the Hydrogenation of Carbon Monoxide over the Co (001) Surface: Implications for the Fischer–Tropsch Process




Abstract
1. Introduction
2. Results and Discussion
2.1. Thermodynamic Analysis
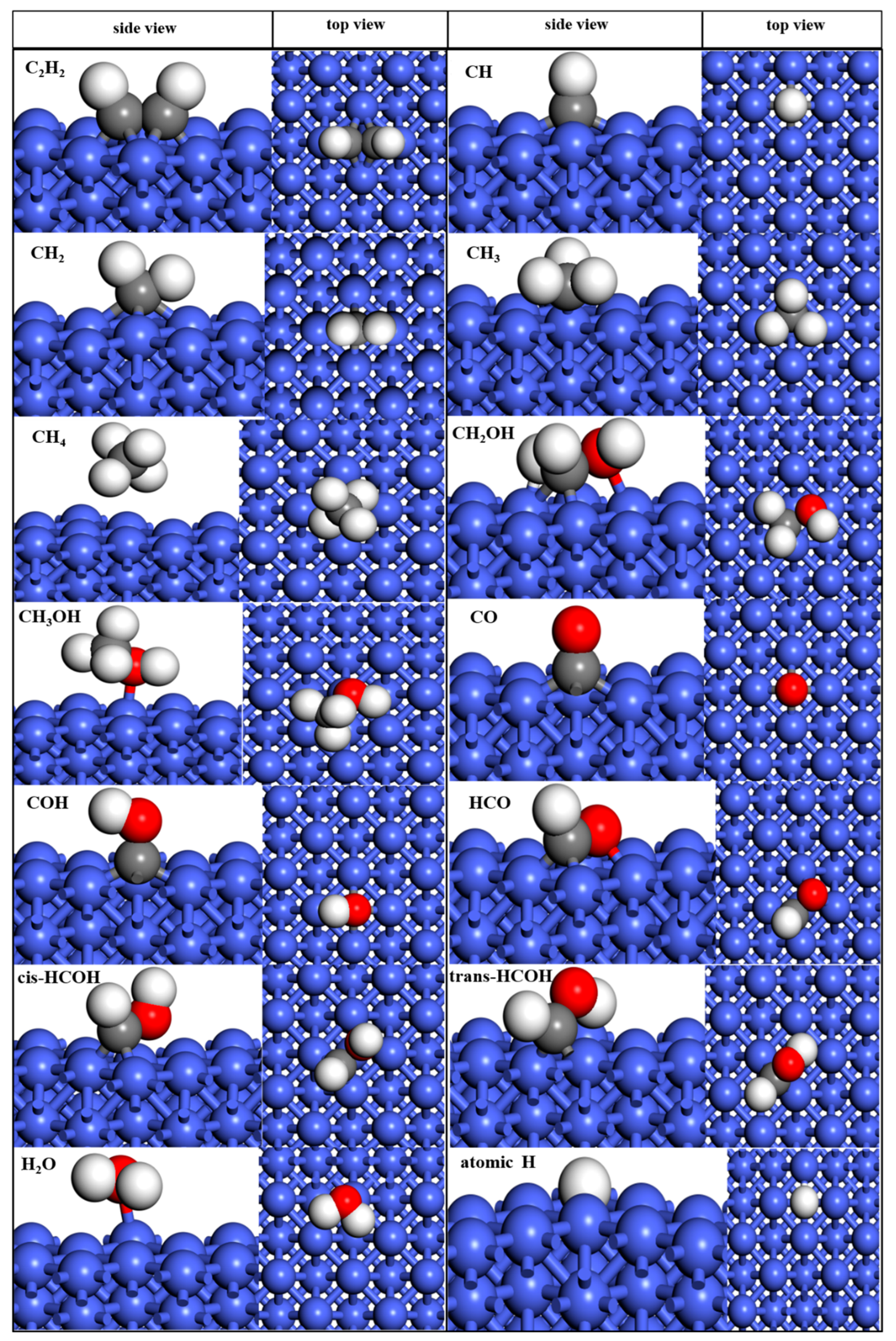
2.1.1. Adsorption of Molecules

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Site, Atom, Bond Length (Å) | Eads (eV) | Eads in Literature (eV) |
|---|---|---|---|
| CO | hollow, carbon, 2.03 | −2.268 | −2.34(Pt(111)) [63], −1.91 (Ni(110)) [64], −2.00 (Fe(100)) [65], −1.92 (Ni(111)) [66] |
| COH | hollow, carbon, 1.960 | −5.696 | −5.64(Pt(111)) [63], −4.01 (Ni(110)) [64], −6.21(Fe(100)) [65], |
| HCO | hollow, carbon,1.917 | −4.03 | −2.60 (Ni(110)) [64], −6.49 (Fe(100)) [65] |
| Cis-HCOH | hollow, carbon,1.970 | −3.919 | −3.51 (Ni(110)) [64], −4.04 (Fe(100)) [65] |
| Trans-HCOH | bridge, carbon, 1.930 | −3.583 | −3.25 Ni(110) [64], −4.04 (Fe(100)) [65] |
| CH | hollow, carbon, 1.942 | −7.946 | −6.43 (Ni(111)) [66], −7.55(Pt(111)) [63] |
| CH2 | hollow, carbon, 2.091 | −5.611 | −4.01 (Ni(111)) [66], −4.56(Pt(111)) [63] |
| CH3 | hollow, carbon, 1.967 | −2.972 | −2.40(Pt(111)) [63] |
| CH4 | hollow, carbon, 3.645 | −0.204 | −0.17 (Ru(0001) [31] |
| H2O | top, oxygen, 2.141 | −0.746 | −0.29 (Ni(111)) [66] |
| CH2OH | hollow, oxygen, 2.110 carbon, 1.941 | −2.74 | −2.79(Pt(111)) [63], −1.68 (Ni(110)) [64] |
| CH3OH | top, oxygen, 2.117 | −0.718 | −0.45 (Ni(110)) [64] |
| C2H2 | hollow, carbon, 1.843 | −3.241 | −2.957 (Ni(111)) [67] |
2.1.2. Hydrogenation
2.1.3. Reactions
| Reaction | Ereaction (eV) | Ereaction (eV) in Literature |
|---|---|---|
| 0.853 | 0.85(InZr3(110)) [68], 1.04(PdCu3(111)) [70] | |
| 0.574 | 0.80(Ni(110)) [64], 0.53(Cu3Ag(211)) [69], 0.75(Cu(211)) [69] | |
| 0.858 | −0.37(PdCu3(111)) [70], 0.14(Cu(111)) [71] | |
| 1.004 | ||
| 1.393 | ||
| 1.539 | ||
| −0.773 | ||
| 0.39 | 0.01(PdCu3(111)) [70], 0.84(Cu(111)) [71] | |
| −1.283 | ||
| −0.12 | 0.01(PdCu3(111)) [70], 0.77(Cu(111)) [71] | |
| 0.019 | 0.35(InZr3(110)) [68] | |
| 0.679 | 0.36(InZr3(110)) [68] | |
| 0.491 | ||
| 0.089 | 0.49(Ni(110)) [64], 0.90(Cu(111)) [71] | |
| 1.511 |
2.2. Analysis of the Kinetics
2.2.1. Transition States
| Reaction | Ea (eV) | Ea (eV) In literature |
|---|---|---|
| 1.804 | 1.55(Co(0001)) [72], 1.07(Fe(100)) [65], 1.97(Ni(111)) [66] | |
| 1.082 | 1.08(Ni(110)) [64] | |
| 1.231 | 1.38(Fe(100)) [65], 1.522(Pt(111)) [74] | |
| 1.231 | 1.38(Fe(100)) [65], 1.522(Pt(111)) [74] | |
| 1.746 | 1.59(Co(0001)) [75] | |
| 1.727 | 1.59(Co(0001)) [75] | |
| 0.066 | ||
| 0.662 | 0.71(Co(0001)) [72], 0.43(Co(0001)) [75] | |
| 1.581 | ||
| 0.131 | 0.71(Co(0001)) [72], 0.43(Co(0001)) [75] | |
| 1.556 | 0.77(Ru(0001) [31] | |
| 0.065 | 0.69(Ni(111)) [66] | |
| 0.969 | 0.81(Fe5C2(100)) [73], 1.360(Pt(111)) [74] | |
| 1.089 | 1.187(Pt(111)) [74], 0.96(Fe5C2(100)) [73], 0.90(Ni(111)) [66] | |
| 0.725 | 1.04(Ni(110)) [64], 0.69(Ni(111)) [66], 0.82(Co(0001)) [75] |
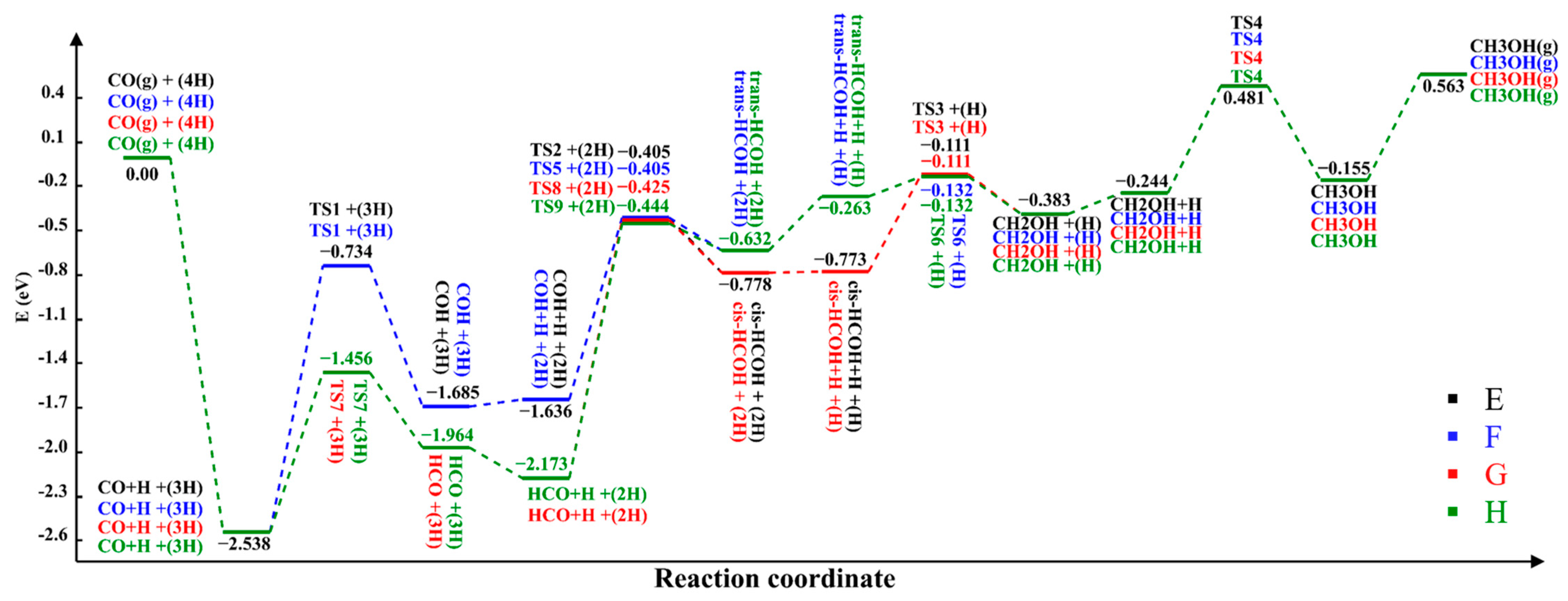
2.2.2. Reaction Pathways
| Path A | Path B | Path C | Path D | ||||
|---|---|---|---|---|---|---|---|
| State | E (eV) | State | E (eV) | State | E (eV) | State | E (eV) |
| CO(g)+(4H) | 0.00 | CO(g)+(4H) | 0.00 | CO(g)+(4H) | 0.00 | CO(g)+(4H) | 0.00 |
| CO+H+(3H) | −2.538 | CO+H+(3H) | −2.538 | CO+H+(3H) | −2.538 | CO+H+(3H) | −2.538 |
| TS1+(3H) | −0.734 | TS1+(3H) | −0.734 | TS7+(3H) | −1.456 | TS7+(3H) | −1.456 |
| COH+(3H) | −1.685 | COH+(3H) | −1.685 | HCO+(3H) | −1.964 | HCO+(3H) | −1.964 |
| COH+H+(2H) | −1.636 | COH+H+(2H) | −1.636 | HCO+H+(2H) | −2.173 | HCO+H+(2H) | −2.173 |
| TS2+(2H) | −0.405 | TS5+(2H) | −0.405 | TS8+(2H) | −0.425 | TS9+(2H) | −0.444 |
| cis-HCOH+(2H) | −0.778 | trans-HCOH+(2H) | −0.632 | cis-HCOH+(2H) | −0.778 | trans-HCOH+(2H) | −0.632 |
| cis-HCOH+H+(H) | −0.773 | trans-HCOH+H+(H) | −0.263 | cis-HCOH+H+(H) | −0.773 | trans-HCOH+H+(H) | −0.263 |
| TS10+(H) | −0.707 | TS14+(H) | 1.318 | TS10+(H) | −0.707 | TS14+(H) | 1.318 |
| CH+H2O+(H) | −1.546 | CH+H2O+(H) | −1.546 | CH+H2O+(H) | −1.546 | CH+H2O+(H) | −1.546 |
| CH+H+(H2O) | −0.991 | CH+H+(H2O) | −0.991 | CH+H+(H2O) | −0.991 | CH+H+(H2O) | −0.991 |
| TS11+(H2O) | −0.93 | TS11+(H2O) | −0.93 | TS11+(H2O) | −0.93 | TS11+(H2O) | −0.93 |
| CH2+(H2O) | −0.976 | CH2+(H2O) | −0.976 | CH2+(H2O) | −0.976 | CH2+(H2O) | −0.976 |
| CH2+H+(OH) | −2.249 | CH2+H+(OH) | −2.249 | CH2+H+(OH) | −2.249 | CH2+H+(OH) | −2.249 |
| TS12+(OH) | −1.28 | TS12+(OH) | −1.28 | TS12+(OH) | −1.28 | TS12+(OH) | −1.28 |
| CH3+(OH) | −1.57 | CH3+(OH) | −1.57 | CH3+(OH) | −1.57 | CH3+(OH) | −1.57 |
| CH3+H+(O) | −2.225 | CH3+H+(O) | −2.225 | CH3+H+(O) | −2.225 | CH3+H+(O) | −2.225 |
| TS13+(O) | −1.136 | TS13+(O) | −1.136 | TS13+(O) | −1.136 | TS13+(O) | −1.136 |
| CH4+(O) | −1.734 | CH4+(O) | −1.734 | CH4+(O) | −1.734 | CH4+(O) | −1.734 |
| CH4(g)+(O) | −1.53 | CH4(g)+(O) | −1.53 | CH4(g)+(O) | −1.53 | CH4(g)+(O) | −1.53 |
| Path E | Path F | Path G | Path H | ||||
|---|---|---|---|---|---|---|---|
| State | E (eV) | State | E (eV) | State | E (eV) | State | E (eV) |
| CO(g)+(4H) | 0.00 | CO(g)+(4H) | 0.00 | CO(g)+(4H) | 0.00 | CO(g)+(4H) | 0.00 |
| CO+H+(3H) | −2.538 | CO+H+(3H) | −2.538 | CO+H+(3H) | −2.538 | CO+H+(3H) | −2.538 |
| TS1+(3H) | −0.734 | TS1+(3H) | −0.734 | TS7+(3H) | −1.456 | TS7+(3H) | −1.456 |
| COH+(3H) | −1.685 | COH+(3H) | −1.685 | HCO+(3H) | −1.964 | HCO+(3H) | −1.964 |
| COH+H+(2H) | −1.636 | COH+H+(2H) | −1.636 | HCO+H+(2H) | −2.173 | HCO+H+(2H) | −2.173 |
| TS2+(2H) | −0.405 | TS5+(2H) | −0.405 | TS8+(2H) | −0.425 | TS9+(2H) | −0.444 |
| cis-HCOH+(2H) | −0.778 | trans-HCOH+(2H) | −0.632 | cis-HCOH+(2H) | −0.778 | trans-HCOH+(2H) | −0.632 |
| cis-HCOH+H+(H) | −0.773 | trans-HCOH+H+(H) | −0.263 | cis-HCOH+H+(H) | −0.773 | trans-HCOH+H+(H) | −0.263 |
| TS3+(H) | −0.111 | TS6+(H) | −0.132 | TS3+(H) | −0.111 | TS6+(H) | −0.132 |
| CH2OH+(H) | −0.383 | CH2OH+(H) | −0.383 | CH2OH+(H) | −0.383 | CH2OH+(H) | −0.383 |
| CH2OH+H | −0.244 | CH2OH+H | −0.244 | CH2OH+H | −0.244 | CH2OH+H | −0.244 |
| TS4 | 0.481 | TS4 | 0.481 | TS4 | 0.481 | TS4 | 0.481 |
| CH3OH | −0.155 | CH3OH | −0.155 | CH3OH | −0.155 | CH3OH | −0.155 |
| CH3OH (g) | 0.563 | CH3OH (g) | 0.563 | CH3OH (g) | 0.563 | CH3OH (g) | 0.563 |
| Path I | Path J | Path K | Path L | ||||
|---|---|---|---|---|---|---|---|
| state | E (eV) | state | E (eV) | State | E (eV) | state | E (eV) |
| CO(g)+(H)+(CH2) | 0.00 | CO(g)+(H)+(CH2) | 0.00 | CO(g)+(H)+(CH2) | 0.00 | CO(g)+(H)+(CH2) | 0.00 |
| CO+H+(CH2) | −2.538 | CO+H+(CH2) | −2.538 | CO+H+(CH2) | −2.538 | CO+H+(CH2) | −2.538 |
| TS1+(CH2) | −0.734 | TS1+(CH2) | −0.734 | TS7+(CH2) | −1.456 | TS7+(CH2) | −1.456 |
| COH+(CH2) | −1.685 | COH+(CH2) | −1.685 | HCO+(CH2) | −1.964 | HCO+(CH2) | −1.964 |
| COH+H+(CH) | −2.298 | COH+H+(CH) | −2.298 | HCO+H+(CH) | −2.833 | HCO+H+(CH) | −2.833 |
| TS2+(CH) | −1.067 | TS5+(CH) | −1.077 | TS8+(CH) | −1.087 | TS9+(CH) | −1.106 |
| cis-HCOH+(CH) | −1.44 | trans-HCOH+(CH) | −1.294 | cis-HCOH+(CH) | −1.44 | trans-HCOH+(CH) | −1.294 |
| cis-HCOH+H+(C) | −2.178 | trans-HCOH+H+(C) | −1.668 | cis-HCOH+H+(C) | −2.178 | trans-HCOH+H+(C) | −1.668 |
| TS10+(C) | −2.112 | TS14+(C) | −0.087 | TS10+(C) | −2.112 | TS14+(C) | −0.087 |
| CH+H2O+(C) | −2.951 | CH+H2O+(C) | −2.951 | CH+H2O+(C) | −2.951 | CH+H2O+(C) | −2.951 |
| CH+CH+(OH) | −3.761 | CH+CH+(OH) | −3.761 | CH+CH+(OH) | −3.761 | CH+CH+(OH) | −3.761 |
| TS15+(OH) | −2.205 | TS15+(OH) | −2.205 | TS15+(OH) | −2.205 | TS15+(OH) | 2.205 |
| C2H2+(OH) | −2.25 | C2H2+(OH) | −2.25 | C2H2+(OH) | −2.25 | C2H2+(OH) | −2.25 |
| C2H2(g)+(OH) | 0.991 | C2H2(g)+(OH) | 0.991 | C2H2(g)+(OH) | 0.991 | C2H2(g)+(OH) | 0.991 |
3. Computational Detail
3.1. Methods
3.2. Model
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Byard, R.W. Carbon Monoxide – the Silent Killer. Forensic Sci. Med. Pathol. 2019, 15, 1–2. [Google Scholar] [CrossRef]
- Can, G.; Sayılı, U.; Aksu Sayman, Ö.; Kuyumcu, Ö.F.; Yılmaz, D.; Esen, E.; Yurtseven, E.; Erginöz, E. Mapping of Carbon Monoxide Related Death Risk in Turkey: A Ten-Year Analysis Based on News Agency Records. BMC Public. Health 2019, 19, 9. [Google Scholar] [CrossRef]
- Kinoshita, H.; Türkan, H.; Vucinic, S.; Naqvi, S.; Bedair, R.; Rezaee, R.; Tsatsakis, A. Carbon Monoxide Poisoning. Toxicol. Rep. 2020, 7, 169–173. [Google Scholar] [CrossRef]
- Fujimori, S.; Inoue, S. Carbon Monoxide in Main-Group Chemistry. J. Am. Chem. Soc. 2022, 144, 2034–2050. [Google Scholar] [CrossRef]
- Elenhorn, M.J. Medical Toxicology Diagnosis and Treatment of Human Poisoning; Williams & Wilkins: Baltimore, MD, USA, 1997. [Google Scholar]
- Baselt, R.C.; Cravey, R.H. Disposition of Toxic Drugs and Chemicals in Man; Biomedical publications: Davis, CA, USA, 1982; Volume 33. [Google Scholar]
- Jager, B.; Espinoza, R. Advances in Low Temperature Fischer-Tropsch Synthesis. Catal. Today 1995, 23, 17–28. [Google Scholar] [CrossRef]
- Biloen, P. On the Activity of Fischer-Tropsch and Methanation Catalysts: A Study Utilizing Isotopic Transients. J. Catal. 1983, 81, 450–463. [Google Scholar] [CrossRef]
- Dry, M.E. Practical and Theoretical Aspects of the Catalytic Fischer-Tropsch Process. Appl. Catal. A Gen. 1996, 138, 319–344. [Google Scholar] [CrossRef]
- Geerlings, J.J.C.; Wilson, J.H.; Kramer, G.J.; Kuipers, H.P.C.E.; Hoek, A.; Huisman, H.M. Fischer–Tropsch Technology — from Active Site to Commercial Process. Appl. Catal. A Gen. 1999, 186, 27–40. [Google Scholar] [CrossRef]
- Dry, M.E. The Fischer–Tropsch Process: 1950–2000. Catal. Today 2002, 71, 227–241. [Google Scholar] [CrossRef]
- Schweicher, J.; Bundhoo, A.; Frennet, A.; Kruse, N.; Daly, H.; Meunier, F.C. DRIFTS/MS Studies during Chemical Transients and SSITKA of the CO/H2 Reaction over Co-MgO Catalysts. J. Phys. Chem. C 2010, 114, 2248–2255. [Google Scholar] [CrossRef]
- Roldan, A.; De Leeuw, N.H. Selective Hydrogenation of CO on Fe 3 S 4 {111}: A Computational Study. Faraday Discuss. 2017, 197, 325–336. [Google Scholar] [CrossRef]
- Cheng, J.; Hu, P.; Ellis, P.; French, S.; Kelly, G.; Lok, C. A DFT Study of the Chain Growth Probability in Fischer–Tropsch Synthesis. J. Catal. 2008, 257, 221–228. [Google Scholar] [CrossRef]
- Liu, J.; Chen, Y.; Wei, J.; Duyar, M.S.; Ordomsky, V.V.; Khodakov, A.Y. Chem Soc Rev Chemical Society Reviews Carbon-Based Catalysts for Fischer-Tropsch Synthesis. Chem. Soc. Rev. 2021, 50, 2337. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, X.; Kang, J.; Yu, Z.; Tian, J.; Gong, Z.; Jia, A.; You, R.; Qian, K.; He, S.; et al. The Active Sites of Cu–ZnO Catalysts for Water Gas Shift and CO Hydrogenation Reactions. Nat. Commun. 2021, 12, 4331. [Google Scholar] [CrossRef]
- Ye, A.; Li, Z.; Ding, J.; Xiong, W.; Huang, W. Synergistic Catalysis of Al and Zn Sites of Spinel ZnAl2O4 Catalyst for CO Hydrogenation to Methanol and Dimethyl Ether. ACS Catal. 2021, 11, 10014–10019. [Google Scholar] [CrossRef]
- Fang, W.; Wang, C.; Liu, Z.; Wang, L.; Liu, L.; Li, H.; Xu, S.; Zheng, A.; Qin, X.; Liu, L.; et al. Physical Mixing of a Catalyst and a Hydrophobic Polymer Promotes CO Hydrogenation through Dehydration. Science 2022, 377, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, X.; Liu, J.; Shi, R.; Waterhouse, G.I.N.; Wen, X.; Zhang, T. Titania-Supported Ni 2 P/Ni Catalysts for Selective Solar-Driven CO Hydrogenation. Adv. Mater. 2021, 33, 2103248. [Google Scholar] [CrossRef]
- Zhang, C.; Li, S.; Zhong, L.; Sun, Y. Theoretical Insights into Morphologies of Alkali-Promoted Cobalt Carbide Catalysts for Fischer–Tropsch Synthesis. J. Phys. Chem. C 2021, 125, 6061–6072. [Google Scholar] [CrossRef]
- Chan, A.Y.; Perry, I.B.; Bissonnette, N.B.; Buksh, B.F.; Edwards, G.A.; Frye, L.I.; Garry, O.L.; Lavagnino, M.N.; Li, B.X.; Liang, Y.; et al. Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 2022, 122, 1485–1542. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Song, T.; Hu, P.; Lok, C.M.; Ellis, P.; French, S. A Density Functional Theory Study of the α-Olefin Selectivity in Fischer-Tropsch Synthesis. J. Catal. 2008, 255, 20–28. [Google Scholar] [CrossRef]
- Cheng, J.; Gong, X.Q.; Hu, P.; Lok, C.M.; Ellis, P.; French, S. A Quantitative Determination of Reaction Mechanisms from Density Functional Theory Calculations: Fischer-Tropsch Synthesis on Flat and Stepped Cobalt Surfaces. J. Catal. 2008, 254, 285–295. [Google Scholar] [CrossRef]
- Zijlstra, B.; Broos, R.J.P.; Chen, W.; Filot, I.A.W.; Hensen, E.J.M. First-Principles Based Microkinetic Modeling of Transient Kinetics of CO Hydrogenation on Cobalt Catalysts. Catal. Today 2020, 342, 131–141. [Google Scholar] [CrossRef]
- Studt, F.; Abild-Pedersen, F.; Wu, Q.; Jensen, A.D.; Temel, B.; Grunwaldt, J.D.; Norskov, J.K. CO Hydrogenation to Methanol on Cu-Ni Catalysts: Theory and Experiment. J. Catal. 2012, 293, 51–60. [Google Scholar] [CrossRef]
- Loveless, B.T.; Buda, C.; Neurock, M.; Iglesia, E. CO Chemisorption and Dissociation at High Coverages during CO Hydrogenation on Ru Catalysts. J. Am. Chem. Soc. 2013, 135, 6107–6121. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, M.; Fei Tan, K.; Borgna, A.; Saeys, M. Density Functional Theory Study of the CO Insertion Mechanism for Fischer−Tropsch Synthesis over Co Catalysts. J. Phys. Chem. C 2009, 113, 8357–8365. [Google Scholar] [CrossRef]
- Lin, S.; Ma, J.; Ye, X.; Xie, D.; Guo, H. CO Hydrogenation on Pd(111): Competition between Fischer-Tropsch and Oxygenate Synthesis Pathways. J. Phys. Chem. C 2013, 117, 14667–14674. [Google Scholar] [CrossRef]
- Zhang, M.; Wu, Y.; Dou, M.; Yu, Y. A DFT Study of Methanol Synthesis from CO2 Hydrogenation on the Pd(111) Surface. Catal. Lett. 2018, 148, 2935–2944. [Google Scholar] [CrossRef]
- Ge, Q.; Neurock, M. Adsorption and Activation of CO over Flat and Stepped Co Surfaces: A First Principles Analysis. J. Phys. Chem. B 2006, 110, 15368–15380. [Google Scholar] [CrossRef]
- Zhang, S.T.; Yan, H.; Wei, M.; Evans, D.G.; Duan, X. Hydrogenation Mechanism of Carbon Dioxide and Carbon Monoxide on Ru(0001) Surface: A Density Functional Theory Study. RSC Adv. 2014, 4, 30241–30249. [Google Scholar] [CrossRef]
- Dragutan, V.; Dragutan, I.; Xiong, G.; You, L.; Sun, Y.; Ding, F. Recent Developments on Carbon-Carbon Cross-Coupling Reactions Using Rare-Earth Metals-Derived Coordination Polymers as Efficient and Selective Pd Catalytic Systems. Resour. Chem. Mater. 2022, 1, 325–338. [Google Scholar] [CrossRef]
- Molnár, Á. Efficient, Selective, and Recyclable Palladium Catalysts in Carbon− Carbon Coupling Reactions. Chem. Rev. 2011, 111, 2251–2320. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Ikeda, Y.; Kuroda, A.; Tanaka, S.; Minakata, S. Pd/NHC-Catalyzed Enantiospecific and Regioselective Suzuki–Miyaura Arylation of 2-Arylaziridines: Synthesis of Enantioenriched 2-Arylphenethylamine Derivatives. J. Am. Chem. Soc. 2014, 136, 8544–8547. [Google Scholar] [CrossRef] [PubMed]
- Masson-Makdissi, J.; Vandavasi, J.K.; Newman, S.G. Switchable Selectivity in the Pd-Catalyzed Alkylative Cross-Coupling of Esters. Org. Lett. 2018, 20, 4094–4098. [Google Scholar] [CrossRef] [PubMed]
- Molander, G.A.; Trice, S.L.J.; Kennedy, S.M. Scope of the Two-Step, One-Pot Palladium-Catalyzed Borylation/Suzuki Cross-Coupling Reaction Utilizing Bis-Boronic Acid. J. Org. Chem. 2012, 77, 8678–8688. [Google Scholar] [CrossRef]
- Hoshi, T.; Honma, T.; Mori, A.; Konishi, M.; Sato, T.; Hagiwara, H.; Suzuki, T. An Active, General, and Long-Lived Palladium Catalyst for Cross-Couplings of Deactivated (Hetero) Aryl Chlorides and Bromides with Arylboronic Acids. J. Org. Chem. 2013, 78, 11513–11524. [Google Scholar] [CrossRef]
- Li, L.; Zhao, S.; Joshi-Pangu, A.; Diane, M.; Biscoe, M.R. Stereospecific Pd-Catalyzed Cross-Coupling Reactions of Secondary Alkylboron Nucleophiles and Aryl Chlorides. J. Am. Chem. Soc. 2014, 136, 14027–14030. [Google Scholar] [CrossRef] [PubMed]
- Martínez, R.; Pastor, I.M.; Yus, M. Biscarboxy-Functionalized Imidazole and Palladium as Highly Active Catalytic System in Protic Solvents: Methanol and Water. Synthesis 2014, 46, 2965–2975. [Google Scholar] [CrossRef]
- Tu, T.; Sun, Z.; Fang, W.; Xu, M.; Zhou, Y. Robust Acenaphthoimidazolylidene Palladium Complexes: Highly Efficient Catalysts for Suzuki–Miyaura Couplings with Sterically Hindered Substrates. Org. Lett. 2012, 14, 4250–4253. [Google Scholar] [CrossRef]
- Tang, J.-S.; Tian, M.; Sheng, W.-B.; Guo, C.-C. Efficient Palladium-Catalyzed Cross-Coupling Reaction of Alkynyl Halides with Organoboronic Acids under Aerobic Conditions. Synthesis 2012, 44, 541–546. [Google Scholar] [CrossRef]
- Ben Halima, T.; Zhang, W.; Yalaoui, I.; Hong, X.; Yang, Y.-F.; Houk, K.N.; Newman, S.G. Palladium-Catalyzed Suzuki–Miyaura Coupling of Aryl Esters. J. Am. Chem. Soc. 2017, 139, 1311–1318. [Google Scholar] [CrossRef]
- Torres Galvis, H.M.; Bitter, J.H.; Khare, C.B.; Ruitenbeek, M.; Dugulan, A.I.; de Jong, K.P. Supported Iron Nanoparticles as Catalysts for Sustainable Production of Lower Olefins. Science 2012, 335, 835–838. [Google Scholar] [CrossRef]
- Ghogia, A.C.; Nzihou, A.; Serp, P.; Soulantica, K.; Pham Minh, D. Cobalt Catalysts on Carbon-Based Materials for Fischer-Tropsch Synthesis: A Review. Appl. Catal. A Gen. 2021, 609, 117906. [Google Scholar] [CrossRef]
- Pedersen, E.Ø.; Svenum, I.H.; Blekkan, E.A. Mn Promoted Co Catalysts for Fischer-Tropsch Production of Light Olefins – An Experimental and Theoretical Study. J. Catal. 2018, 361, 23–32. [Google Scholar] [CrossRef]
- Borji, F.; Pour, A.N.; Karimi, J.; Izadyar, M.; Keyvanloo, Z.; Hashemian, M. The Molecular Adsorption of Carbon Monoxide on Cobalt Surfaces: A DFT Study. Prog. React. Kinet. Mech. 2017, 42, 89–98. [Google Scholar] [CrossRef]
- Yao, Z.; Guo, C.; Mao, Y.; Hu, P. Quantitative Determination of C-C Coupling Mechanisms and Detailed Analyses on the Activity and Selectivity for Fischer-Tropsch Synthesis on Co(0001): Microkinetic Modeling with Coverage Effects. ACS Catal. 2019, 9, 5957–5973. [Google Scholar] [CrossRef]
- Chen, C.; Wang, Q.; Wang, G.; Hou, B.; Jia, L.; Li, D. Mechanistic Insight into the C 2 Hydrocarbons Formation from Syngas on Fcc-Co(111) Surface: A DFT Study. J. Phys. Chem. C 2016, 120, 9132–9147. [Google Scholar] [CrossRef]
- Petersen, M.A.; Van Den Berg, J.A.; Ciobîcǎ, I.M.; Van Helden, P. Revisiting CO Activation on Co Catalysts: Impact of Step and Kink Sites from DFT. ACS Catal. 2017, 7, 1984–1992. [Google Scholar] [CrossRef]
- Van Helden, P.; Van Den Berg, J.A.; Petersen, M.A.; Janse Van Rensburg, W.; Ciobîcǎ, I.M.; Van De Loosdrecht, J. Computational Investigation of the Kinetics and Mechanism of the Initial Steps of the Fischer-Tropsch Synthesis on Cobalt. Faraday Discuss. 2017, 197, 117–151. [Google Scholar] [CrossRef]
- Ciobica, I.M.; van Rensburg, W.J. Hydrogen-Assisted CO Dissociation on FCC-Co {100}: DFT Analysis and Microkinetic Interpretation. In Proceeding of the EuropaCat IX, Catalysis for a Sustainable World, Salamanca, Spain, 30 August–4 September 2009. [Google Scholar]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. A Dimer Method for Finding Saddle Points on High Dimensional Potential Surfaces Using Only First Derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- Heyden, A.; Bell, A.T.; Keil, F.J. Efficient Methods for Finding Transition States in Chemical Reactions: Comparison of Improved Dimer Method and Partitioned Rational Function Optimization Method. J. Chem. Phys. 2005, 123, 224101. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Bolton, E.E.; Chen, J.; Kim, S.; Han, L.; He, S.; Shi, W.; Simonyan, V.; Sun, Y.; Thiessen, P.A.; Wang, J.; et al. PubChem3D: A New Resource for Scientists. J. Cheminform 2011, 3, 32. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary; National Center for Biotechnology Information: Bethesda, MD, USA, 2022. [Google Scholar]
- NCBI National Center for Biotechnology Information. PubChem Substance Record for SID 10512399, HCOH (Hydroxymethylene) Source: NIST Chemistry WebBook. 2022. Available online: https://pubchem.ncbi.nlm.nih.gov/substance/10512399 (accessed on 24 August 2022).
- BIOVIA, Dassault Systèmes. Materials Studio, 17.1.0.48; Dassault Systèmes: San Diego, CA, USA, 2022. [Google Scholar]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Dubay, O.; Dubay, O. P4vasp. 2018. Available online: http://Www.P4vasp.At/ (accessed on 20 August 2022).
- Ganose, A.M.; Jackson, A.J.; Scanlon, D.O. Sumo: Command-Line Tools for Plotting and Analysis of Periodic Ab Initio Calculations. J. Open. Source Softw. 2018, 3, 717. [Google Scholar] [CrossRef]
- Psofogiannakis, G.; St-Amant, A.; Ternan, M. Methane Oxidation Mechanism on Pt(111): A Cluster Model DFT Study. J. Phys. Chem. B 2006, 110, 24593–24605. [Google Scholar] [CrossRef]
- Ashwell, A.P.; Lin, W.; Hofman, M.S.; Yang, Y.; Ratner, M.A.; Koel, B.E.; Schatz, G.C. Hydrogenation of CO to Methanol on Ni(110) through Subsurface Hydrogen. J. Am. Chem. Soc. 2017, 139, 17582–17589. [Google Scholar] [CrossRef] [PubMed]
- Amaya-Roncancio, S.; Linares, D.H.; Duarte, H.A.; Sapag, K. DFT Study of Hydrogen-Assisted Dissociation of CO by HCO, COH, and HCOH Formation on Fe(100). J. Phys. Chem. C 2016, 120, 10830–10837. [Google Scholar] [CrossRef]
- Zhu, Y.A.; Chen, D.; Zhou, X.G.; Yuan, W.K. DFT Studies of Dry Reforming of Methane on Ni Catalyst. Catal. Today 2009, 148, 260–267. [Google Scholar] [CrossRef]
- Will Medlin, J.; Allendorf, M.D. Theoretical Study of the Adsorption of Acetylene on the (111) Surfaces of Pd, Pt, Ni, and Rh. J. Phys. Chem. B 2002, 107, 217–223. [Google Scholar] [CrossRef]
- Zhang, M.; Dou, M.; Yu, Y. DFT Study of CO2 Conversion on InZr3(110) Surface. Phys. Chem. Chem. Phys. 2017, 19, 28917–28927. [Google Scholar] [CrossRef]
- Hirunsit, P. Electroreduction of Carbon Dioxide to Methane on Copper, Copper–Silver, and Copper–Gold Catalysts: A DFT Study. J. Phys. Chem. C 2013, 117, 8262–8268. [Google Scholar] [CrossRef]
- Liu, L.; Yao, H.; Jiang, Z.; Fang, T. Theoretical Study of Methanol Synthesis from CO2 Hydrogenation on PdCu3(111) Surface. Appl. Surf. Sci. 2018, 451, 333–345. [Google Scholar] [CrossRef]
- Zhao, Y.F.; Yang, Y.; Mims, C.; Peden, C.H.F.; Li, J.; Mei, D. Insight into Methanol Synthesis from CO2 Hydrogenation on Cu(1 1 1): Complex Reaction Network and the Effects of H2O. J. Catal. 2011, 281, 199–211. [Google Scholar] [CrossRef]
- Qi, Y.; Yang, J.; Duan, X.; Zhu, Y.A.; Chen, D.; Holmen, A. Discrimination of the Mechanism of CH4formation in Fischer-Tropsch Synthesis on Co Catalysts: A Combined Approach of DFT, Kinetic Isotope Effects and Kinetic Analysis. Catal. Sci. Technol. 2014, 4, 3534–3543. [Google Scholar] [CrossRef]
- Cheng, J.; Hu, P.; Ellis, P.; French, S.; Kelly, G.; Martin Lok, C. Density Functional Theory Study of Iron and Cobalt Carbides for Fischer−Tropsch Synthesis. J. Phys. Chem. C 2009, 114, 1085–1093. [Google Scholar] [CrossRef]
- Niu, J.; Du, X.; Ran, J.; Wang, R. Dry (CO2) Reforming of Methane over Pt Catalysts Studied by DFT and Kinetic Modeling. Appl. Surf. Sci. 2016, 376, 79–90. [Google Scholar] [CrossRef]
- Cheng, J.; Hu, P.; Ellis, P.; French, S.; Kelly, G.; Martin Lok, C. A First-Principles Study of Oxygenates on Co Surfaces in Fischer−Tropsch Synthesis. J. Phys. Chem. C 2008, 112, 9464–9473. [Google Scholar] [CrossRef]
- Parr, R.G. Density Functional Theory. Annu. Rev. Phys. Chem. 1983, 34, 631–656. [Google Scholar] [CrossRef]
- Orio, M.; Pantazis, D.A.; Neese, F. Density Functional Theory. Photosynth. Res. 2009, 102, 443–453. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Challenges for Density Functional Theory. Chem. Rev. 2012, 112, 289–320. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Open-Shell Transition Metals. Phys. Rev. B 1993, 48, 13115–13118. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef]
- Olsen, R.A.; Kroes, G.J.; Henkelman, G.; Arnaldsson, A.; Jónsson, H. Comparison of Methods for Finding Saddle Points without Knowledge of the Final States. J. Chem. Phys. 2004, 121, 9776–9792. [Google Scholar] [CrossRef]
- Kästner, J.; Sherwood, P. Superlinearly Converging Dimer Method for Transition State Search. J. Chem. Phys. 2008, 128, 014106. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Sheppard, D.; Rogal, J.; Henkelman, G. Solid-State Dimer Method for Calculating Solid-Solid Phase Transitions. J. Chem. Phys. 2014, 140, 174104. [Google Scholar] [CrossRef] [PubMed]
- Aroyo, M.I.; Orobengoa, D.; De La Flor, G.; Tasci, E.S.; Perez-Mato, J.M.; Wondratschek, H. Brillouin-Zone Database on the Bilbao Crystallographic Server. Acta Crystallogr. A Found. Adv. 2014, 70, 126–137. [Google Scholar] [CrossRef] [PubMed]
- de la Peña O’Shea, V.A.; Moreira, I. de P.R.; Roldán, A.; Illas, F. Electronic and Magnetic Structure of Bulk Cobalt: The α, β, and ε-Phases from Density Functional Theory Calculations. J. Chem. Phys. 2010, 133, 024701. [Google Scholar] [CrossRef] [PubMed]
- Hatzor, A.; Weiss, P.S. Molecular Rulers for Scaling Down Nanostructures. Science 2001, 291, 1019–1020. [Google Scholar] [CrossRef]
- de la Peña O′Shea, V.A.; de la Piscina, P.R.; Homs, N.; Aromí, G.; Fierro, J.L.G. Development of Hexagonal Closed-Packed Cobalt Nanoparticles Stable at High Temperature. Chem. Mater. 2009, 21, 5637–5643. [Google Scholar] [CrossRef]
- Sun, S.; Murray, C.B. Synthesis of Monodisperse Cobalt Nanocrystals and Their Assembly into Magnetic Superlattices (Invited). J. Appl. Phys. 1999, 85, 4325–4330. [Google Scholar] [CrossRef]
- Materials Project. Available online: https://materialsproject.org/ (accessed on 3 July 2022).
- Liu, J.-X.; Su, H.-Y.; Sun, D.-P.; Zhang, B.-Y.; Li, W.-X. Crystallographic Dependence of CO Activation on Cobalt Catalysts: HCP versus FCC. J. Am. Chem. Soc. 2013, 135, 16284–16287. [Google Scholar] [CrossRef]
- Skriver, H.L.; Rosengaard, N.M. Surface Energy and Work Function of Elemental Metals. Phys. Rev. B 1992, 46, 7157. [Google Scholar] [CrossRef] [PubMed]










| Number of Layers | |
|---|---|
| 3 | 3.590 |
| 4 | 3.647 |
| 5 | 3.639 |
| 6 | 3.626 |
| 7 | 3.639 |
| 8 | 3.632 |
| Number of the Fixed Layers | |
|---|---|
| 1 | 4.492 |
| 2 | 4.495 |
| 3 | 4.4955 |
| 4 | 4.4955 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torkashvand, M.; Sarabadani Tafreshi, S.; de Leeuw, N.H. Density Functional Theory Study of the Hydrogenation of Carbon Monoxide over the Co (001) Surface: Implications for the Fischer–Tropsch Process. Catalysts 2023, 13, 837. https://doi.org/10.3390/catal13050837
Torkashvand M, Sarabadani Tafreshi S, de Leeuw NH. Density Functional Theory Study of the Hydrogenation of Carbon Monoxide over the Co (001) Surface: Implications for the Fischer–Tropsch Process. Catalysts. 2023; 13(5):837. https://doi.org/10.3390/catal13050837
Chicago/Turabian StyleTorkashvand, Mostafa, Saeedeh Sarabadani Tafreshi, and Nora H. de Leeuw. 2023. "Density Functional Theory Study of the Hydrogenation of Carbon Monoxide over the Co (001) Surface: Implications for the Fischer–Tropsch Process" Catalysts 13, no. 5: 837. https://doi.org/10.3390/catal13050837
APA StyleTorkashvand, M., Sarabadani Tafreshi, S., & de Leeuw, N. H. (2023). Density Functional Theory Study of the Hydrogenation of Carbon Monoxide over the Co (001) Surface: Implications for the Fischer–Tropsch Process. Catalysts, 13(5), 837. https://doi.org/10.3390/catal13050837