Abstract
Aniline, a vital component in various chemical industries, is known to be a hazardous persistent organic pollutant that can cause environmental pollution through its manufacturing, processing, and transportation. In this study, the microcosms were established using sediment with a history of aniline pollution as an inoculum to analyze the aniline biodegradation under aerobic conditions through stable isotope probing (SIP) and isopycnic density gradient centrifugation technology. During the degradation assay, aniline that was 13C-labeled in all six carbons was utilized to determine the phylogenetic identity of the aniline-degrading bacterial taxa that incorporate 13C into their DNA. The results revealed that aniline was completely degraded in the microcosm after 45 and 69 h respectively. The bacteria affiliated with Acinetobacter (up to 34.6 ± 6.0%), Zoogloea (up to 15.8 ± 2.2%), Comamonas (up to 2.6 ± 0.1%), and Hydrogenophaga (up to 5.1 ± 0.6%) genera, which are known to degrade aniline, were enriched in the heavy fractions (the DNA buoyant density was 1.74 mg L−1) of the 13C-aniline treatments. Moreover, some rarely reported aniline-degrading bacteria, such as Prosthecobacter (up to 16.0 ± 1.6%) and Curvibacter (up to 3.0 ± 1.6%), were found in the DNA-SIP experiment. Gene families affiliated with atd, tdn, and dan were speculated to be key genes for aniline degradation based on the abundance in functional genes and diversity in different treatments as estimated using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States version 2 (PICRUSt2) and the Kyoto Encyclopedia of Genes and Genomes (KEGG). This study revealed the functional bacteria and possible degradation genes for aniline degradation in simulated polluted environments through SIP. These findings suggest that important degrading bacteria for the transformation of aniline and potential degradation pathways may be useful in the effective application of bioremediation technologies to remediate aniline-contaminated sites.
1. Introduction
Aniline, an industrial feedstock used in the production of dyes, pesticides, and pharmaceutical compounds, has become a serious environmental pollutant in soil and water []. Aniline can be released into the environment during chemical manufacturing, spill accidents, or biodegradation of azo dyes, nitroaromatic compounds, and chloroaniline pesticides in soils, thereby causing environmental damage [,]. As a priority environmental pollutant, aniline is considered a potential carcinogen that is toxic to aquatic organisms and humans []. For example, the sudden water pollution accident in 2012 led to more than 39 tons of aniline entering the Zhuozhang River, causing a water crisis for more than one million residents in several downstream provinces []. In recent years, aniline contamination has posed significant threat to environmental safety. For example, in 2019, the explosion of a chemical tank at a chemical plant in Jiangsu Province caused aniline levels in downstream water samples to exceed 39 times the environmental standard and an economic loss of more than CNY 1.986 billion []. Therefore, it is important to investigate the environmental fate of aniline.
Remediation technologies for sites contaminated with aniline include biological and nonbiological methods. Various physical and chemical methods such as adsorption, filtration, coagulation, precipitation, and chemical oxidation have been explored to remove aniline. However, they are not widely used due to their low efficiency, high cost, and high energy requirements [,]. The biodegradation of aniline is considered a cost-effective, efficient, and environmentally friendly alternative method that has attracted increasing attention in recent decades and is an important repair process in various environmental media [,]. Currently, most studies on aniline degradation focus on the degradation effects, microbial communities, and isolation and identification of degrading bacteria. For example, Zhang et al. [] used exogenous auto-inducers to stimulate aniline degradation, but they could only identify changes in the microbial community and dominant bacteria. Peng et al. [] isolated Delftia tsuruhatensis from activated sludge, which could effectively remove aniline in a concentration range between 200 and 800 mg/L within 72 h. Moreover, studies on the biodegradation genes and pathways of aniline remain at the level of pure microbial isolates [].
However, since many functional bacteria are difficult to separate and identify, there is an urgent need to accurately identify functional aniline-degrading bacteria and potential degradation pathways using novel in situ analytical methods. Stable isotope probing (SIP) is a molecular ecology method that uses stable isotopes to detect microbial genomic DNA or RNA in complex environments []. By isolating stable isotope (13C)-labeled DNA, RNA, proteins, or phospholipid fatty acids from substrates with isopycnic density gradient centrifugation, SIP can be used to identify active microorganisms in the environment and reveal functional genes at the community level []. In this study, DNA-SIP was combined with high-throughput sequencing to identify aniline-degrading bacteria, and PICRUST was used to predict the metabolic potential of aniline-degrading bacteria. This study solved the problem of the inability to lock aniline-degrading functional bacteria in situ, and it will provide a theoretical reference for subsequent aniline pollution remediation.
2. Results
2.1. Variation in Aniline Concentration
Aniline degradation without a delay was observed in the microcosms containing 13C/12C under aerobic conditions (Figure 1). It was found that the labeling of 13C molecules did not affect the degradation rate of aniline at all. The aniline-degradation rate in the control microcosm with sterile soil was very slow during the cultivation process. After 69 h of incubation, only a 21.2% decrease in aniline content was observed in the sterilized samples, which could be attributed to other physical and chemical factors, such as volatilization. In contrast, depletion of aniline was observed in the 12C-aniline and the 13C-aniline treatments at both 45 h and 69 h, respectively. This indicates that the decrease in aniline was primarily due to the biodegradation of aniline in microcosms amended with nonsterilized sediment. It took 45 h to degrade aniline for the first time completely, but it only took 24 h for the second time, which was significantly shorter (p < 0.05). The degradation rates of aniline for the first and second cycles in the microcosms reached 1.11 mg/L/h and 2.08 mg/L/h, respectively.
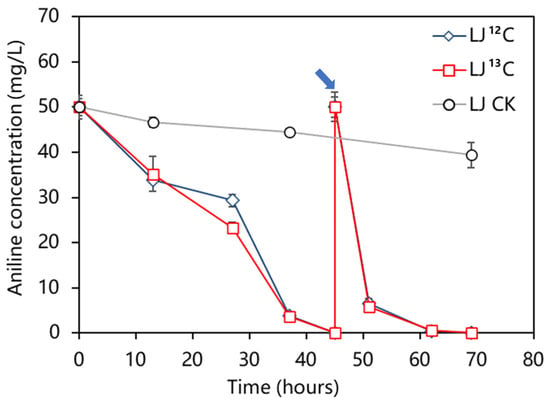
Figure 1.
Aniline concentration in the treatments throughout the cultivation period. The soil in the control microcosms was autoclaved before incubation. The arrows indicate the recovery of the aniline reamendment. Data are expressed as mean ± standard error (n = 3).
2.2. Isopycnic Density Gradient Centrifugation of DNA
According to the buoyant density (BD) of the fractions obtained through isopycnic density gradient centrifugation, genomic DNA extracted from 12C-aniline and 13C-aniline treatments at the corresponding time points (45 h and 69 h, respectively) were separated into “heavy” fractions (BD = 1.74 g mL−1) and “light” fractions (BD = 1.71 g mL−1). The relative abundance in the 16S rRNA genes across the CsCl density gradient fractions of the 12C-aniline and 13C-aniline treatments is shown in Figure 2. The maximum relative abundance in the 16S rRNA gene in the 12C-aniline treatments was observed in the light fractions during SIP incubation (Figure 2a,c). However, in addition to the first peak, a second peak appeared in the heavier fractions in the 13C-aniline treatment (Figure 2b,d), indicating that 13C was incorporated into the aniline-degrading bacteria during cultivation.
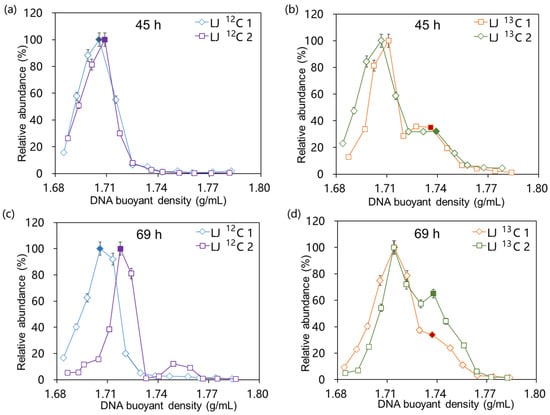
Figure 2.
Relative abundance in the 16S rRNA gene across CsCl density gradient fractions of 13C-aniline (b,d) and 12C-aniline (a,c) treatments after 45 and 69 h of incubation. The figures show all the fractions with a density range of 1.68 to 1.80 g mL−1. The relative abundance was expressed as the ratio of 16S rRNA gene copy number in each fraction to the highest 16S rRNA gene copy number in the gradient fractions. Then, the filled-symbol-corresponded DNA fractions were analyzed for subsequent microbial communities.
2.3. Distribution of Microorganisms in Different Fractions
In order to identify the 13C-labeled aniline-degrading bacteria and isolate them from the microcosmos, the light fractions (BD = 1.71 g mL−1) containing the highest abundance in the 16S rRNA gene from the treatments with 12C-aniline and the heavy DNA fractions (BD = 1.74 g mL−1) from the treatments with 13C-aniline at 45 and 69 h were selected for 16S rRNA amplicon sequencing (Figure 3). The dominant phylum was Proteobacteria in all treatments throughout the culture stage. As shown in Figure 3a, bacteria belonging to Proteobacteria (86.2 ± 1.4%) dominated the heavy DNA fractions (BD = 1.74 g mL−1) in the 13C-aniline treatment at 45 h, followed by Actinobacteria (5.4 ± 1.3%) and Firmicutes (2.1 ± 0.9%). Moreover, in the 13C-aniline treatment at 69 h, bacteria affiliated with Proteobacteria (75.7 ± 0.2%) dominated the heavy DNA fractions (BD = 1.74 g mL−1), followed by Verrucomicrobia (16.6 ± 1.6%) and Actinobacteria (2.2 ± 0.1%).
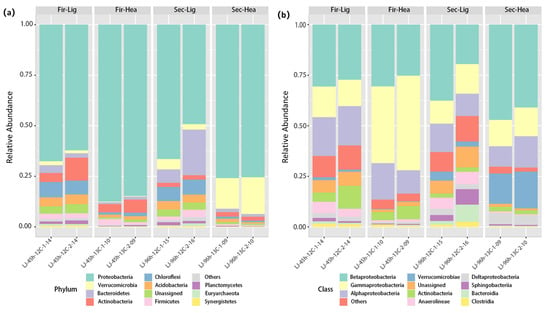
Figure 3.
Bar plot illustrating the microbial community composition of the top 11 taxonomic groups at the phylum (a) and class (b) levels for 13C-aniline and 12C-aniline treatments at 45 and 69 h. “Fir” and “Sec” stand for samples at 45 and 69 h, while “Lig” and “Hec” stand for light and heavy fractions, respectively (the same applies below).
At the class level, Betaproteobacteria (19.57–47.17%), Gammaproteobacteria (11.24–46.73%), and Alphaproteobacteria (10.19–19.53%) accounted for a high proportion in all treatments (Figure 3b). Gammaproteobacteria were the dominant bacteria in the 13C-aniline treatments at 45 h, whereas Betaproteobacteria were the dominant bacteria in the other treatments. In addition, Verrucomicrobiae were significantly enriched in the microbial communities of the 13C-aniline treatments at 69 h (p < 0.05).
The sample–genera relationship was presented using a Circos plot. As shown in Figure 4, the most of the reads were affiliated with Acinetobacter members. This genus accounted for 9.6% of the total reads (2.0–36.5% in the DNA libraries) and was more abundant in the 13C-aniline-degraded samples. Zoogloea was the second most abundant genus (5.2% of total reads) and accounted for 0.3–16.4% of the total valid reads in each sample. Other important genera were Dechloromonas (0.9–8.8%), Prosthecobacter (0.1–16.2%), Novosphingobium (3.3–4.7%), and Aquabacterium (0.8–5.9%).
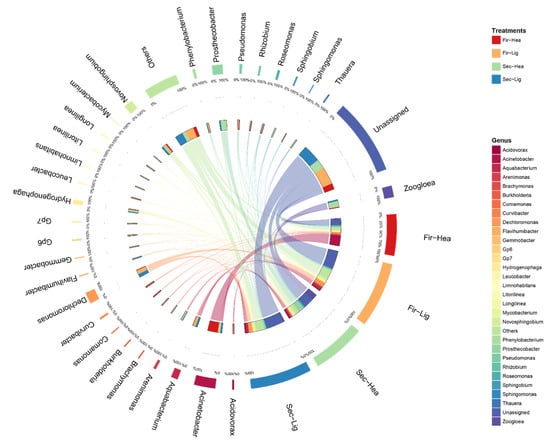
Figure 4.
Distribution in dominant genera in DNA samples from 13C-SIP/12C incubations. The data were visualized using the Circos software (http://circos.ca/, accessed on 6 January 2021). Absolute abundances are given above the inner segment, whereas relative abundances are given above the outer segment. Different colored bands represent different genera.
The light fractions of the 12C-aniline treatments contained the most valid reads for Mycobacterium (75.4%), Litorilinea (56.4%), Brachymonas (55.7%), and Aquabacterium (54.8%) at 45 h. Most valid reads of Comamonas (96.8%), Acinetobacter (71.0%), Curvibacter (65.5%), and Rhizobium (51.2%) were distributed in the heavy fractions of the 13C-aniline treatments at 45 h. It was indicated that these genera probably degraded and assimilated 13C from aniline. Compared with 45 h, the distribution in valid reads of bacteria in different treatments changed at 69 h. For example, most valid reads of Prosthecobacter (80.4%), Zoogloea (75.1%), Sphingobium (74.2%), and Hydrogenophaga (58.4%) were distributed in the heavy fractions at 69 h in the 13C-aniline treatment.
2.4. Enrichment of Functional Bacteria in Heavy Fractions
As shown in Figure 5a and Table S1, in the 12C-aniline treatments, bacteria affiliated with Dechloromonas (7.7 ± 1.8%) dominated the light DNA fractions (BD = 1.71 g mL−1) at 45 h, followed by Aquabacterium (5.3 ± 0.2%), Novosphingobium (3.9 ± 1.8%), and Phenylobacterium (2.0 ± 0.2%). Notably, at 45 h of the 13C-aniline treatment, Acinetobacter-associated bacteria (34.6 ± 6.0%) dominated the heavy DNA fractions (BD = 1.74 g mL−1), followed by Novosphingobium (3.3 ± 1.7%), Curvibacter (3.0 ± 1.6%), Comamonas (2.6 ± 0.1%), and Rhizobium (2.3 ± 0.3%). As shown in Figure 5b, Dechloromonas (7.1 ± 1.6%) was the most abundant genus in the light fractions of the 12C-aniline treatments at 69 h (Figure 5b), followed by Aquabacterium (3.8 ± 0.7%), Acinetobacter (3.6 ± 3.0%), Novosphingobium (3.4 ± 0.4%), and Prosthecobacter (2.6 ± 0.8%). In addition, Prosthecobacter (16.0 ± 1.6%) was the most abundant genus in the heavy fractions of the 13C-aniline treatments at 69 h (Figure 5b), followed by Zoogloea (15.8 ± 2.2%), Hydrogenophaga (5.1 ± 0.6%), Acinetobacter (4.8 ± 0.1%), and Novosphingobium (4.5 ± 2.1%).
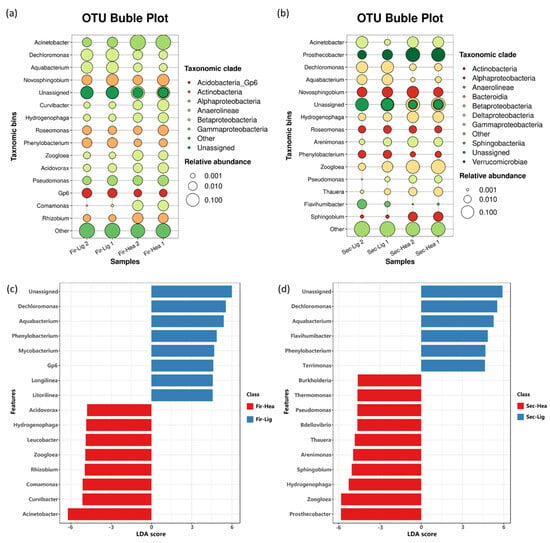
Figure 5.
The difference in the bacterial community composition shown by the most abundant genera (top 15) in the heavy fractions of the 13C-aniline treatments and the light fractions of the 12C-aniline treatments at 45 h (a) and 69 h (b). The bubble plots show the relative abundances between genera. Each bubble represents a representative fraction of the culture. Three cultures were sequenced for each treatment. Linear discriminant analysis effect size (LEFSe) revealed differentially abundant genera between the heavy fractions of the 13C-aniline and light fractions of the 12C-aniline treatments at 45 h (c) and 69 h (d) (p < 0.05 and LDA score > 2.0).
In addition, linear discriminant analysis effect size (LEfSe) identified 15 genera with significantly different abundances between the light fractions of the 12C-aniline treatments and the heavy fractions of the 13C-aniline treatments at 45 and 69 h (p < 0.05 and LDA score > 3.0) (Figure 5b,c). In particular, Acinetobacter, Curvibacter, Comamonas, Rhizobium, and Zoogloea were significantly enriched in the heavy fractions of the 13C-aniline treatments at 45 h, indicating that bacteria affiliated with these genera might be putative aniline-degrading bacteria. However, Prosthecobacter, Zoogloea, Hydrogenophaga, Sphingobium, and Arenimonas were significantly enriched in the heavy fractions of the 13C-aniline treatments at 69 h, indicating that the recontamination of the aniline led to the succession in degrading bacteria.
2.5. Co-Occurrence Network of Microbial Community
In the view of the entire microbial community level, co-occurrence networks were constructed to investigate microbe–microbe interactions in the 13C-aniline samples (Figure 6). Only strong and significant Spearman correlations (0.8 < |r| < 1; p < 0.05) between pairwise operational taxonomic units (OTUs) were visualized in the network. Strong correlations between the nodes determined the size of each node. The size of each node was proportional to the number of strong correlations with other nodes. A strong interaction was observed between the two networks when the taxonomy of each node was considered. At 45 h, OTUs were mainly correlated with the genera Zoogloea, Acinetobacter, Rhizobium, and Novosphingobium, accounting for 7.55%, 7.55%, 5.66%, and 5.66% of the total number of nodes, respectively, while Zoogloea became the most correlated genus at 69 h, accounting for 11.32% of the total number of nodes, followed by Prosthecobacter (7.55%), Acinetobacter (5.66%), and Sphingobium (3.77%). Notably, three unique correlating genera (Zoogloea, Acinetobacter, and Roseomonas) were identified in both networks in the 13C-aniline-degrading treatments at 45 and 69 h.
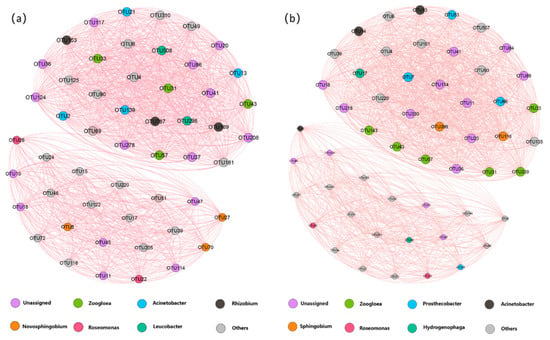
Figure 6.
Co-occurrence network showing correlations between the most abundant OTUs (relative abundance > 0.2% in each group) in the heavy fractions from the 13C-aniline treatments at 45 h (a) and 69 h (b). One connection represents a significant Spearman correlation (0.8 < |r| < 1; p < 0.05). The size of each node is proportional to the number of strong correlations with other nodes. The color of a node indicates its association with the phylum. Aniline-degrading bacteria identified using DNA-SIP are shown in the network diagram.
Although the number of nodes in the microbial co-occurrence networks at both 45 h and 69 h was 54, the network at 69 h had a greater number of links than the other networks at 45 h. The number of links/nodes, closeness centrality, and betweenness are typically used to evaluate the interactions between OTUs within a network—the higher the value, the stronger the interaction. This finding supported our expectation that the potential interactions within the network at 69 h were stronger than those at 45 h, as indicated by the number of links. The centralization in closeness and betweenness, which represented the number of paths through a node, was higher in the network at 69 h than at 45 h.
2.6. Prediction of Functional Genes
Functional gene abundance and diversity in different treatment groups were estimated using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States version 2 (PICRUSt2) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Figure 7). Many studies have predicted that functional genes are linked to aniline metabolism [,]. Figure 7 visualizes the ratios of the gene prediction results to the OTU numbers. Functional gene prediction showed that a high proportion of genes, including glutamine synthetase (atdA1, K01915), glutamine amidotransferase (atdA2, K02501), LysR family transcriptional regulator (danR, K03566), and 4-oxalocrotonate decarboxylase (tdnK, K01617) were present in most samples. Catechol 1,2-dioxygenase(catA2, K03381) and LysR family transcriptional regulator (danR, K03566) genes were enriched in the 13C-labeled heavy fractions at 46 h. The proportion of aniline-degradation genes in the 13C-labeled heavy fractions gradually increased with an increasing culture time. After 96 h of incubation, almost all aniline-degradation-related genes were higher in the heavy fractions of the 13C-aniline treatments than in the light fractions of the 12C-aniline treatment.
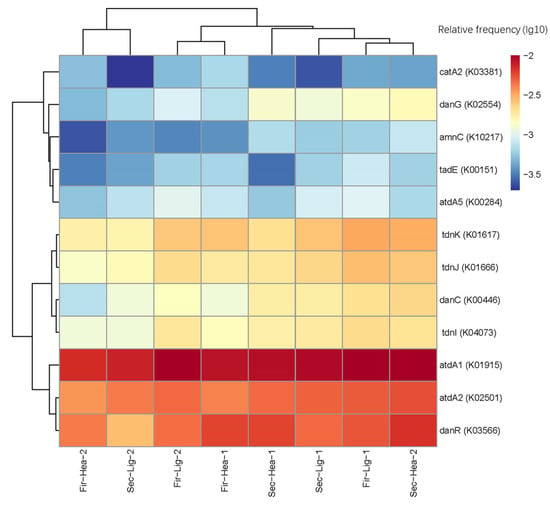
Figure 7.
Heat map of the proportion of functional genes involved in aniline degradation relative to OTUs.
3. Discussion
3.1. Degradation in Aniline and Uptake of 13C by the Microorganisms
In this study, DNA-SIP was used to identify functional microorganisms from microbial communities in the microcosms using 13C-labeled aniline. The experimental results showed that 13C had no effect on the aniline-degradation rate of the microorganisms (Figure 1). Due to small change in the aniline concentration in the sterilization group, we concluded that the degradation in the aniline concentration in the experimental groups was mainly due to microbial degradation []. Moreover, the aniline-degradation rate increased with an increasing culture time, which may have been due to the gradual accumulation in functional bacteria []. Therefore, after the degradation in 13C-aniline, a clear peak appeared in the heavy DNA fractions during isopycnic density gradient centrifugation. This indicated that 13C atoms entered the cells of the functional microorganisms during the degradation and assimilation of the aniline and were incorporated into the DNA []. Therefore, the composition of the functional bacteria associated with aniline degradation was determined through 16S rRNA gene sequencing of the centrifuged heavy components.
3.2. Aniline-Degrading Bacteria Identified in Aerobic Microcosm
As shown in Figure 5, several genera (including Acinetobacter, Curvibacter, and Comamonas, etc.) were significantly enriched in the heavy fractions of the 45 h 13C-aniline treatments and were therefore identified as aniline-degrading bacteria. Acinetobacter [] has been shown to degrade aniline and/or aniline derivatives []. These gene clusters are responsible for the complete conversion of aniline to intermediates of the TCA cycle that were cloned from the aniline-degrading plasmid pYA1 of Acinetobacter sp. YAA [,,]. Although there are few reports on aniline degradation by Curvibacter, it has been demonstrated that Curvibacter can degrade several benzenoid compounds, such as phthalates and catechol []. In a previous study on aniline-degrading bacteria, Acinetobacter, Comamonas, and Sphingobium were detected in biofilters after aniline acclimation []. In addition, some species of Acinetobacter sp., Comamonas sp., Sphingobium sp., Acidovorax sp., and Pseudomonas sp. have been isolated and identified as aniline-degrading bacteria [,,].
The genera Prosthecobacter and Zoogloea were important biodegraders of aniline in the 13C-labeled treatments at 96 h. Chen et al. [] found that there was a homogeneous microbial community structure in the up-flow anaerobic sludge blanket (UASB) reactor with bio-electrochemical properties and chloronitrobenzenes in the seeding sludge, including Prosthecobacter (5.3%) and Thauera (4.6%), and they believed that Prosthecobacter, as an aniline-degrading bacterium, was worthy of in-depth investigation. Zoogloea was found to adhere to the surface of the ceramic particles of a biotrickling filter used to treat aniline [] and was shown to degrade phenols, nitrogenous heterocyclic compounds [] and nitrobenzene []. In addition, the genera Zoogloea, Acinetobacter, and Roseomonas are all presented in two groups of the network plots (Figure 6). Thus, the co-occurrence of the above microbial communities confirmed that a “small world” was established in the aniline-spiked microcosm, as the previous studies suggested []. In addition, some bacteria only had a high proportion in the heavy fractions at 45 or 69 h. This change was considered to be a natural succession in microbial communities involved in aniline degradation over time [].
3.3. Aniline-Degradation Genes Possessed by Microorganisms
The 16S rRNA gene sequencing and functional prediction of DNA extracted from the aniline-degradation system showed that there were a large number of biodegraders carrying rich and diverse genes for aniline degradation, such as atd, tdn, and dan gene clusters []. These key genes were predicted not only in the 13C-labeled heavy fractions of the samples but also in the corresponding components of the 12C aniline treatments. The prediction results showed that many microorganisms are involved in the degradation of aniline and play an important role in various steps in aniline degradation [].
Takeo et al. [] indicated that the atdA1 gene (encoding a GS-like protein) forms γ-glutamyl anilide (γ-GA) from aniline and L-glutamate in the aniline oxidation reaction, whereupon the dioxygenase proteins AtdA3, AtdA4, and AtdA5 convert γ-GA to catechol. In addition, Acinetobacter sp. YAA has a gene cluster for aniline degradation in the plasmid pYA1; the cluster consists of 14 genes (atdA1-A5RSBCDEFGH) responsible for the conversion of aniline to intermediates of the TCA cycle [,]. Similar multicomponent aniline dioxygenase gene clusters (atdA1-A5) encoding proteins involved in the initial oxidation of aniline to catechol have been cloned from Pseudomonas sp. UCC22 [] and Comamonas testosteroni I2 []. Several genera, such as Comamonas sp., also carry a tad gene for aniline degradation, which may be the main reason for the rapid start-up and subsequent reliable efficiency of aniline degradation [].
Aniline-degrading genes often occur in different gene clusters in different strains. These gene clusters often have similar sequences and encode similar degradative enzymes [,]. The tdn gene cluster for aniline degradation shown in Figure 7 is very similar to the tad gene cluster described by Shin et al. []. In addition, the tad gene cluster showed considerable similarity in nucleotide sequence and genetic organization with the plasmid-encoded aniline-degradation gene cluster (atd genes) of Pseudomonas putida UCC22 []. Therefore, our study revealed that the atd, tdn and dan gene families play crucial roles in the degradation of aniline individually or collaboratively in aerobic environments.
4. Materials and Methods
4.1. Soil Microcosms Incubations
The inoculum of aniline-degrading microcosms was sediment collected from the Puning area of the Lijiang River, which has a history of aniline pollution. The microcosms were established through mixed collected sediment (2 g), 50 mg/L aniline, and 40 mL of autoclaved mineral salt medium (MSM) with pH 7.0 in 60 mL serum bottles. The MSM mainly contained Na2HPO4·7H2O (7.9 g L−1), MgSO4·7H2O (0.1 g L−1), KH2PO4 (1.5 g L−1), NH4Cl (0.3 g L−1), a vitamin solution (10 mL L−1), and a trace elements solution (5 mL L−1) []. Three groups of microcosm experiments were performed: (i) unlabeled 12C-aniline-degradation treatments; (ii) labeled 13C-aniline (Sigma-Alrdich, MO, USA)-degradation treatments; and (iii) sterilized 12C-aniline-degradation treatments. After adding 10 mM 13C-aniline or 12C-aniline to the cultures, they were sealed with a breathable membrane and incubated at room temperature in the dark with shaking at 120 rpm. The concentration in aniline was determined in triplicate using an Agilent 1100 HPLC (Agilent, Santa Clara, CA, USA) with 0.1 mL supernatant liquid taken from the degradation system at intervals []. Then the degradation rate of the aniline in the microcosms was calculated []. When the aniline in the system was completely degraded, the cultures were again spiked with 50 mg/L 13C-aniline or 12C-aniline. All cultures were incubated under the above conditions, and a certain number of bottles were sacrificially sampled and centrifuged at 6000 rpm to obtain sediment for the microbial community analysis after the aniline degradation was completed (45 h and 96 h of incubation). The significant differences in raw data were analyzed using an analysis of variance (ANOVA).
4.2. DNA Extraction and Quantitative PCR of 16S rRNA Gene
Genomic DNA was extracted from 0.25 g of soil collected from aniline-degradation incubation (45 h and 69 h) using the DNeasy PowerSoil DNA Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions []. Copies of the 16S rRNA gene were quantified via quantitative PCR using the primer sets 515F (5′-GTG CCA GCM GCC GCG GTA A-3′) and 806R (5′-GGA CTA CVS GGG TAT CTA AT-3′), which can amplify the hypervariable region V3-V4 of the 16S rRNA gene in almost all eubacteria [].
4.3. SIP Gradient Fractionation
Genomic DNAs from the 12C-aniline- and 13C-aniline-degradation systems (after 45 h and 69 h, respectively) were separated into “heavy” (i.e., 13C-DNA) and “light” (i.e., 12C-DNA) fractions using isopycnic density gradient centrifugation as previously described []. Briefly, approximately 2 μg of DNA and CsCl solution with a buoyant density (BD) of 1.402 g mL−1 were added to 4.9 mL OptiSeal polyallomer tubes (Beckman Coulter, Inc., Brea, CA, USA). An Optima XPN-100 ultracentrifuge (Beckman Coulter Inc., Brea, CA, USA) was used to centrifuge the mixture at 470,000× g for 48 h at 20 °C with a VTi 90 vertical rotor []. The resulting DNA gradients were fractionated into 24 density fractions using a fraction recovery system (Beckman Coulter, Inc., Brea, CA, USA). The BD values of each fraction were determined immediately by measuring the refractive index using a digital palette refractometer (Atago, Tokyo, Japan). The nucleic acids in each CsCl gradient fraction were precipitated with 6 μL glycogen (Zomanbio, Beijing, China) dissolved in 1100 μL of 27% ethanol []. DNA was harvested via centrifugation, air-dried, and eluted with 30 μL of TE buffer (pH 8.0). Finally, quantitative PCR was performed as described by Sun et al. [] to determine the copy number of the 16S rRNA gene in the eluted DNA of the 24 fractions.
4.4. Illumina MiSeq Sequencing and Analysis
Genomic DNA from the light fractions of the 12C-aniline treatments and the heavy fractions of the 13C-aniline treatments (45 and 69 h, respectively) were used for amplicon sequencing of the partial 16S rRNA gene sequencing using the primer set 515F/802R []. Amplicon sequencing was performed using the Illumina MiSeq platform (Personalbio, Shanghai, China) []. Paired-end reads were analyzed using the QIIME2-201904 toolkit []. Briefly, all raw reads were qualified, merged, and cleared of chimeras. The filtered reads were clustered into amplicon sequence variants (ASV) using DADA2 []. The obtained ASV features with a proportion > 0.01% were assigned to a taxonomy using the SILVA database [].
4.5. Microbial Functional Prediction Using PICRUSt2
Phylogenetic Investigation of Communities by Reconstruction of Unobserved States version 2 (PICRUSt2) was used to predict the potential of microbial communities in representative fractions [,]. PICRUSt2 was used to predict the functional composition of the metagenome using the 16S rRNA gene sequencing and a reference genome database. Sequences used for PICRUSt2 prediction were clustered into OTUs (97% similarity) using QIIME2 software (version 2.9.1) against the Greengenes 13.5 database []. Subsequently, the copy numbers of the 16S rRNA genes were normalized using the rarefied OTU table. The normalized OTU table and KEGG database were compared to obtain the different functional files for each sample. Finally, PICRUSt2 was used to obtain information on metabolic pathways at the three levels. The KEGG genes involved in aniline degradation in the KEGG database were compared with the PICRUSt2 functional prediction results, and the copy numbers of genes associated with the aniline metabolic pathway in each of the samples were selected for analysis [].
5. Conclusions
In this study, aerobic bacteria capable of degrading aniline were analyzed using DNA-SIP with 13C-labeled aniline. The rate of the aniline degradation was found to increase in the microcosm. Through isopycnic density gradient centrifugation and microbial community analysis, it was observed that bacteria such as Acinetobacter (up to 34.6 ± 6.0%), Zoogloea (up to 15.8 ± 2.2%), Comamonas (up to 2.6 ± 0.1%), and Hydrogenophaga (up to 5.1 ± 0.6%) were enriched in the 13C-labeled heavy fractions. These bacteria were confirmed to degrade aniline at the molecular level. Additionally, Prosthecobacter (up to 16.0 ± 1.6%) and Curvibacter (up to 3.0 ± 1.6%) were also found to be labeled by 13C in the aniline-degrading system and were likely to have the potential to degrade anilines. Through functional prediction of the microbial communities in the heavy fractions, the atd, tdn, and dan gene families were identified as the key genes for aniline degradation in aerobic microcosms. The results of this study can provide theoretical and technical reference for understanding the biodegradation process and for developing effective aniline compound pollution control programs.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal14010064/s1, Table S1: Microbial community composition at the genus level in samples of different treatment groups (Top 11).
Author Contributions
Conceptualization and methodology, W.S.; software and validation, X.S.; formal analysis and investigation, H.L.; resources and data curation, G.Y.; writing—original draft preparation, R.Y.; writing—review and editing, B.S. and M.U.G.; visualization, Y.H.; supervision, Y.R.; project administration, B.S. and B.L.; funding acquisition, W.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (Grant Nos. 42007224, U21A2035, 32161143018, 42107285, 42277247, and 42107230), the Guangdong Basic and Applied Basic Research Foundation (Grant Nos. 2023B1515040007, 2022B1515120033, 2021A1515110282, and 2023A1515012283), GDAS’s Project of Science and Technology Development (Grant Nos. 2020GDASYL-20200103082, 2022GDASZH-2022010203, and 2023GDASZH-2023010103), and the Guangdong Foundation for Programs of Science and Technology Research (Grant No. 2023B1212060044).
Data Availability Statement
The data presented in this study are available.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- O’Neill, F.J.; Bromley-Challenor, K.C.A.; Greenwood, R.J.; Knapp, J.S. Bacterial growth on aniline: Implications for the biotreatment of industrial wastewater. Water Res. 2000, 34, 4397–4409. [Google Scholar] [CrossRef]
- Zhu, L.; Lv, M.L.; Dai, X.; Xu, X.Y.; Qi, H.Y.; Yu, Y.W. Reaction kinetics of the degradation of chloroanilines and aniline by aerobic granule. Biochem. Eng. J. 2012, 68, 215–220. [Google Scholar] [CrossRef]
- Yang, L.H.; Zhang, Y.M.; Bai, Q.; Yan, N.; Xu, H.; Rittmann, B.E. Intimately coupling of photolysis accelerates nitrobenzene biodegradation, but sequential coupling slows biodegradation. J. Hazard. Mater. 2015, 287, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Barrington, S.; Kim, J.W. Biodegradation of pentyl amine and aniline from petrochemical wastewater. J. Environ. Manag. 2007, 83, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Jiang, J.P.; Liu, R.T.; Khan, A.U.; Wang, P. Engineering risk assessment for emergency disposal projects of sudden water pollution incidents. Environ. Sci. Pollut. Res. 2017, 24, 14819–14833. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Ding, X. Xiangshui Explosion Accident: Anilines in the Surface Water of Some Rivers in the Park Exceeded the Standard. The Paper, 20:15. Available online: https://www.thepaper.cn/newsDetail_forward_%203182916 (accessed on 22 March 2019).
- Franciscon, E.; Zille, A.; Dias Guirnaro, F.; Ragagnin de Menezes, C.; Durrant, R.L.; Cavaco-Paulo, A. Biodegradation of textile azo dyes by a facultative Staphylococcus arlettae strain VN-11 using a sequential microaerophilic/aerobic process. Int. Biodeterior. Biodegrad. 2009, 63, 280–288. [Google Scholar]
- Vikrant, K.; Giri, B.S.; Raza, N.; Roy, K.; Kim, K.H.; Rai, B.N.; Singh, R.S. Recent advancements in bioremediation of dye: Current status and challenges. Bioresour. Technol. 2018, 253, 355–367. [Google Scholar] [CrossRef]
- Asad, U.K.; Mujaddad, U.R.; Muhammad, Z.; Abdul, B.S.; Ivar, Z. Biodegradation of Brown 706 Dye by Bacterial Strain Pseudomonas aeruginosa. Water 2021, 13, 2959. [Google Scholar]
- Chen, H.; Sun, C.R.; Liu, R.H.; Yuan, M.Z.; Mao, Z.H.; Wang, Q.; Zhou, H.B.; Cheng, H.N.; Zhan, W.H.; Wang, Y.G. Enrichment and domestication of a microbial consortium for degrading aniline. J. Water Process Eng. 2021, 42, 102108. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Zhang, Q.; Peng, H.J.; Wei, H.; Feng, J.P.; Su, J.H.; He, J. An attempt to stimulate aniline degrading bioreactor by exogenous auto-inducer: Decontamination performance, sludge characteristics, and microbial community structure response. Bioresour. Technol. 2022, 347, 126675. [Google Scholar] [CrossRef]
- Peng, H.J.; Zhang, Q.; Li, M.; Li, Y.; Zhang, W.L. Identification and characterization of a highly efficient and resistant aniline-degrading strain AD4. Environ. Eng. Sci. 2021, 38, 742–751. [Google Scholar] [CrossRef]
- Qin, X.M.; Hua, Y.D.; Sun, H.; Xie, J.Y.; Zhao, Y.S. Visualization study on aniline-degrading bacteria AN-1 transport in the aquifer with the low-permeability lens. Water Res. 2020, 186, 116329. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.M.; Li, Y.; McGuinness, L.R.; Luo, S.A.; Huang, W.L.; Kerkhof, L.J.; Mack, E.E.; Häggblom, M.M.; Fennell, D.E. Identification of anaerobic aniline-degrading bacteria at a contaminated industrial site. Environ. Sci. Technol. 2015, 49, 11079–11088. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, R.C.; Hanson, B.T.; Chandra, S.; Madsen, E. Community dynamics and functional characteristics of naphthalene-degrading populations in contaminated surface sediments and hypoxic/anoxic groundwater. Environ. Microbiol. 2018, 20, 3543–3559. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.K. Bacterial degradation of monocyclic aromatic amines. Front. Microbiol. 2015, 6, 820. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, J.L.; Liu, S.J.; Liu, Z.P. A novel and complete gene cluster involved in the degradation of aniline by Delftia sp. AN3. J. Environ. Sci. 2018, 20, 717–724. [Google Scholar] [CrossRef]
- Liu, H.Q.; Lin, H.Z.; Song, B.R.; Sun, X.X.; Xu, R.; Kong, T.L.; Xu, F.Q.; Li, B.Q.; Sun, W.M. Stable-isotope probing coupled with high-throughput sequencing reveals bacterial taxa capable of degrading aniline at three contaminated sites with contrasting pH. Sci. Total Environ. 2021, 771, 144807. [Google Scholar] [CrossRef]
- Kim, S.I.; Leem, S.H.; Choi, J.S.; Chung, Y.H.; Kim, S.; Park, Y.M.; Park, Y.K.; Lee, Y.N.; Ha, K.S. Cloning and characterization of two catA genes in Acinetobacter lwoffii K24. J. Bacteriol. 1997, 179, 5226–5231. [Google Scholar] [CrossRef]
- Liang, Q.F.; Takeo, M.; Chen, M.; Zhang, W.; Xu, Y.Q.; Lin, M. Chromosome-encoded gene cluster for the metabolic pathway that converts aniline to TCA-cycle intermediates in Delftia tsuruhatensis AD9. Microbiology 2005, 151, 3435–3446. [Google Scholar] [CrossRef]
- Fujii, T.; Takeo, M.; Maeda, Y. Plasmid-encoded genes specifying aniline oxidation from Acinetobacter sp. strain YAA. Microbiology 1997, 143, 93–99. [Google Scholar] [CrossRef]
- Boon, N.; Goris, J.; De Vos, P.; Verstraete, W.; Top, E.M. Genetic diversity among 3-chloroaniline- and aniline-degrading strains of the Comamonadaceae. Appl. Environ. Microbiol. 2001, 67, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Hao, Z.Y.; Sun, R.; Bartlam, M.; Wang, Y.Y. Genome sequence of a typical ultramicro bacterium, Curvibacter sp. Strain PAE-UM, capable of phthalate ester degradation. Genome Announc. 2016, 4, e01510–15. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.F.; Wu, Q.P.; Gu, Q.H.; Zhou, Q.; Zhang, J.M. Community structure analysis and biodegradation potential of aniline-degrading bacteria in biofilters. Curr. Microbiol. 2018, 75, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Urata, M.; Uchida, E.; Nojiri, H.; Omori, T.; Obo, R.; Miyaura, N.; Ouchiyama, N. Genes involved in aniline degradation by Delftia acidovorans strain 7N and its distribution in the natural environment. Biosci. Biotechnol. Biochem. 2004, 68, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.M.; Kim, J. Sphingobium aromaticivastans sp. nov. a novel aniline- and benzene-degrading, and antimicrobial compound producing bacterium. Arch. Microbiol. 2019, 201, 155–161. [Google Scholar] [CrossRef]
- Chen, H.; Gao, X.Y.; Wang, C.Q.; Shao, J.J.; Xu, X.Y.; Zhu, L. Efficient 2,4-dichloronitrobenzene removal in the coupled BES-UASB reactor: Effect of external voltage mode. Bioresour. Technol. 2017, 241, 879–886. [Google Scholar] [CrossRef] [PubMed]
- Li, G.Y.; Wan, S.G.; An, T.C. Efficient bio-deodorization of aniline vapor in a biotrickling filter: Metabolic mineralization and bacterial community analysis. Chemosphere 2012, 87, 253–258. [Google Scholar] [CrossRef]
- Lin, Y.Z.; Yin, J.; Wang, J.H.; Tian, W.D. Performance and microbial community in hybrid anaerobic baffled reactor-constructed wetland for nitrobenzene wastewater. Bioresour. Technol. 2012, 118, 128–135. [Google Scholar] [CrossRef]
- Peng, H.; Zhang, Y.; Zhang, Q.; Zhang, W.; Li, M.; Feng, J.; Su, J.; He, J.; Zhong, M. Control of aeration time in the aniline degrading-bioreactor with the analysis of metagenomic: Aniline degradation and nitrogen metabolism. Bioresour. Technol. 2022, 344, 126281. [Google Scholar] [CrossRef]
- Takeo, M.; Ohara, A.; Sakae, S.; Okamoto, Y.; Kitamura, C.; Kato, D.; Negoro, S. Mechanism of bacterial aniline oxidation via γ-glutamylanilide: Function of a glutamine synthetase-like protein. J. Bacteriol. 2013, 195, 4406–4414. [Google Scholar] [CrossRef]
- Fukumori, F.; Saint, C.P. Nucleotide sequences and regulational analysis involved in conversion of aniline to catechol in Pseudomonas putida UCC22 (pTDN1). J. Bacteriol. 1997, 179, 399–408. [Google Scholar] [CrossRef]
- Król, J.E.; Penrod, J.T.; McCaslin, H.; Rogers, L.M.; Yano, H.; Stancik, A.D.; Dejonghe, W.; Brown, C.J.; Parales, R.E.; Wuertz, S.; et al. Role of IncP-1 β plasmids pWDL7::rfp and pNB8c in chloroaniline catabolism as determined by genomic and functional analyses. Appl. Environ. Microbiol. 2012, 78, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, W.L.; He, Q.L.; Li, M.; Li, Y.; Huang, W.S. Effects of dissolved oxygen concentrations on a bioaugmented sequencing batch rector treating aniline-laden wastewater: Reactor performance, microbial dynamics and functional genes. Bioresour. Technol. 2020, 313, 123598. [Google Scholar] [CrossRef]
- Shin, K.A.; Spain, J.C. Pathway and evolutionary implications of diphenylamine biodegradation by Burkholderia sp. Strain JS667. Appl. Environ. Microb. 2009, 75, 2694–2704. [Google Scholar] [CrossRef][Green Version]
- Zhang, J.; Zhou, W.X.; Liu, B.B.; He, J.; Shen, Q.R.; Zhao, F.J. Anaerobic arsenite oxidation by an autotrophic arsenite-oxidizing bacterium from an arsenic-contaminated paddy soil. Environ. Sci. Technol. 2015, 49, 5956–5964. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, Z.; Azmat, U.; Sultan, A.; Mian, M.; Roy, H.S.; Ivar, Z.; Amir, S. Novel magnetite nanocomposites (Fe3O4/C) for efficient immobilization of ciprofloxacin from aqueous solutions through adsorption pretreatment and membrane processes. Water 2022, 14, 724. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.M.; Alves, R.J.E.; Zhang, D.D.; Han, L.L.; He, J.Z.; Zhang, L.M. Time-dependent shifts in populations and activity of bacterial and archaeal ammonia oxidizers in response to liming in acidic soils. Soil Boil. Biochem. 2017, 112, 77–89. [Google Scholar] [CrossRef]
- Li, Y.B.; Guo, L.F.; Häggblom, M.M.; Yang, R.; Li, M.Y.; Sun, X.X.; Chen, Z.; Li, F.B.; Su, X.F.; Yan, G.; et al. Serratia spp. are responsible for nitrogen fixation fueled by As(III) oxidation, a novel biogeochemical process identified in mine tailings. Environ. Sci. Technol. 2022, 56, 2033–2043. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef]
- Zhang, M.M.; Li, Z.; Häggblom, M.M.; Young, L.L.; Li, F.B.; He, Z.J.; Lu, G.M.; Xu, R.; Sun, X.X.; Qiu, L.; et al. Bacteria responsible for nitrate-dependent antimonite oxidation in antimony-contaminated paddy soil revealed by the combination of DNA-SIP and metagenomics. Soil Boil. Biochem. 2021, 156, 108194. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glockner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Guo, X.X.; Song, G.S.; Li, Y.Y.; Zhao, L.; Wang, J. Switch of bacteria community under oxygen depletion in sediment of Bohai Sea. Front. Mar. Sci. 2022, 9, 2296–7745. [Google Scholar] [CrossRef]
- Sun, X.X.; Song, B.R.; Xu, R.; Zhang, M.M.; Gao, P.; Lin, H.Z.; Sun, W.M. Root-associated (rhizosphere and endosphere) microbiomes of the Miscanthus sinensis and their response to the heavy metal contamination. J. Environ. Sci. 2021, 104, 387–398. [Google Scholar] [CrossRef]
- Toole, D.R.; Zhao, J.; Martens-Habbena, W.; Strauss, S.L. Bacterial functional prediction tools detect but underestimate metabolic diversity compared to shotgun metagenomics in southwest Florida soils. Appl. Soil Ecol. 2021, 168, 104129. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).