
Enhancing Catalytic Efficiency in Long-Chain Linear α-Olefin Epoxidation: A Study of CaSnO3-Based Catalysts

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Characterizations of the Catalysts
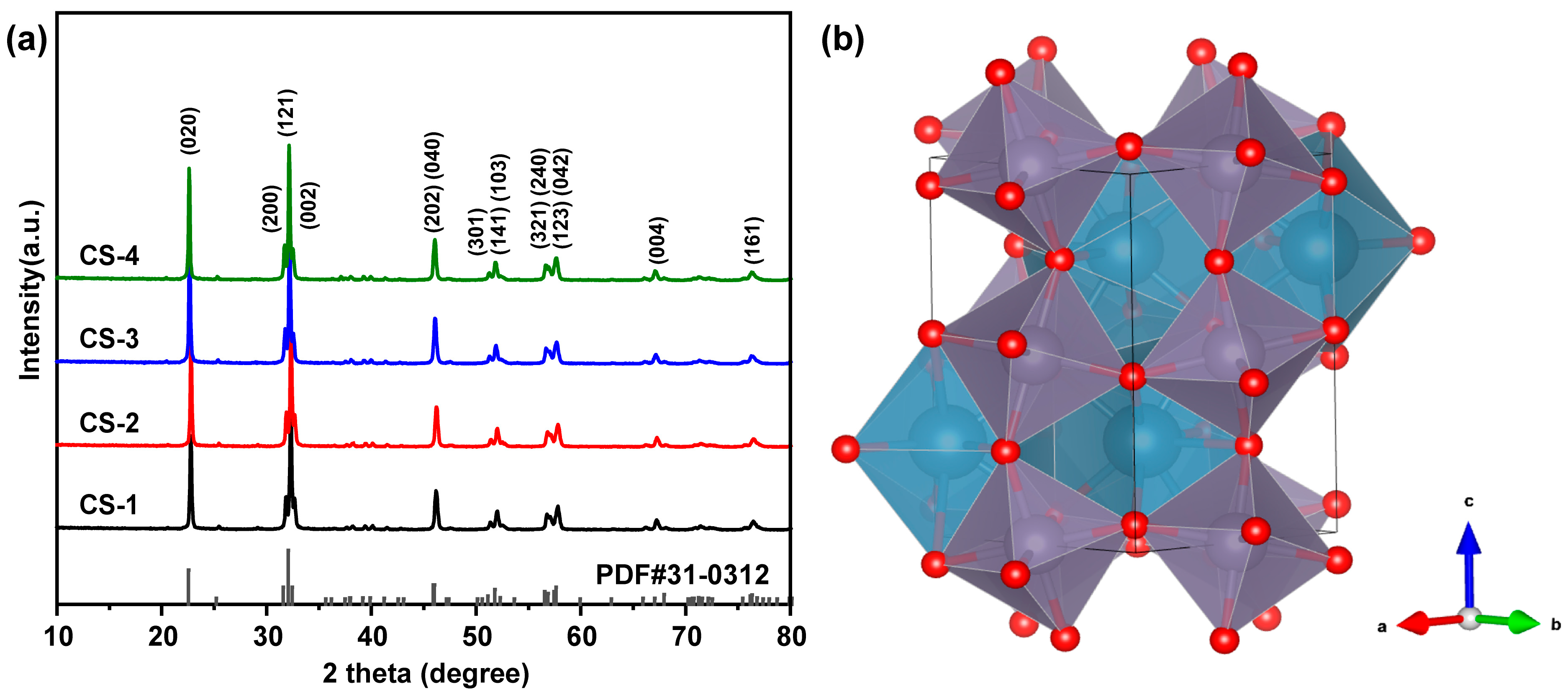
2.1.1. Structures of Catalysts
2.1.2. Morphology and Surface Element Distribution of Catalysts
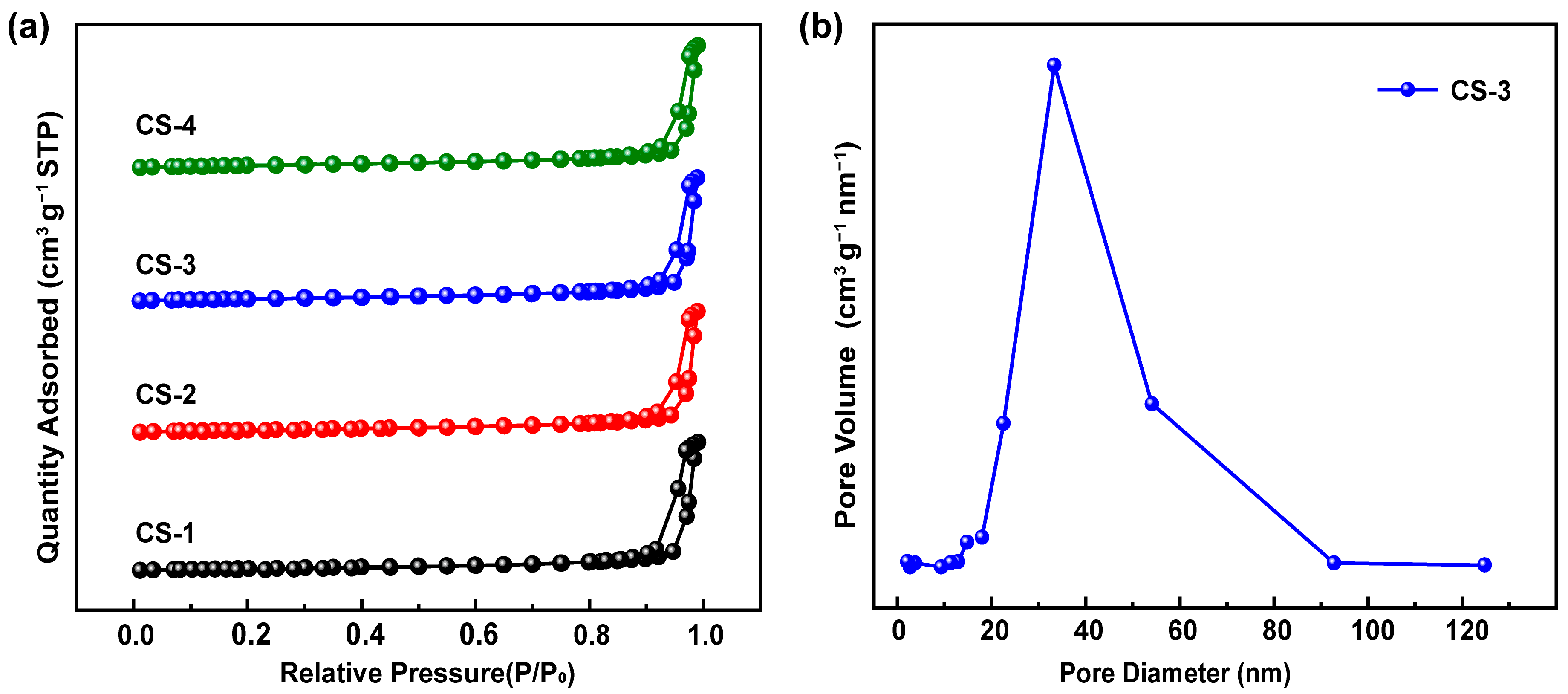
2.1.3. Specific Surface Areas of Catalysts
2.2. Catalytic Performance of the Catalysts
2.2.1. Evaluation of the Different Catalysts for 1-Octene Epoxidation
2.2.2. The Influence of Reaction Time
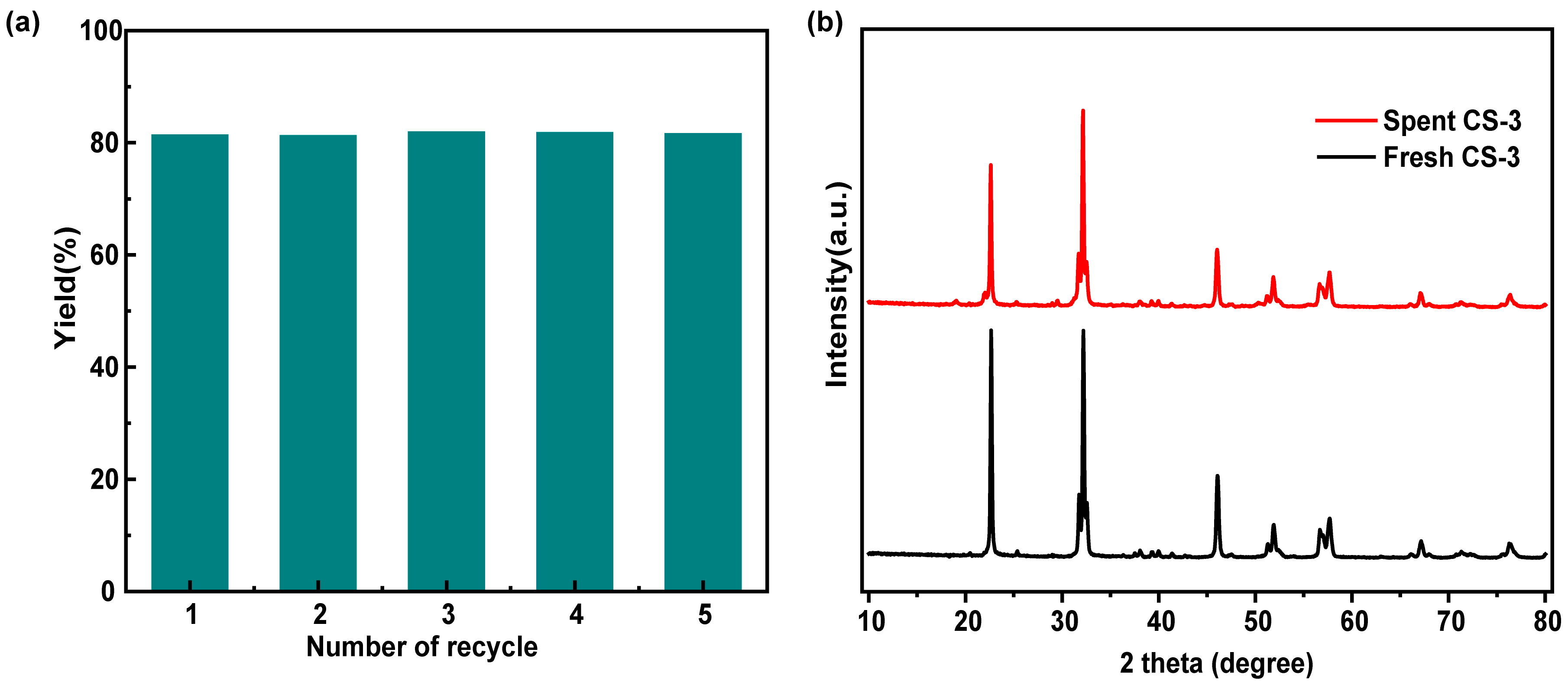
2.2.3. Catalytic Stability
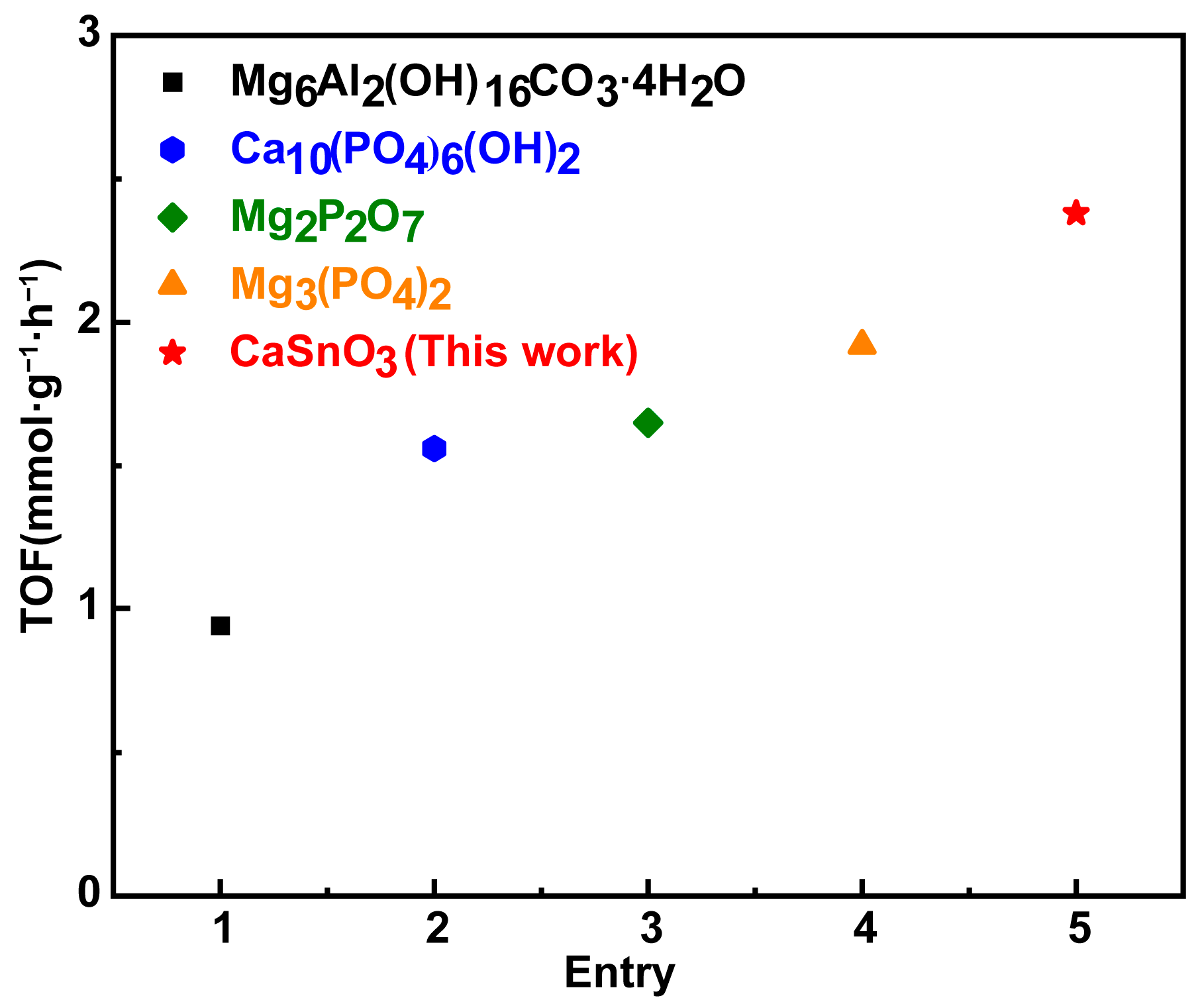
2.2.4. Comparative Results with Different Catalysts for the Epoxidation of 1-Octene
2.2.5. Substrate Range Extension
2.3. Surface Properties of Catalysts
2.3.1. Surface Alkalinity
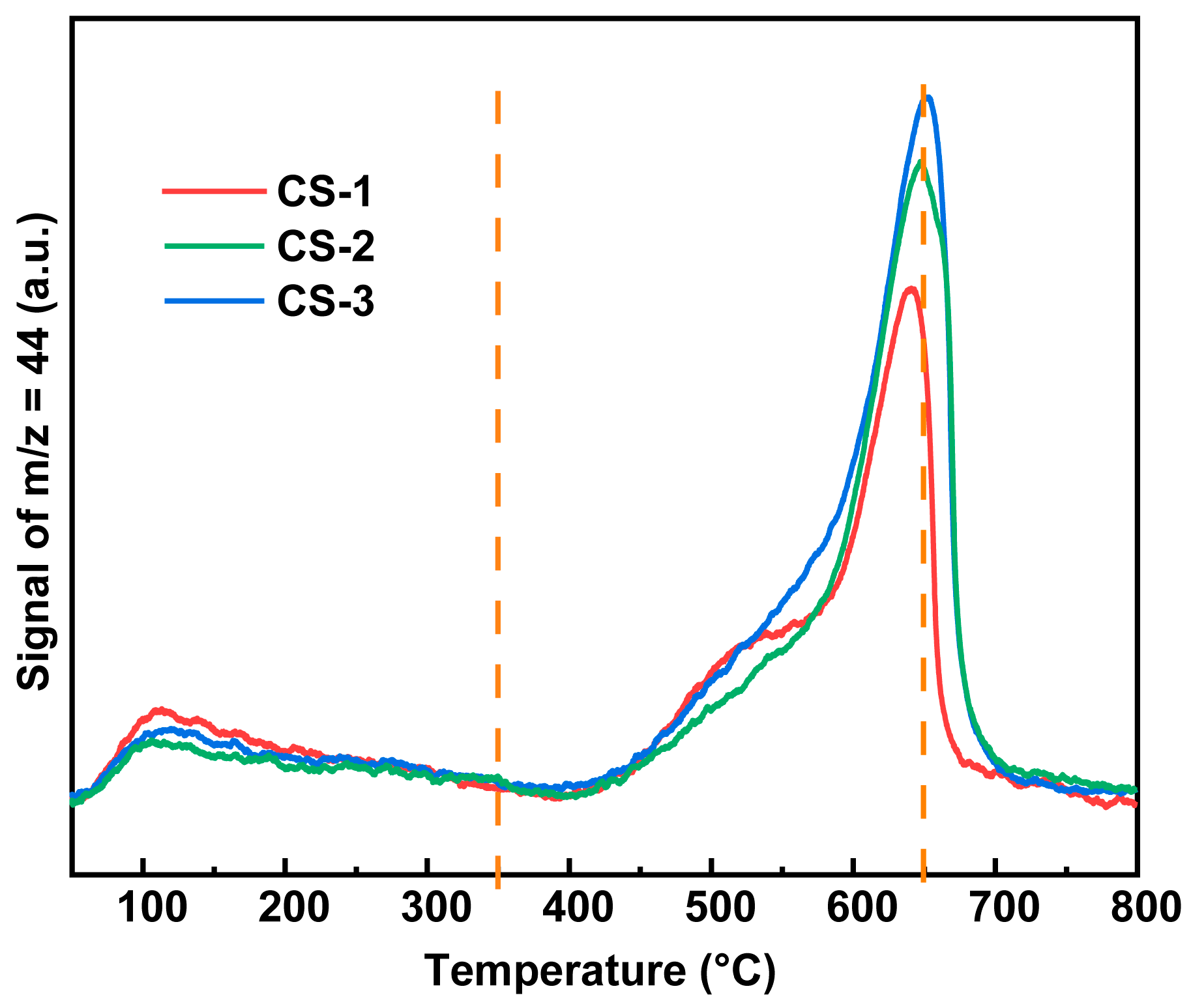
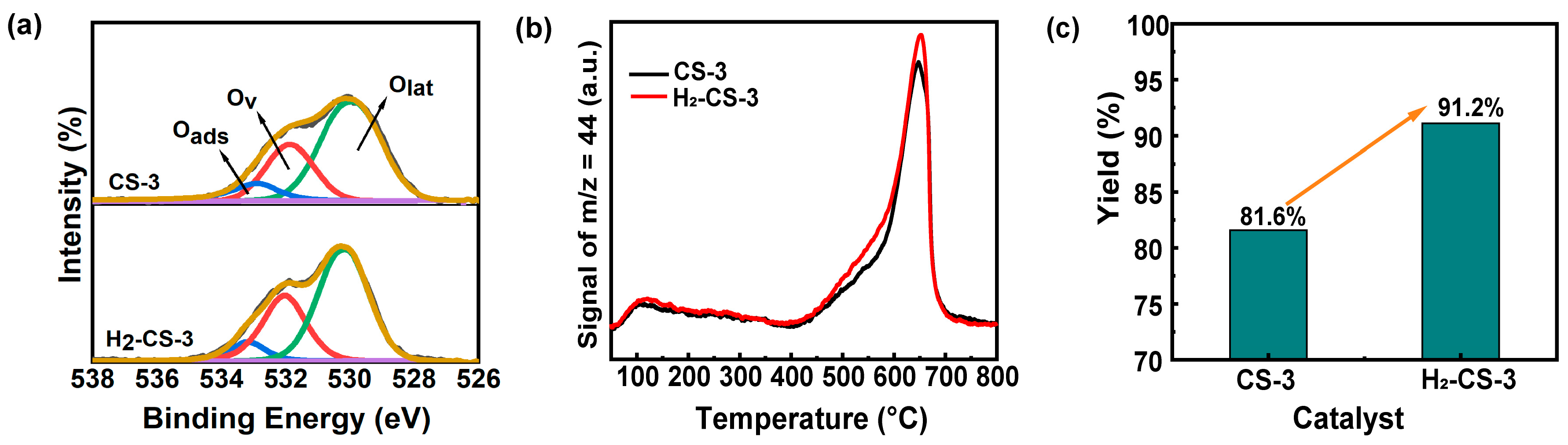
2.3.2. Surface Oxygen Species
2.4. Modification of Catalyst
2.4.1. The Structure, Morphology, and Specific Surface Area of the Modified Catalyst
2.4.2. Catalytic Activity, Surface Alkalinity, and Surface Oxygen Species of the Modified Catalyst
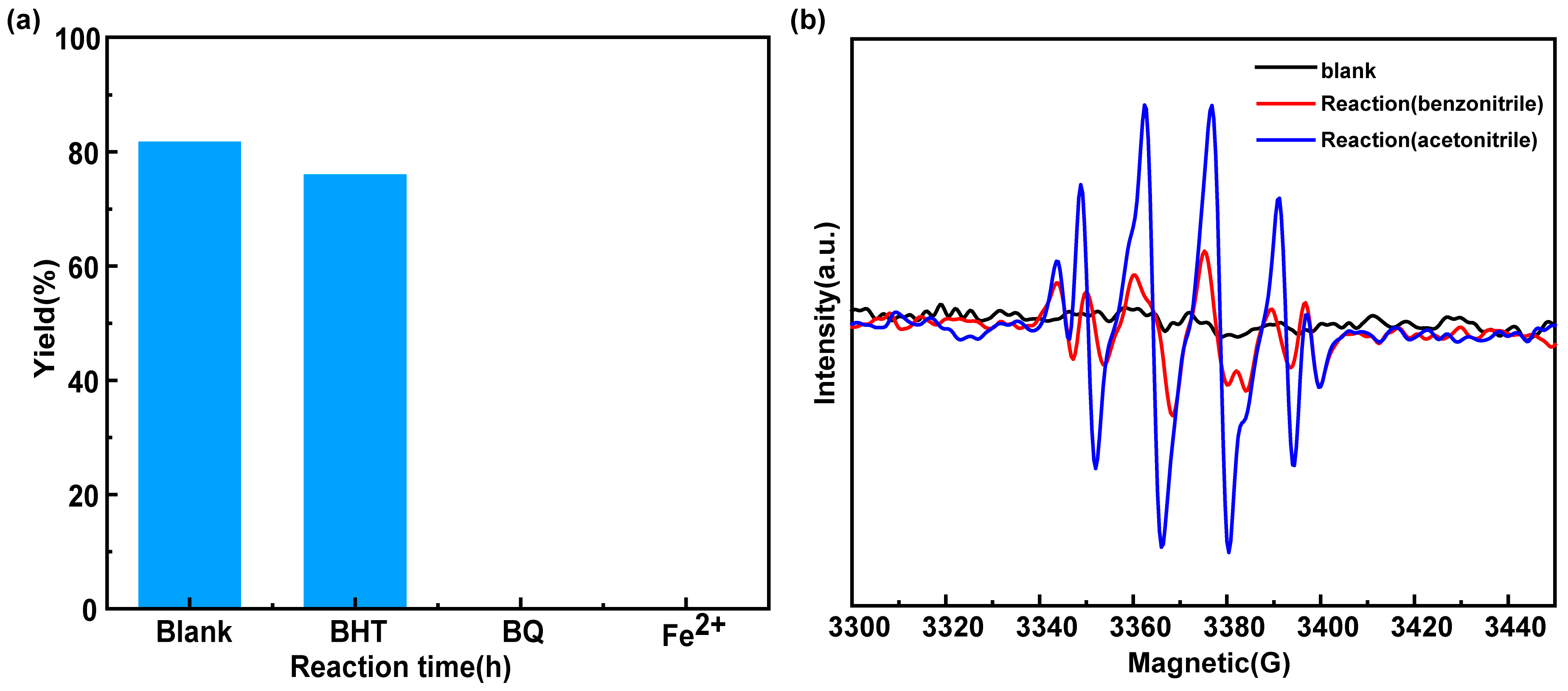
2.5. Reaction Mechanism Study
3. Materials and Methods
3.1. Materials
3.2. Catalysts’ Preparation
3.3. Catalysts’ Characterization
3.4. Catalyst Activity Evaluation
3.5. Catalyst Recycling Experiment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cao, J.; Zhou, Z.; Zhang, M.; Gong, N.; Yin, A.; Cai, Y.; Sun, X.; Wan, H.; Li, Y.; Cao, Z. Tuning the electronic properties of supported Cu catalysts for efficient epoxidation of long-chain α-olefins. Fuel 2023, 342, 127829. [Google Scholar] [CrossRef]
- Li, P.; Gao, J.; Shi, J.; Wang, H.; Xing, X.; Ren, J.; Meng, Y.; Wang, L.; Lv, B. Insights into the effect of oxygen vacancies on the epoxidation of 1-hexene with hydrogen peroxide over WO3−x/SBA-15. Catal. Sci. Technol. 2022, 12, 6827–6837. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, F.; Yan, T.; Wan, H.; Yao, R.; Zhang, K.; Liu, Y.; Wang, S.; Xu, D.; Hou, H.; et al. Boosting the epoxidation of long-chain linear α-olefins via bimetallic CoIr composite. Fuel 2022, 326, 125050. [Google Scholar] [CrossRef]
- Ren, J.; Wang, L.; Li, P.; Xing, X.; Wang, H.; Lv, B. Ag supported on alumina for the epoxidation of 1-hexene with molecular oxygen: The effect of Ag+/Ag0. New J. Chem. 2022, 46, 4792–4799. [Google Scholar] [CrossRef]
- Nesterov, D.S.; Nesterova, O.V. Catalytic Oxidations with Meta-Chloroperoxybenzoic Acid (m-CPBA) and Mono- and Polynuclear Complexes of Nickel: A Mechanistic Outlook. Catalysts 2021, 11, 1148. [Google Scholar] [CrossRef]
- Goud, V.V.; Patwardhan, A.V.; Dinda, S.; Pradhan, N.C. Kinetics of epoxidation of jatropha oil with peroxyacetic and peroxyformic acid catalysed by acidic ion exchange resin. Chem. Eng. Sci. 2007, 62, 4065–4076. [Google Scholar] [CrossRef]
- Shimura, K.; Kanai, H.; Utani, K.; Matsuyama, K.; Imamura, S. Selective epoxidation of allyl acetate with tert-butyl hydroperoxide over MoO3/MgO. Appl. Catal. A Gen. 2005, 283, 117–124. [Google Scholar] [CrossRef]
- Rossi-Fernandez, L.; Dorn, V.; Radivoy, G. A new and efficient methodology for olefin epoxidation catalyzed by supported cobalt nanoparticles. Beilstein J. Org. Chem. 2021, 17, 519–526. [Google Scholar] [CrossRef]
- Yang, H.; Liu, Q.; Li, Y.; Sun, K.; Chen, Z.; Peng, Q.; Chen, C. Isolated Single-Atom Ruthenium Anchored on Beta Zeolite as an Efficient Heterogeneous Catalyst for Styrene Epoxidation. ChemNanoMat 2020, 6, 1647–1651. [Google Scholar] [CrossRef]
- Jameel, U.; Zhu, M.-Q.; Chen, X.-Z.; Liu, Y.; Tong, Z.-F. Green epoxidation of cyclooctene with molecular oxygen over an ecofriendly heterogeneous polyoxometalate-gold catalyst Au/BW11/Al2O3. J. Zhejiang Univ.-Sci. A 2016, 17, 1000–1012. [Google Scholar] [CrossRef]
- Bregante, D.T.; Tan, J.Z.; Schultz, R.L.; Ayla, E.Z.; Potts, D.S.; Torres, C.; Flaherty, D.W. Catalytic Consequences of Oxidant, Alkene, and Pore Structures on Alkene Epoxidations within Titanium Silicates. ACS Catal. 2020, 10, 10169–10184. [Google Scholar] [CrossRef]
- Mahha, Y.; Salles, L.; Piquemal, J.; Briot, E.; Atlamsani, A.; Bregeault, J. Environmentally friendly epoxidation of olefins under phase-transfer catalysis conditions with hydrogen peroxide. J. Catal. 2007, 249, 338–348. [Google Scholar] [CrossRef]
- Yu, X.; Ji, Y.; Jiang, Y.; Lang, R.; Fang, Y.; Qiao, B. Recent Development of Single-Atom Catalysis for the Functionalization of Alkenes. Catalysts 2023, 13, 730. [Google Scholar] [CrossRef]
- Fraile, J.M.; García, J.I.; Mayoral, J.A. Basic solids in the oxidation of organic compounds. Catal. Today 2000, 57, 3–16. [Google Scholar] [CrossRef]
- Pavel, O.D.; Cojocaru, B.; Angelescu, E.; Pârvulescu, V.I. The activity of yttrium-modified Mg,Al hydrotalcites in the epoxidation of styrene with hydrogen peroxide. Appl. Catal. A Gen. 2011, 403, 83–90. [Google Scholar] [CrossRef]
- Ionescu, R.; Pavel, O.D.; Bîrjega, R.; Zăvoianu, R.; Angelescu, E. Epoxidation of Cyclohexene with H2O2 and Acetonitrile Catalyzed by Mg–Al Hydrotalcite and Cobalt Modified Hydrotalcites. Catal. Lett. 2009, 134, 309–317. [Google Scholar] [CrossRef]
- Bian, X.; Gu, Q.; Shi, L.; Sun, Q. Epoxidation of Styrene with Hydrogen Peroxide over MgO Catalyst. Chin. J. Catal. (Chin. Version) 2011, 32, 682–687. [Google Scholar] [CrossRef]
- Mandelli, D.; van Vliet, M.C.A.; Sheldon, R.A.; Schuchardt, U. Alumina-catalyzed alkene epoxidation with hydrogen peroxide. Appl. Catal. A Gen. 2001, 219, 209–213. [Google Scholar] [CrossRef]
- Romero, M.D.; Calles, J.A.; Ocaña, M.A.; Gómez, J.M. Epoxidation of cyclohexene over basic mixed oxides derived from hydrotalcite materials: Activating agent, solvent and catalyst reutilization. Microporous Mesoporous Mater. 2008, 111, 243–253. [Google Scholar] [CrossRef]
- Muthukutty, B.; Krishnapandi, A.; Chen, S.-M.; Abinaya, M.; Elangovan, A. Innovation of Novel Stone-Like Perovskite Structured Calcium Stannate (CaSnO3): Synthesis, Characterization, and Application Headed for Sensing Photographic Developing Agent Metol. ACS Sustain. Chem. Eng. 2020, 8, 4419–4430. [Google Scholar] [CrossRef]
- Sumithra; Victor Jaya, N. Band gap tuning & Room temperature ferromagnetism of hydrothermally prepared Cobalt doped CaSnO3 nanopowders. Mater. Res. Innov. 2018, 23, 375–384. [Google Scholar] [CrossRef]
- Pillai, U.R.; Sahle-Demessie, E. Epoxidation of olefins and α,β-unsaturated ketones over sonochemically prepared hydroxyapatites using hydrogen peroxide. Appl. Catal. A Gen. 2004, 261, 69–76. [Google Scholar] [CrossRef]
- Fernandes, C.I.; Vaz, P.D.; Nunes, T.G.; Nunes, C.D. Zinc biomimetic catalysts for epoxidation of olefins with H2O2. Appl. Clay Sci. 2020, 190, 105562. [Google Scholar] [CrossRef]
- Wexler, R.B.; Gautam, G.S.; Stechel, E.B.; Carter, E.A. Factors Governing Oxygen Vacancy Formation in Oxide Perovskites. J. Am. Chem. Soc. 2021, 143, 13212–13227. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Ma, A.; Yu, Y.; Tan, R.; Liu, C.; Zhang, P.; Liu, D.; Gui, J. Engineering Oxygen Vacancies into LaCoO3 Perovskite for Efficient Electrocatalytic Oxygen Evolution. ACS Sustain. Chem. Eng. 2018, 7, 2906–2910. [Google Scholar] [CrossRef]
- Hou, W.; Feng, P.; Guo, X.; Wang, Z.; Bai, Z.; Bai, Y.; Wang, G.; Sun, K. Catalytic Mechanism of Oxygen Vacancies in Perovskite Oxides for Lithium-Sulfur Batteries. Adv. Mater. 2022, 34, e2202222. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tian, L. Luminescence characteristics and energy transfer of CaSnO3:Pr3+, Bi3+ phosphors. Optik 2021, 242, 166866. [Google Scholar] [CrossRef]
- Park, S.Y.; Abroshan, H.; Shi, X.; Jung, H.S.; Siahrostami, S.; Zheng, X. CaSnO3: An Electrocatalyst for Two-Electron Water Oxidation Reaction to Form H2O2. ACS Energy Lett. 2018, 4, 352–357. [Google Scholar] [CrossRef]
- Sandesh, S.; Kristachar, P.K.R.; Manjunathan, P.; Halgeri, A.B.; Shanbhag, G.V. Synthesis of biodiesel and acetins by transesterification reactions using novel CaSn(OH)6 heterogeneous base catalyst. Appl. Catal. A Gen. 2016, 523, 1–11. [Google Scholar] [CrossRef]
- Li, B.; Li, J.; Rong, Y.; Tian, Y.; Li, J.; Liu, X.; Hao, Q.; Teng, B. Unravelling the surface structure and active site of CaSnO3, as well as H2O2 formation mechanism in two electron water oxidation reaction. Appl. Surf. Sci. 2022, 598, 153832. [Google Scholar] [CrossRef]
- Chen, X.Y.; Ma, C.; Bao, S.P.; Zhang, H.Y. Novel porous CaSnO3:Eu3+ and Ca2SnO4:Eu3+ phosphors by co-precipitation synthesis and postannealing approach: A general route to alkaline-earth stannates. J. Alloys Compd. 2010, 497, 354–359. [Google Scholar] [CrossRef]
- Wang, J.; Asakura, Y.; Hasegawa, T.; Yin, S. Morphology and facet tailoring of CaSnO3 assembled in molten salt with defect-mediated photocatalytic activity. J. Environ. Chem. Eng. 2022, 10, 108169. [Google Scholar] [CrossRef]
- Gu, Q.; Han, D.; Shi, L.; Sun, Q. Styrene epoxidation with hydrogen peroxide over calcium oxide catalysts prepared from various precursors. J. Nat. Gas. Chem. 2012, 21, 452–458. [Google Scholar] [CrossRef]
- Ueno, S.; Yoshida, K.; Ebitani, K.; Kaneda, K. Hydrotalcite catalysis: Heterogeneous epoxidation of olefins using hydrogen peroxide in the presence of nitriles. Chem. Commun. 1998, 3, 295–296. [Google Scholar] [CrossRef]
- Yang, X.; Li, X.; Dong, J. Magnesium Pyrophosphate-Catalyzed Epoxidation of 1-Octene with Aqueous Hydrogen Peroxide. Catal. Lett. 2021, 152, 162–171. [Google Scholar] [CrossRef]
- Yang, X.; Li, X.; Dong, J. Farringtonite as an efficient catalyst for linear-chain α-olefin epoxidation with aqueous hydrogen peroxide. New J. Chem. 2021, 45, 11968–11976. [Google Scholar] [CrossRef]
- Palomeque, J.; Lopez, J.; Figueras, F. Epoxydation of Activated Olefins by Solid Bases. J. Catal. 2002, 211, 150–156. [Google Scholar] [CrossRef]
- Lu, X.; Lin, H.; Yuan, Y. Epoxidation of Olefins Catalyzed by Magnesium-Based Mixed Oxides with Hy-drogen Peroxide. Chin. J. Catal. (Chin. Version) 2011, 31, 1457–1464. [Google Scholar] [CrossRef]
- Zăvoianu, R.; Cruceanu, A.; Pavel, O.D.; Bradu, C.; Florea, M.; Bîrjega, R. Green Epoxidation of Olefins with ZnxAl/MgxAl-LDH Compounds: Influence of the Chemical Composition. Catalysts 2022, 12, 145. [Google Scholar] [CrossRef]
- Xu, J.; Xi, R.; Xiao, Q.; Xu, X.; Liu, L.; Li, S.; Gong, Y.; Zhang, Z.; Fang, X.; Wang, X. Design of strontium stannate perovskites with different fine structures for the oxidative coupling of methane (OCM): Interpreting the functions of surface oxygen anions, basic sites and the structure–reactivity relationship. J. Catal. 2022, 408, 465–477. [Google Scholar] [CrossRef]
- Ou, G.; Xu, Y.; Wen, B.; Lin, R.; Ge, B.; Tang, Y.; Liang, Y.; Yang, C.; Huang, K.; Zu, D.; et al. Tuning defects in oxides at room temperature by lithium reduction. Nat. Commun. 2018, 9, 1302. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Luo, L.; Li, M.; Chen, W.; Liu, Y.; Yang, J.; Xu, S.M.; Zhou, H.; Ma, L.; Xu, M.; et al. Photoelectrocatalytic C-H halogenation over an oxygen vacancy-rich TiO2 photoanode. Nat. Commun. 2021, 12, 6698. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Zhao, M.; Yang, Z.; Duan, X.; Jiang, G.; Li, G.; Zhang, F.; Hao, Z. Oxygen vacancy-engineered titanium-based perovskite for boosting H2O activation and lower-temperature hydrolysis of organic sulfur. Proc. Natl. Acad. Sci. USA 2023, 120, e2217148120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gao, Y.; Gao, D.; Wang, Y. Effect of oxide defect on photocatalytic properties of MSnO3 (M = Ca, Sr, and Ba) photocatalysts. Appl. Catal. B Environ. 2019, 243, 428–437. [Google Scholar] [CrossRef]
- Zhang, Z.; Yu, F.; Huang, L.; Jiatieli, J.; Li, Y.; Song, L.; Yu, N.; Dionysiou, D.D. Confirmation of hydroxyl radicals (*OH) generated in the presence of TiO2 supported on AC under microwave irradiation. J. Hazard. Mater. 2014, 278, 152–157. [Google Scholar] [CrossRef]
- Jiang, F.; Zhang, L.; Yue, T.; Tang, H.; Wang, L.; Sun, W.; Zhang, C.; Chen, J. Defect-boosted molybdenite-based co-catalytic Fenton reaction. Inorg. Chem. Front. 2021, 8, 3440–3449. [Google Scholar] [CrossRef]
- Li, H.; McKay, G. Fluorescence Quenching of Humic Substances and Natural Organic Matter by Nitroxide Free Radicals. Environ. Sci. Technol. 2023, 57, 719–729. [Google Scholar] [CrossRef]
- Peng, S.; Li, R.; Rao, Y.; Huang, Y.; Zhao, Y.; Xiong, M.; Cao, J.; Lee, S. Tuning the unsaturated iron sites in MIL-101(Fe) nanoparticles for reactive oxygen species-mediated bacterial inactivation in the dark. Appl. Catal. B Environ. 2022, 316, 121693. [Google Scholar] [CrossRef]
- Zhang, L.; Mao, S.; Liu, Y.; Lu, B.; Wang, Y.; Li, H.; Wang, Y. Tandem catalytic efficient olefin epoxidation with integrated production of nicotinamide derivatives. Chem. Catal. 2023, 3, 100691. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Solvent | Conversion (%) | Selectivity (%) | Yield (%) |
|---|---|---|---|---|
| CS-1 | Methanol | 65.1 | 98.7 | 64.2 |
| CS-2 | Methanol | 74.3 | 98.8 | 73.4 |
| CS-3 | Methanol | 82.5 | 98.9 | 81.6 |
| CS-4 | Methanol | 83.6 | 98.3 | 82.2 |
| SS-3 | Methanol | 45.8 | 97.6 | 44.7 |
| BS-3 | Methanol | 36.1 | 97.2 | 35.1 |
| CaO | Methanol | 17.2 | 85.4 | 14.7 |
| SnO2 | Methanol | 7.4 | 99.3 | 7.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, M.; Xiang, H.; Wen, X. Enhancing Catalytic Efficiency in Long-Chain Linear α-Olefin Epoxidation: A Study of CaSnO3-Based Catalysts. Catalysts 2024, 14, 70. https://doi.org/10.3390/catal14010070
Zhang M, Xiang H, Wen X. Enhancing Catalytic Efficiency in Long-Chain Linear α-Olefin Epoxidation: A Study of CaSnO3-Based Catalysts. Catalysts. 2024; 14(1):70. https://doi.org/10.3390/catal14010070
Chicago/Turabian StyleZhang, Min, Hongwei Xiang, and Xiaodong Wen. 2024. "Enhancing Catalytic Efficiency in Long-Chain Linear α-Olefin Epoxidation: A Study of CaSnO3-Based Catalysts" Catalysts 14, no. 1: 70. https://doi.org/10.3390/catal14010070