
Water Oxidation over Au-Pd/TiO2 as a Substitute for Iridium-Based Catalysts



Abstract
:1. Introduction
2. Results
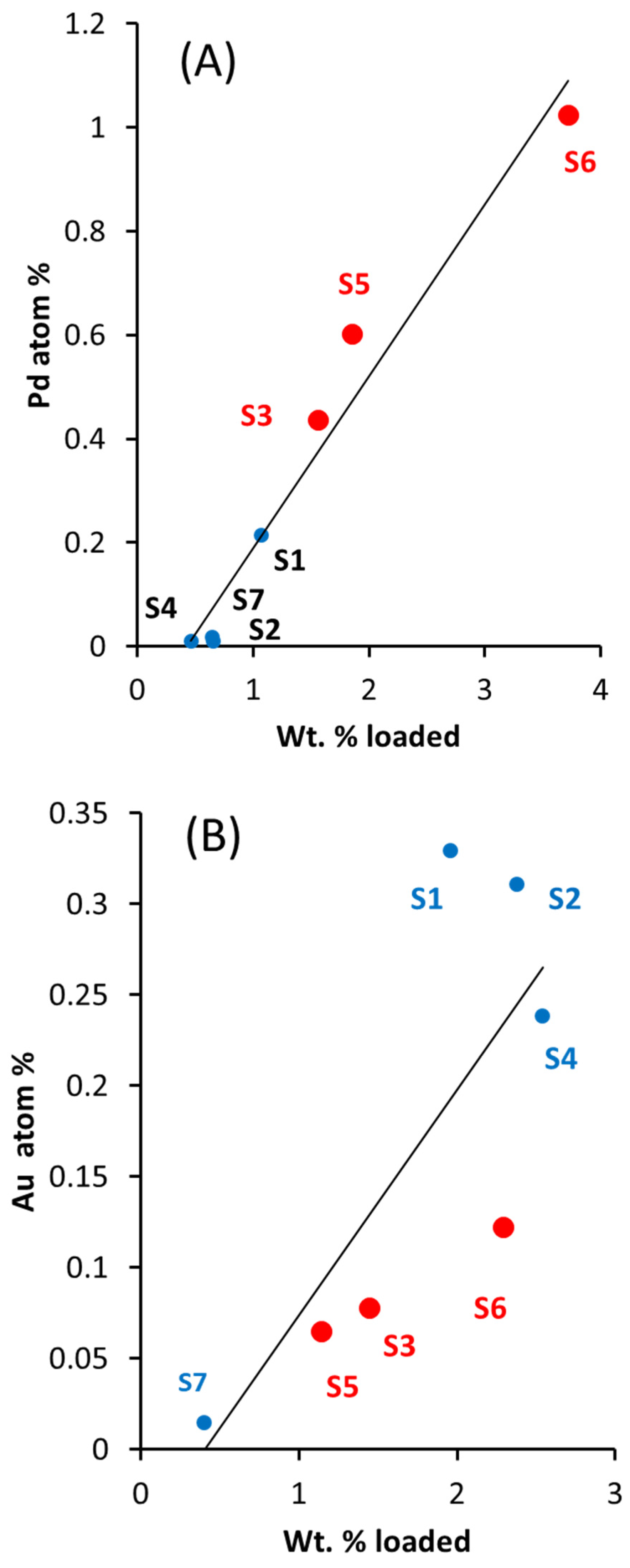
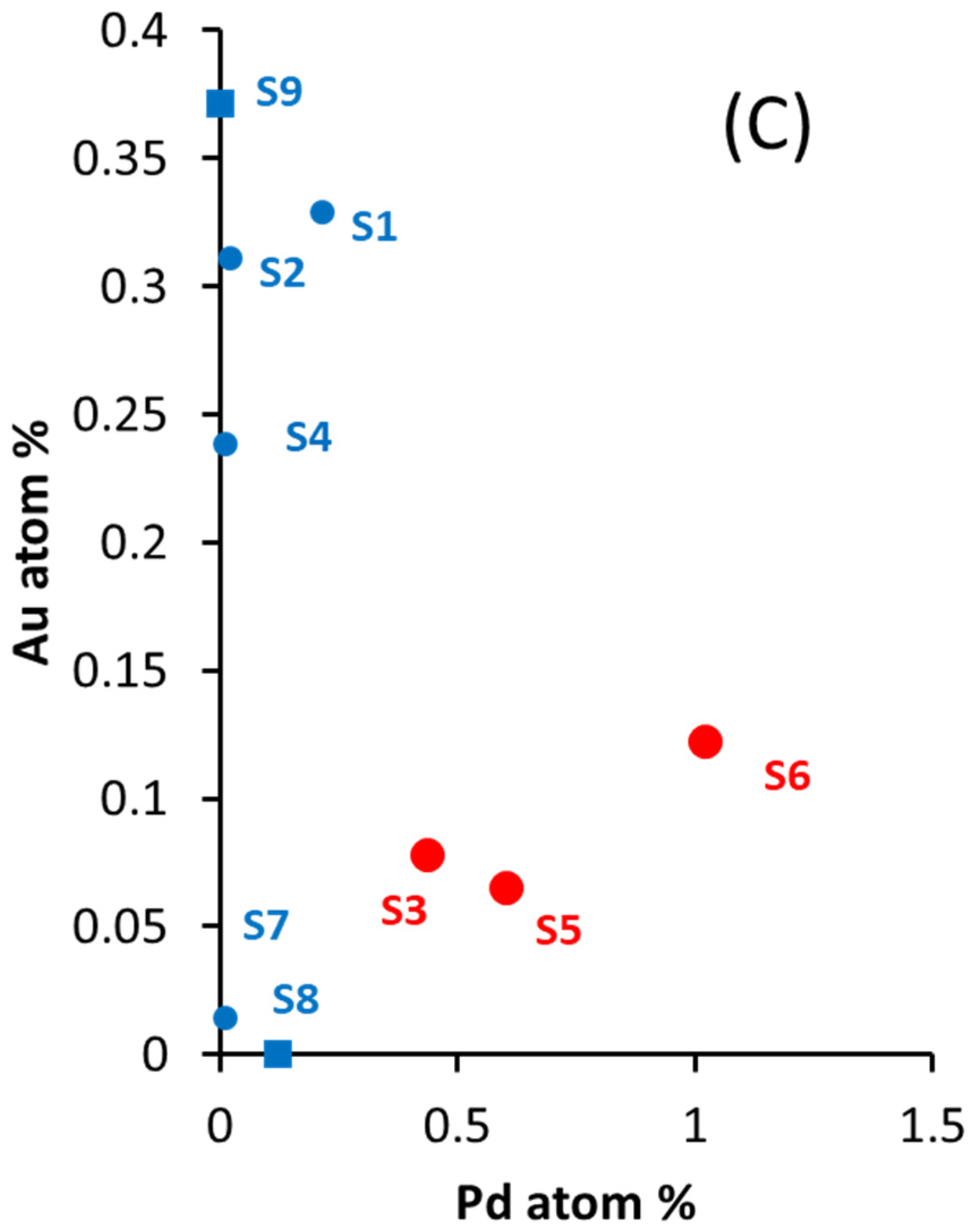
2.1. Catalysts Composition
2.2. Catalytic Activity of Au-Pd Catalysts
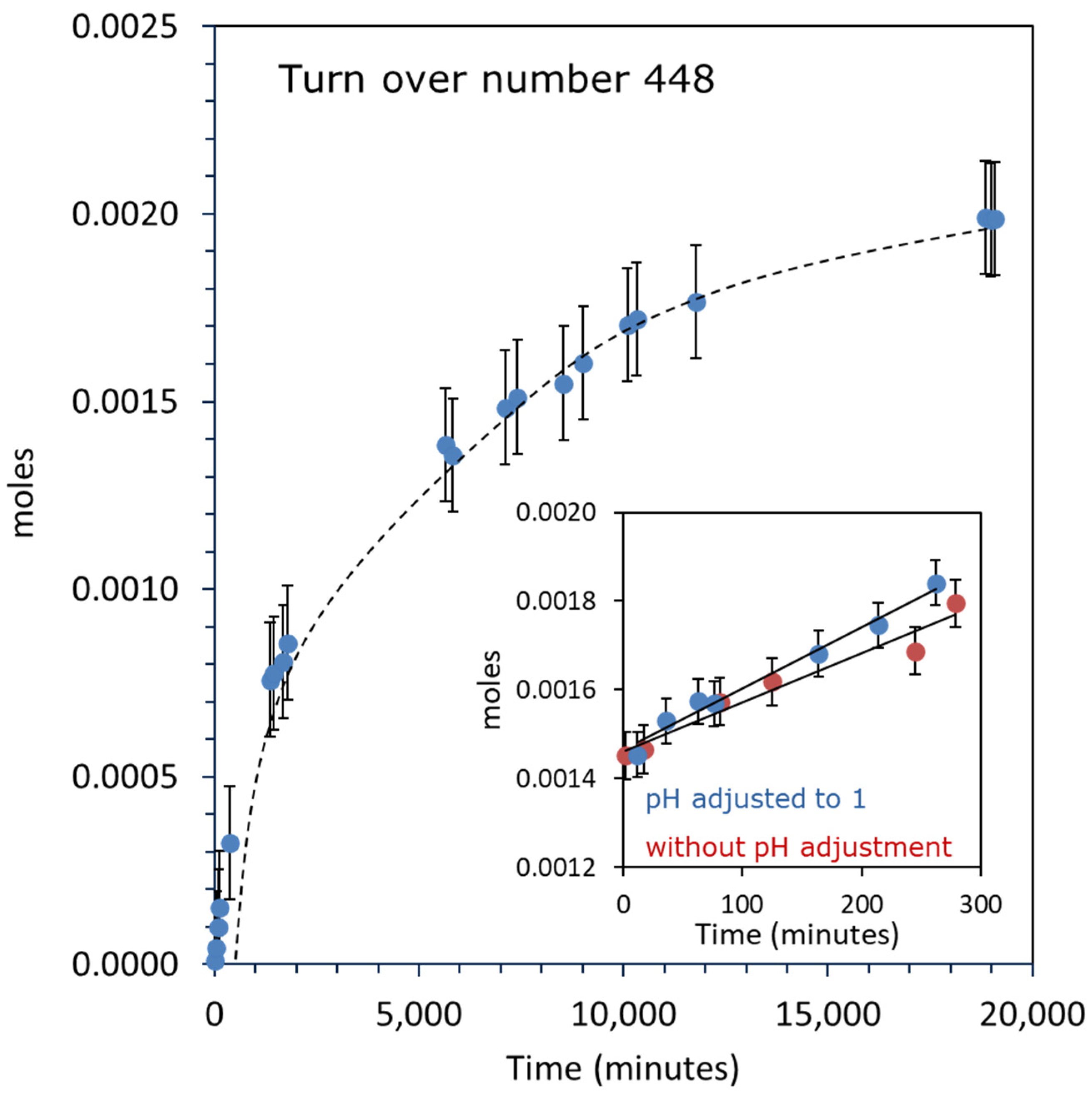
2.2.1. Oxygen Production over Au-Pd/TiO2
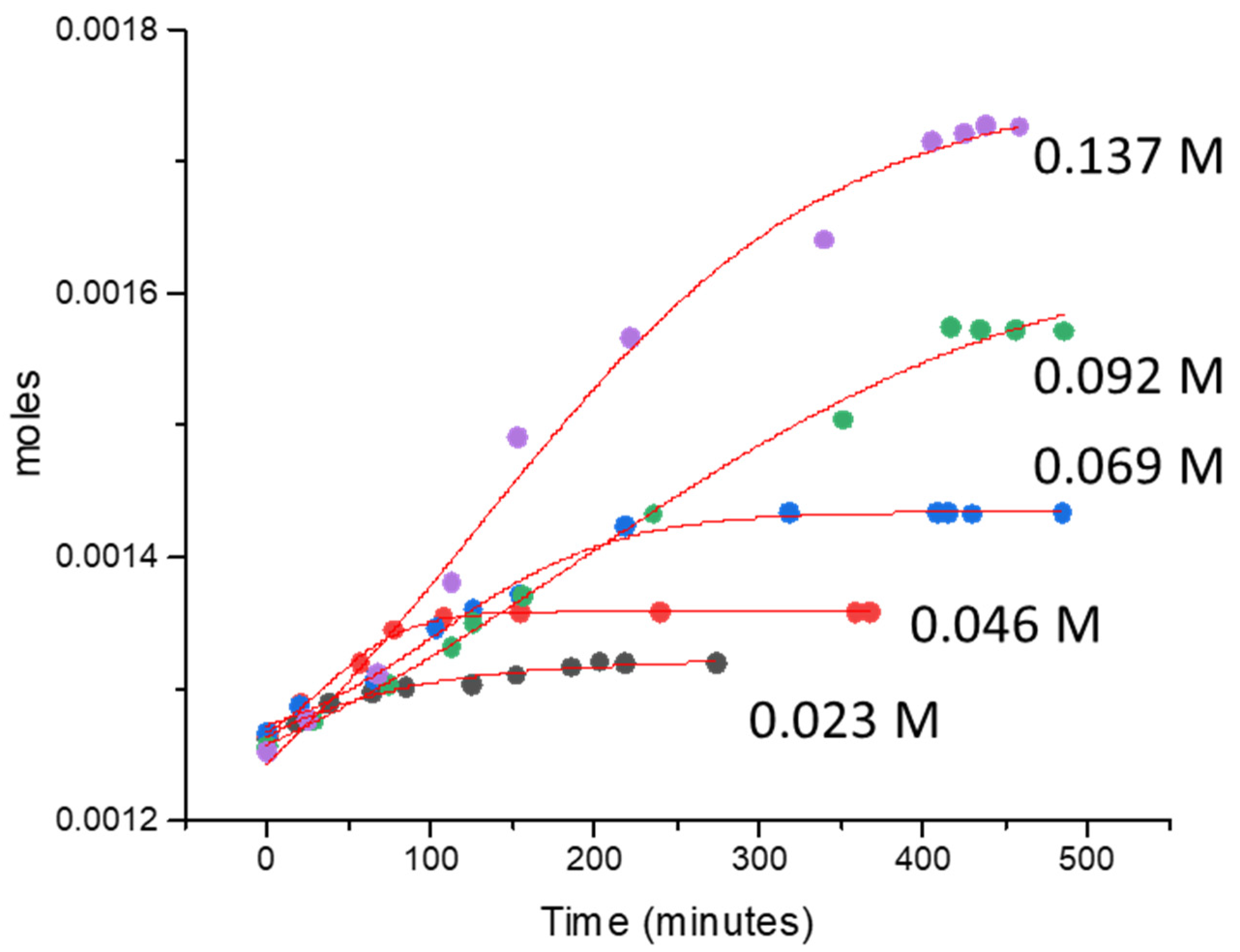
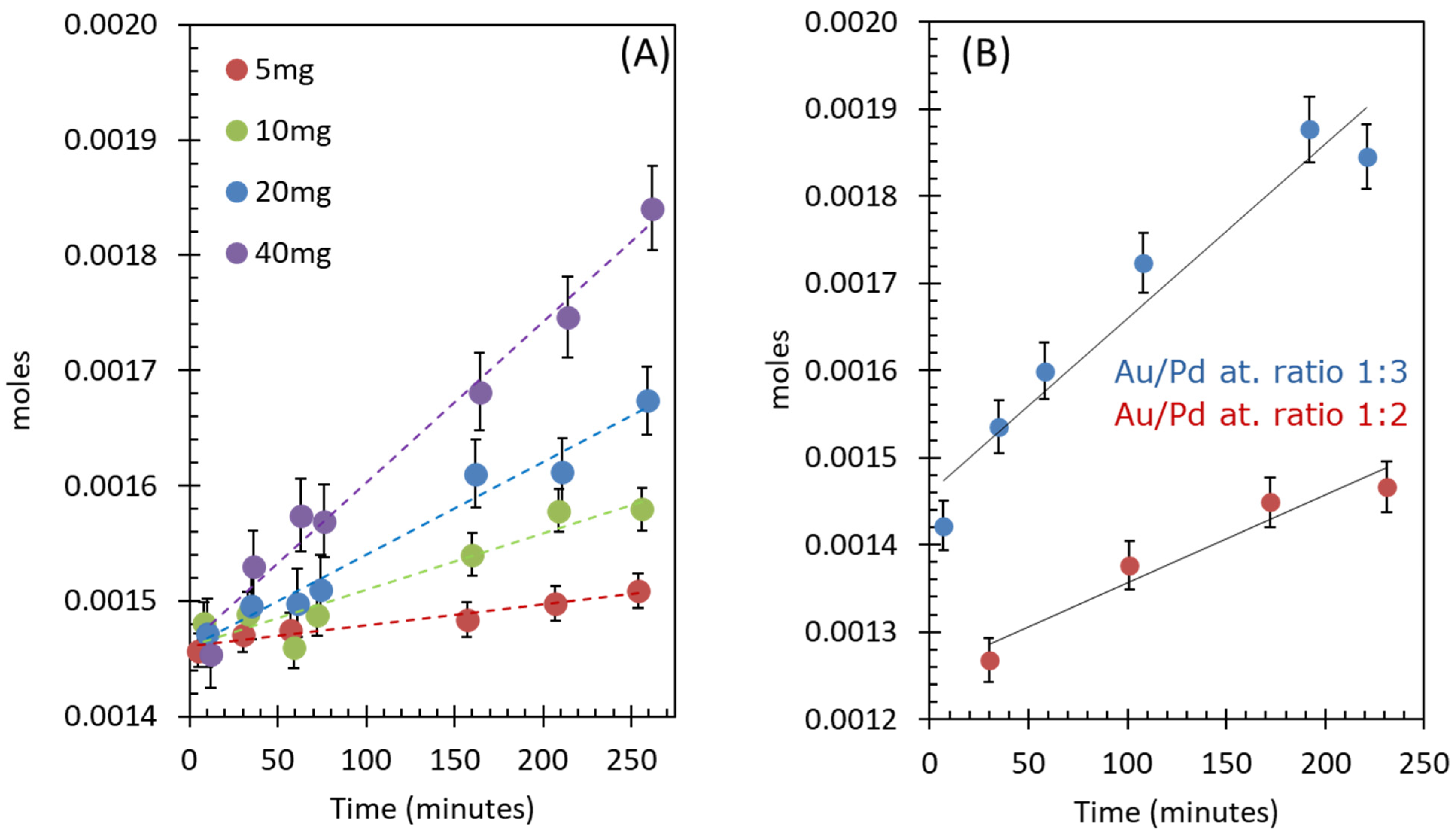
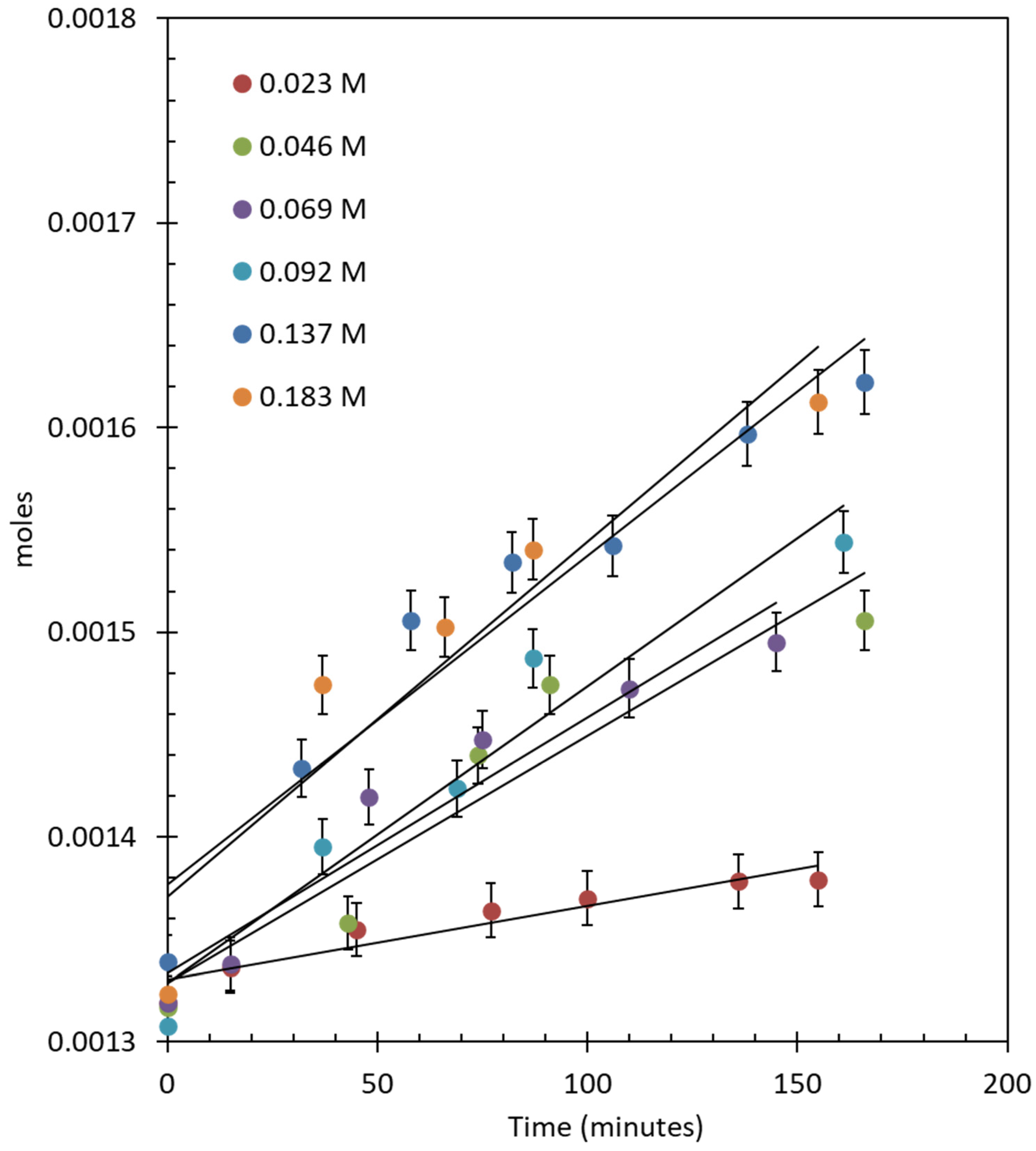
2.2.2. Effect of Catalyst Concentration
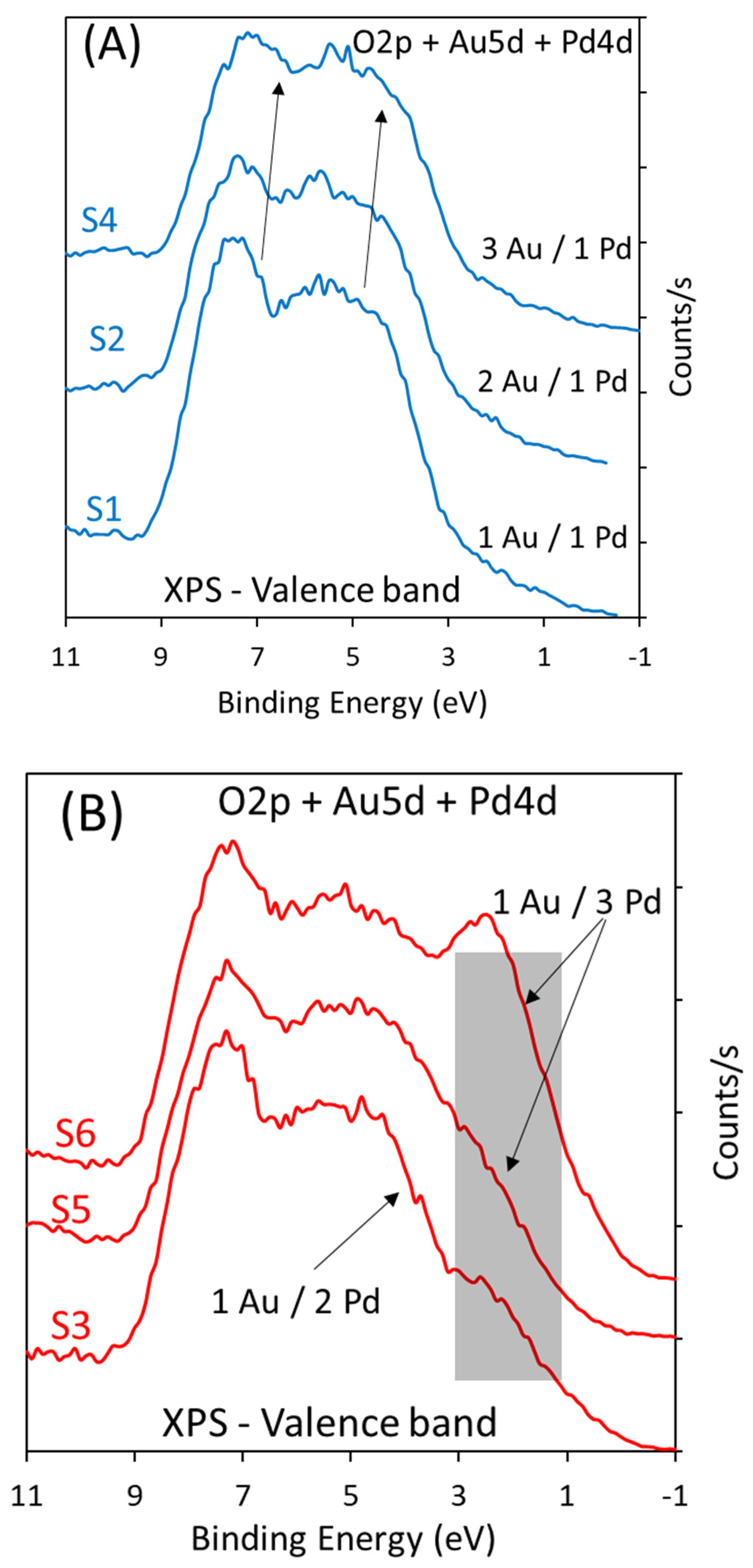
2.2.3. Effect of Catalyst Composition (Au-to-Pd Ratios)
2.3. UV-Vis Absorbance Spectroscopy of the Liquid Phase
2.4. Oxygen Production over IrO2 Catalysts
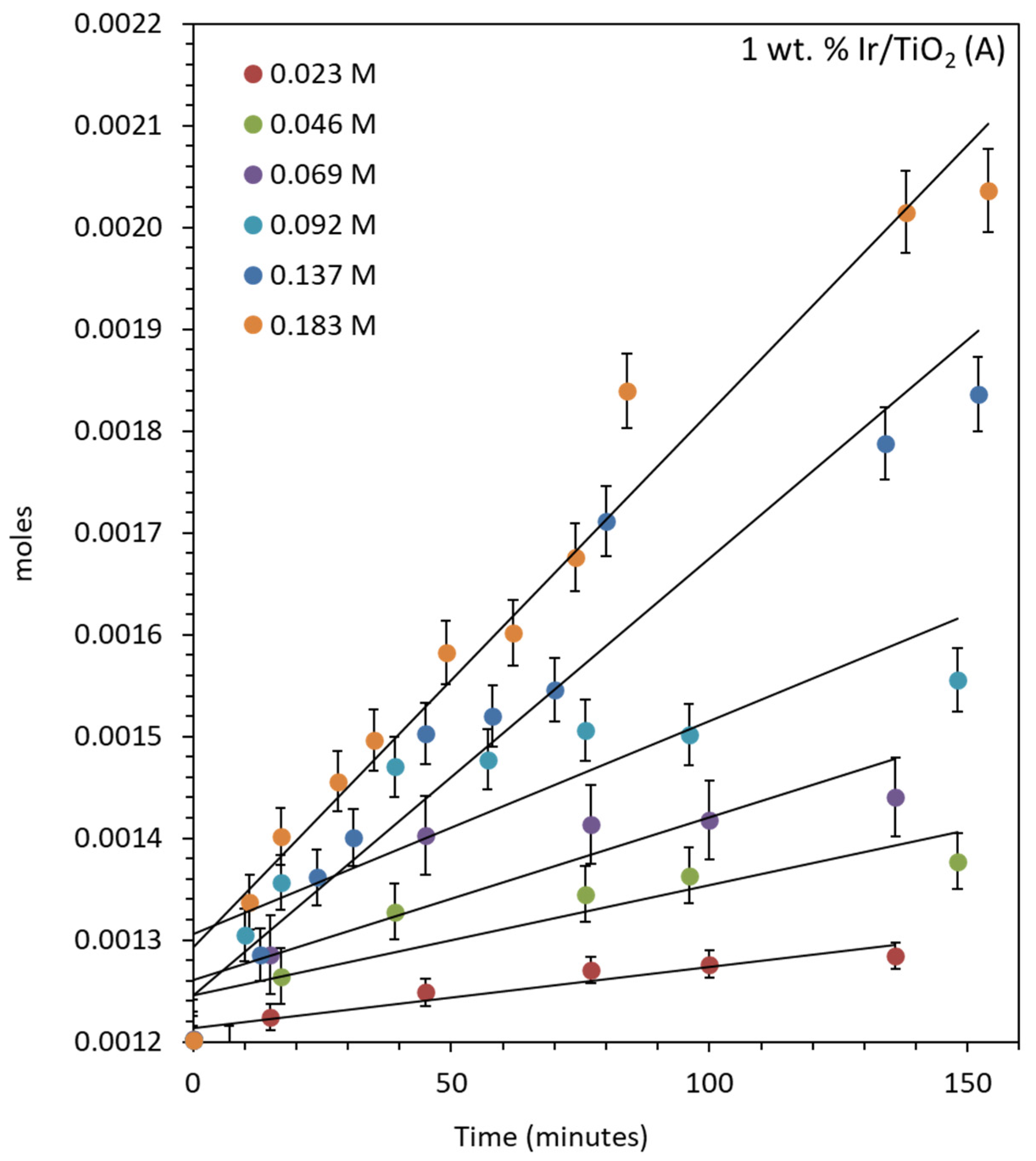
2.4.1. Oxygen Production over IrO2/TiO2 (Anatase)
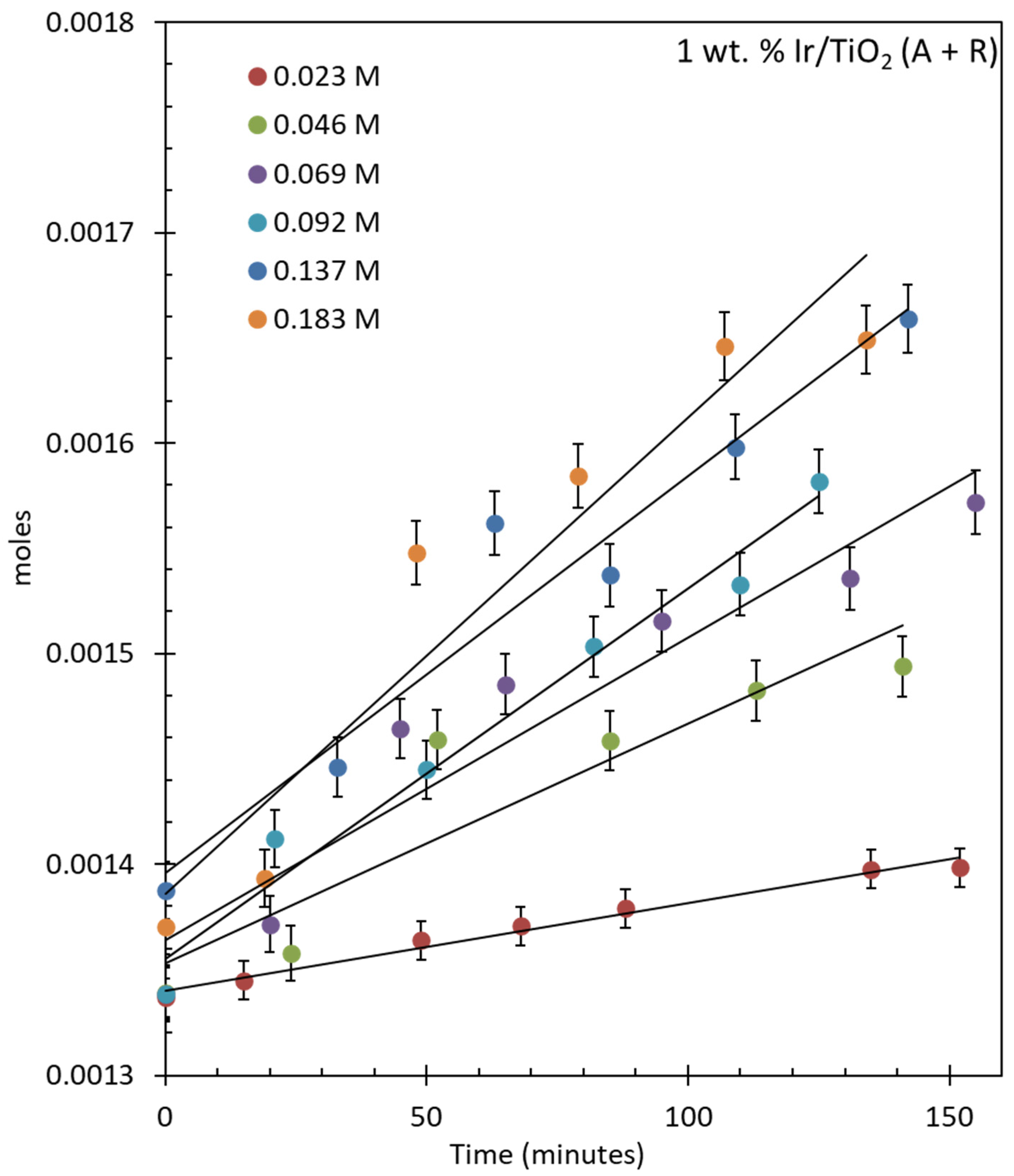
2.4.2. Oxygen Production over IrO2/TiO2 (Anatase + Rutile)
2.4.3. Oxygen Production over IrO2/CeO2
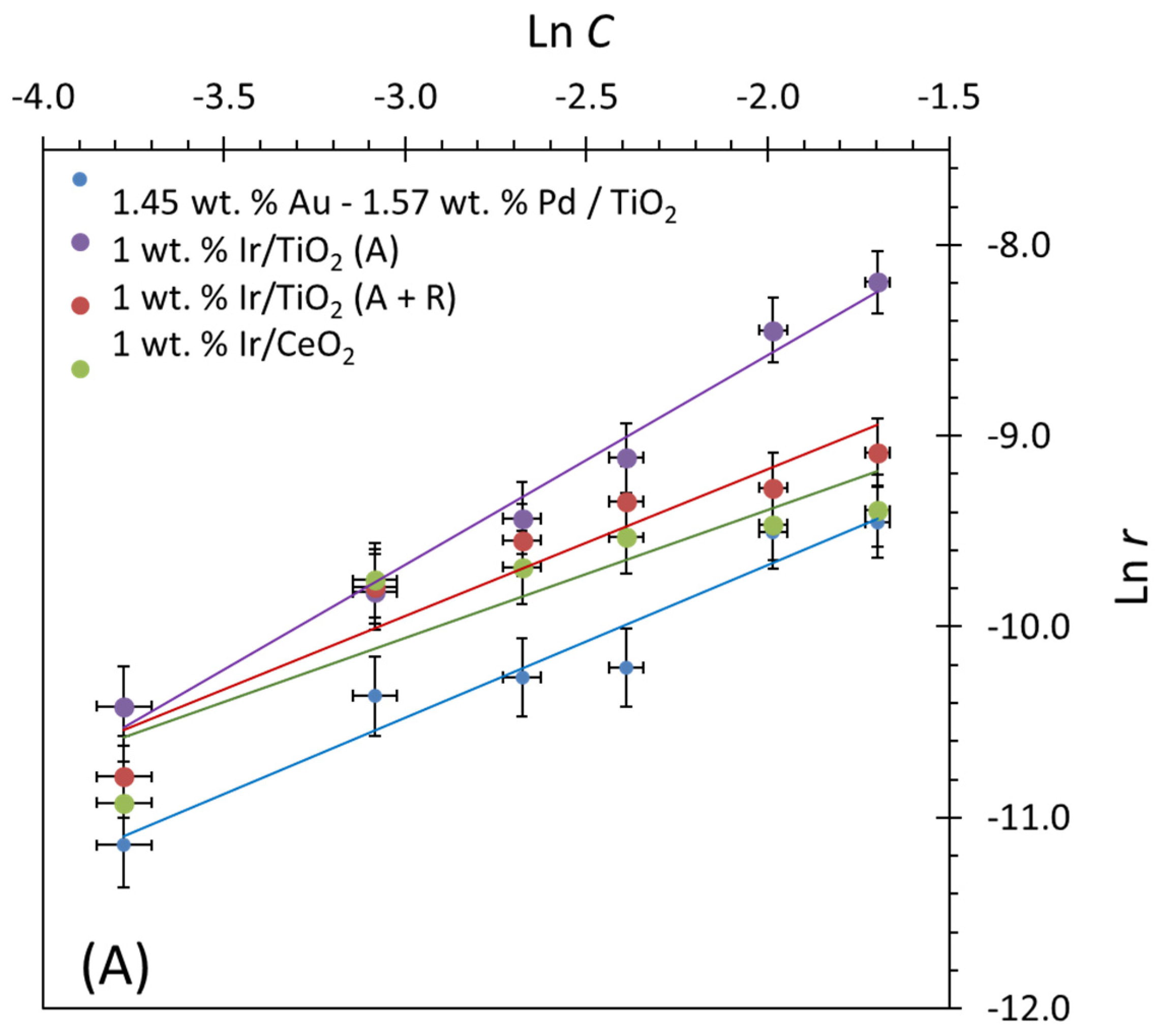
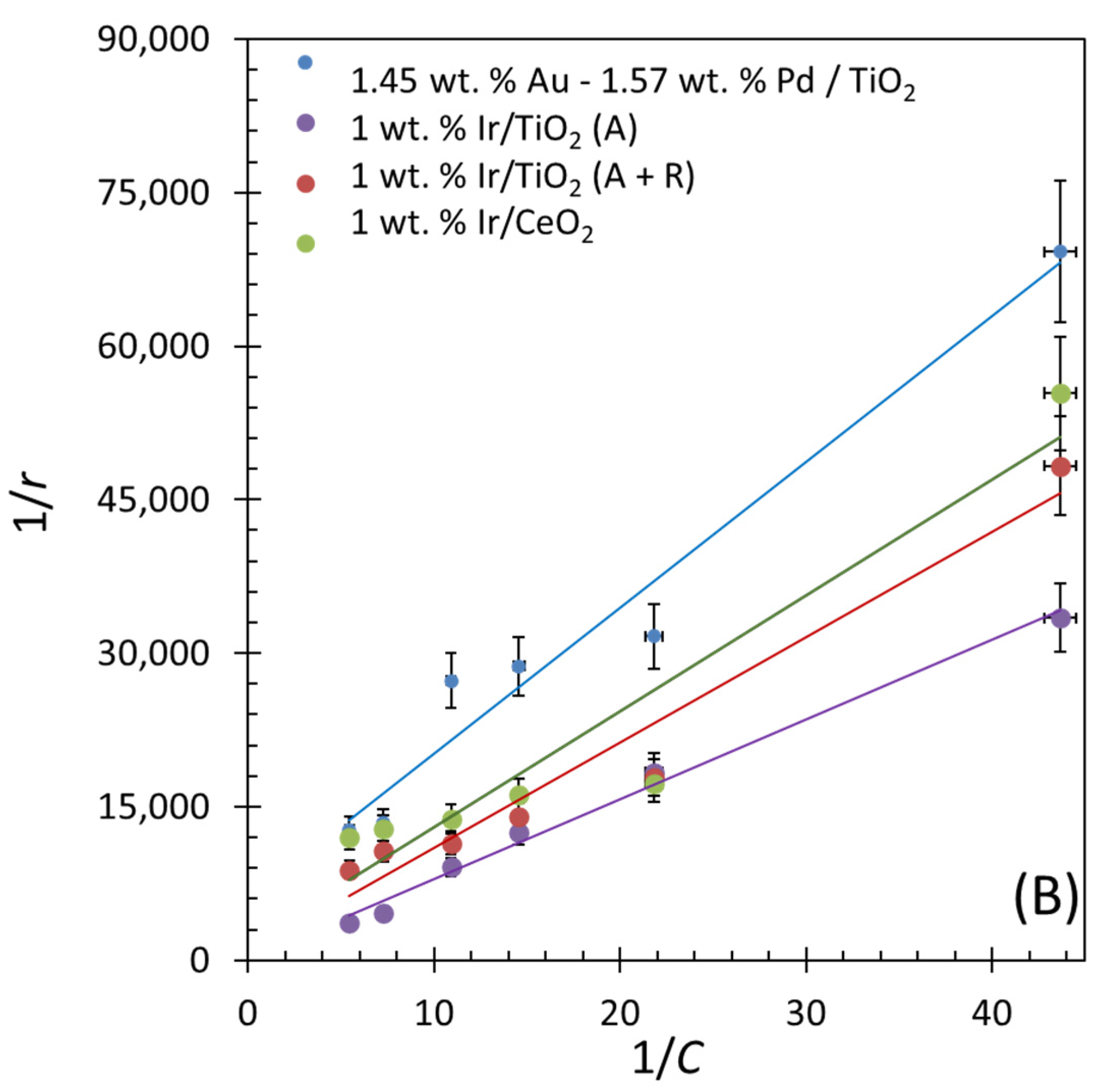
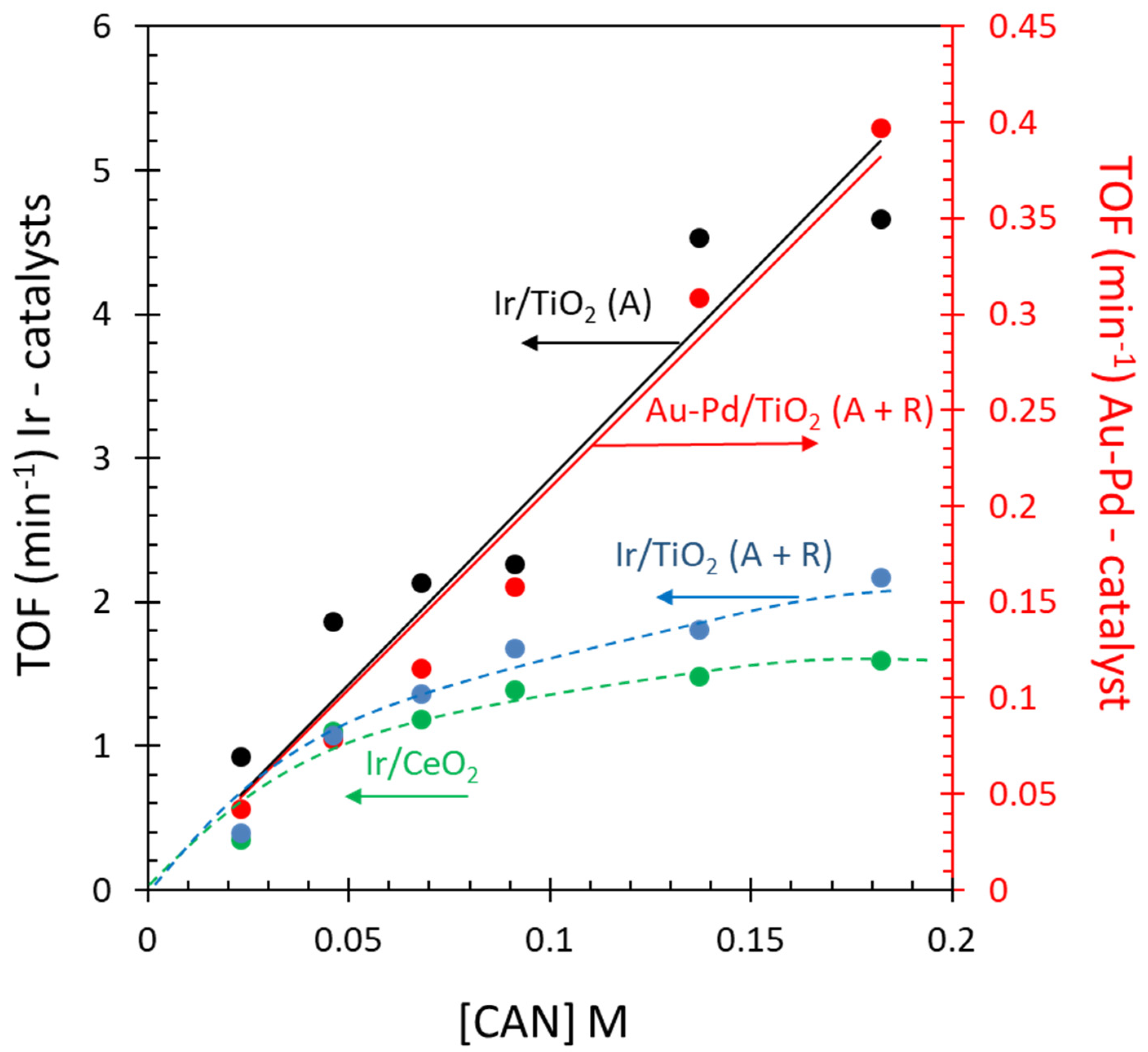
2.5. Reaction Kinetics
reactant catalyst reactant/product
3. Experimental
3.1. Catalyst Preparation
3.2. Reaction Setup
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fukuzumi, S.; Hong, D. Homogeneous versus Heterogeneous Catalysts in Water Oxidation. Eur. J. Inorg. Chem. 2014, 2014, 645–659. [Google Scholar] [CrossRef]
- Huang, Y.; Li, M.; Yang, W.; Yu, Y.; Hao, S. Ce-Doped Ordered Mesoporous Cobalt Ferrite Phosphides as Robust Catalysts for Water Oxidation. Chem. Eur. J. 2020, 26, 13305–13310. [Google Scholar] [CrossRef] [PubMed]
- Concepcion, J.J.; Jurss, J.W.; Templeton, J.L.; Meyer, T.J. One Site is Enough. Catalytic Water Oxidation by [Ru(tpy)(bpm)(OH2)]2+ and [Ru(tpy)(bpz)(OH2)]2+. J. Am. Chem. Soc. 2008, 130, 16462–16463. [Google Scholar] [CrossRef] [PubMed]
- Kiyota, J.; Yokoyama, J.; Yoshida, M.; Masaoka, S.; Sakai, K. Electrocatalytic O2 Evolution from Water at an ITO Electrode Modified with [Ru(terpy){4,4′-(CH2PO3H2)2-2,2′-bpy}(OH2)]2+: Evidence for a Unimolecular Pathway. Chem. Lett. 2010, 39, 1146–1148. [Google Scholar] [CrossRef]
- Wasylenko, D.J.; Ganesamoorthy, C.; Henderson, M.A.; Koivisto, B.D.; Osthoff, H.D.; Berlinguette, C.P. Electronic Modification of the [RuII(tpy)(bpy)(OH2)]2+ Scaffold: Effects on Catalytic Water Oxidation. J. Am. Chem. Soc. 2010, 132, 16094–16106. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Baik, M.-H. cis,cis-[(bpy)2RuVO]2O4+ Catalyzes Water Oxidation Formally via in Situ Generation of Radicaloid RuIV−O•. J. Am. Chem. Soc. 2006, 128, 7476–7485. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Masaoka, S.; Abe, J.; Sakai, K. Catalysis of mononuclear aquaruthenium complexes in oxygen evolution from water: A new radical coupling path using hydroxocerium (IV) species. Chem. Asian J. 2010, 5, 2369–2378. [Google Scholar] [CrossRef]
- Grotjahn, D.B.; Brown, D.B.; Martin, J.K.; Marelius, D.C.; Abadjian, M.-C.; Tran, H.; Kalyuzhny, G.; Vecchio, K.S.; Specht, Z.G.; Cortes-Llamas, S.A. Evolution of iridium-based molecular catalysts during water oxidation with ceric ammonium nitrate. J. Am. Chem. Soc. 2011, 133, 19024–19027. [Google Scholar] [CrossRef]
- Zhao, Y.; Hernandez-Pagan, E.A.; Vargas-Barbosa, N.M.; Dysart, J.L.; Mallouk, T.E. A high yield synthesis of ligand-free iridium oxide nanoparticles with high electrocatalytic activity. J. Phys. Chem. Lett. 2011, 2, 402–406. [Google Scholar] [CrossRef]
- Bucci, A.; Menendez Rodriguez, G.; Bellachioma, G.; Zuccaccia, C.; Poater, A.; Cavallo, L.; Macchioni, A. An alternative reaction pathway for iridium-catalyzed water oxidation driven by cerium ammonium nitrate (CAN). ACS Catal. 2016, 6, 4559–4563. [Google Scholar] [CrossRef]
- Pastori, G.; Wahab, K.; Bucci, A.; Bellachioma, G.; Zuccaccia, C.; Llorca, J.; Idriss, H.; Macchioni, A. Heterogenized water oxidation catalysts prepared by immobilizing Kläui-type organometallic precursors. Chem. A Eur. J. 2016, 22, 13459–13463. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Murakami, M.; Yamada, Y.; Fukuzumi, S. Efficient water oxidation by cerium ammonium nitrate with [IrIII(Cp*)(4, 4′-bishydroxy-2, 2′-bipyridine)(H2O)]2+ as a precatalyst. Energy Environ. Sci. 2012, 5, 5708–5716. [Google Scholar] [CrossRef]
- Harriman, A.; Richoux, M.-C.; Christensen, P.A.; Mosseri, S.; Neta, P. Redox reactions with colloidal metal oxides. Comparison of radiation-generated and chemically generated RuO2 2H2O. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1987, 83, 3001–3014. [Google Scholar] [CrossRef]
- Harriman, A.; Pickering, I.J.; Thomas, J.M.; Christensen, P.A. Metal oxides as heterogeneous catalysts for oxygen evolution under photochemical conditions. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1988, 84, 2795–2806. [Google Scholar] [CrossRef]
- Morris, N.D.; Suzuki, M.; Mallouk, T.E. Kinetics of Electron Transfer and Oxygen Evolution in the Reaction of [Ru(bpy)3]3+ with Colloidal Iridium Oxide. J. Phys. Chem. A 2004, 108, 9115–9119. [Google Scholar] [CrossRef]
- Ping, Y.; Galli, G.; Goddard, W.A., III. Electronic Structure of IrO2: The Role of the Metal d Orbitals. J. Phys. Chem. C 2015, 119, 11570–11577. [Google Scholar] [CrossRef]
- He, J.; Zhou, X.; Xu, P.; Sun, J. Regulating Electron Redistribution of Intermetallic Iridium Oxide by Incorporating Ru for Efficient Acidic Water Oxidation. Adv. Energy Mater. 2021, 11, 2102883. [Google Scholar] [CrossRef]
- Kawasaki, S.; Takahashi, R.; Akagi, K.; Yoshinobu, J.; Komori, F.; Horiba, K.; Kumigashira, H.; Iwashina, K.; Kudo, A.; Lippmaa, M. Electronic Structure and Photoelectrochemical Properties of an Ir-Doped SrTiO3 Photocatalyst. J. Phys. Chem. C 2014, 118, 20222–20228. [Google Scholar] [CrossRef]
- Sun, W.; Liu, J.-Y.; Gong, X.-Q.; Zaman, W.Q.; Cao, L.-M.; Yang, J. OER activity manipulated by IrO6 coordination geometry: An insight from pyrochlore iridates. Sci. Rep. 2016, 6, 38429. [Google Scholar] [CrossRef]
- Kahk, J.M.; Poll, C.G.; Oropeza, F.E.; Ablett, J.M.; Céolin, D.; Rueff, J.-P.; Agrestini, S.; Utsumi, Y.; Tsuei, K.D.; Liao, Y.F.; et al. Understanding the Electronic Structure of IrO2 Using Hard-X-ray Photoelectron Spectroscopy and Density-Functional Theory. Phys. Rev. Lett. 2014, 112, 117601. [Google Scholar] [CrossRef]
- Alrushaid, M.; Nadeem, M.A.; Wahab, K.A.; Idriss, H. Extracting Turnover Frequencies of Electron Transfer in Heterogeneous Catalysis: A Study of IrO2-TiO2 Anatase for Water Oxidation Using Ce4+ Cations. Catalysts 2021, 11, 1030. [Google Scholar] [CrossRef]
- Liu, B.; Yang, H.; Hu, P.; Wang, G.-S.; Guo, Y.; Zhao, H. Dimension Engineering in Noble-Metal-Based Nanocatalysts. Catalysts 2024, 14, 9. [Google Scholar] [CrossRef]
- Reinecke, B.N.; Kuhl, K.P.; Ogasawara, H.; Li, L.; Voss, J.; Abild-Pedersen, F.; Nilsson, A.; Jaramillo, T.F. Elucidating the electronic structure of supported gold nanoparticles and its relevance to catalysis by means of hard X-ray photoelectron spectroscopy. Surf. Sci. 2016, 650, 24–33. [Google Scholar] [CrossRef]
- Boyen, H.-G.; Kästle, G.; Weigl, F.; Koslowski, B.; Dietrich, C.; Ziemann, P.; Spatz, J.P.; Riethmüller, S.; Hartmann, C.; Möller, M.; et al. Oxidation-Resistant Gold-55 Clusters. Science 2002, 297, 1533. [Google Scholar] [CrossRef] [PubMed]
- McGuirk, G.M.; Ledieu, J.; Gaudry, É.; de Weerd, M.-C.; Fournée, V. Surface structures of In-Pd intermetallic compounds. I. Experimental study of In thin films on Pd(111) and alloy formation. J. Chem. Phys. 2014, 141, 084702. [Google Scholar] [CrossRef] [PubMed]
- McClure, J.P.; Boltersdorf, J.; Baker, D.R.; Farinha, T.G.; Dzuricky, N.; Villegas, C.E.P.; Rocha, A.R.; Leite, M.S. Structure–Property-Performance Relationship of Ultrathin Pd–Au Alloy Catalyst Layers for Low-Temperature Ethanol Oxidation in Alkaline Media. ACS Appl. Mater. Interfaces 2019, 11, 24919–24932. [Google Scholar] [CrossRef]
- Dash, P.; Bond, T.; Fowler, C.; Hou, W.; Coombs, N.; Scott, R.W.J. Rational Design of Supported PdAu Nanoparticle Catalysts from Structured Nanoparticle Precursors. J. Phys. Chem. C 2009, 113, 12719–12730. [Google Scholar] [CrossRef]
- Sellidj, A.; Koel, B.E. Electronic and CO chemisorption properties of ultrathin Pd films vapor deposited on Au(111). Phys. Rev. B 1994, 49, 8367–8376. [Google Scholar] [CrossRef]
- Nahm, T.U.; Jung, R.; Kim, J.-Y.; Park, W.-G.; Oh, S.-J.; Park, J.-H.; Allen, J.W.; Chung, S.-M.; Lee, Y.S.; Whang, C.N. Electronic structure of disordered Au-Pd alloys studied by electron spectroscopies. Phys. Rev. B 1998, 58, 9817–9825. [Google Scholar] [CrossRef]
- Hutchings, G.J. Nanocrystalline gold and gold–palladium alloy oxidation catalysts: A personal reflection on the nature of the active sites. Dalton Trans. 2008, 5523–5553. [Google Scholar] [CrossRef]
- Marx, S.; Baiker, A. Beneficial Interaction of Gold and Palladium in Bimetallic Catalysts for the Selective Oxidation of Benzyl Alcohol. J. Phys. Chem. C 2009, 113, 6191–6201. [Google Scholar] [CrossRef]
- Boltersdorf, J.; Leff, A.C.; Forcherio, G.T.; Baker, D.R. Plasmonic Au–Pd Bimetallic Nanocatalysts for Hot-Carrier-Enhanced Photocatalytic and Electrochemical Ethanol Oxidation. Crystals 2021, 11, 226. [Google Scholar]
- Idriss, H.; Khaja, A.W. Photocatalyst Comprising Gold-Palladium Alloy, Method for Preparation, Photolysis System. PCT Patents WO2014033645A1, 6 March 2014. [Google Scholar]
- Waterhouse, G.I.N.; Wahab, A.K.; Al-Oufi, M.; Jovic, V.; Sun-Waterhouse, D.; Anjum, D.; Llorca, J.; Idriss, H. Photonic Band Gap Au/TiO2 materials as highly active and stable Photocatalysts for Hydrogen production from water. Sci. Rep. 2013, 3, 2849. [Google Scholar] [CrossRef]
- Kaczur, J.J.; Yang, H.; Liu, Z.; Sajjad, S.D.; Masel, R.I. Carbon Dioxide and Water Electrolysis Using New Alkaline Stable Anion Membranes. Front. Chem. 2018, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Idriss, H. Towards large scale hydrogen production from water, what have we learned and what are the main hurdles to cross for commercialization. Energy Technol. 2021, 9, 2000843. [Google Scholar] [CrossRef]
- Yang, M.; Wang, Z.; Wang, W.; Liu, C.-J. Synthesis of AuPd alloyed nanoparticles via room-temperature electron reduction with argon glow discharge as electron source. Nanoscale Res. Lett. 2014, 9, 405. [Google Scholar]
- Xiao, Q.; Jaatinen, S.E.; Jia, J.; Arnold, D.P.; Liu, H.; Zhu, H. Efficient photocatalytic Suzuki cross-coupling reactions on Au–Pd alloy nanoparticles under visible light irradiation. Green Chem. 2014, 16, 4272–4285. [Google Scholar] [CrossRef]
- Tsuji, M.; Uto, K.; Hayashi, J.-I.; Yoshiwara, A. Synthesis of Flower-Like AuPd@SiO2 Nanoparticles with a Broad Light Extinction for Application to Efficient Dye-Sensitized Solar Cells. Part. Part. Syst. Charact. 2018, 35, 1700396. [Google Scholar] [CrossRef]
- Damasceno, J.P.V.; Maroneze, C.M.; Mathias Strauss, M.; Sigolia, F.A.; Mazali, I.O. Preparation of supported AuPd nanoalloys mediated by ionic liquid-like functionalized SBA-15: Structural correlations concerning its catalytic activity. New J. Chem. 2016, 40, 6636–6642. [Google Scholar] [CrossRef]
- Zhong, J.B.; Lu, Y.; Jiang, W.D.; Meng, Q.M.; He, X.Y.; Li, J.Z.; Chen, Y.Q. Characterization and photocatalytic property of Pd/TiO2 with the oxidation of gaseous benzene. J. Hazard. Mater. 2009, 168, 1632–1635. [Google Scholar] [CrossRef]
- Duan, K.; Liu, Z.; Li, J.; Yuan, L.; Hu, H.; Woo, S.I. Novel Pd–Au/TiO2 catalyst for the selective catalytic reduction of NOx by H2. Catal. Commun. 2014, 57, 19–22. [Google Scholar] [CrossRef]
- Bashir, S.; Wahab, A.K.; Anjum, D.; Al-Salik, Y.; Idriss, H. Ethanol photoreactions over Au-Pd/TiO2. Appl. Petrochem. Res. 2014, 4, 55–62. [Google Scholar]
- Bashir, S.; Wahab, A.K.; Idriss, H. Synergism and photocatalytic water splitting to hydrogen over Pt/TiO2 catalysts: Effect particle size. Catal. Today 2015, 240, 242–247. [Google Scholar] [CrossRef]
- Fadley, C.S. X-ray photoelectron spectroscopy: Progress and perspectives. J. Electron Spectrosc. Relat. Phenom. 2010, 178–179, 2–32. [Google Scholar] [CrossRef]
- Wang, D.; Cui, X.; Xiao, Q.; Hu, Y.; Wang, Z.; Yiu, Y.M.; Sham, T.K. Electronic behaviour of Au-Pt alloys and the 4f binding energy shift anomaly in Au bimetallics-X-ray spectroscopy studies. AIP Adv. 2018, 8, 065210. [Google Scholar] [CrossRef]
- Caux, M.; Menard, H.; Alsalik, Y.; Irvine, J.T.S.; Idriss, H. g-C3N4 influence on gold nanoparticles composition and morphology for visible light-driven hydrogen formation. Phys. Chem. Chem. Phys. 2019, 21, 15974–15987. [Google Scholar] [CrossRef]
- Heidt, L.J.; Berestecki, J. Optical Studies of Cerous Solutions. J. Am. Chem. Soc. 1955, 77, 2049–2054. [Google Scholar] [CrossRef]
- Najafpour, M.M.; Kompany-Zareh, M.; Zahraei, A.; Sedigh, D.J.; Jaccard, H.; Khoshkam, M.; Britt, R.D.; Casey, W.H. Mechanism, decomposition pathway and new evidence for self-healing of manganese oxides as efficient water oxidizing catalysts: New insights. Dalton Trans. 2013, 42, 14603–14611. [Google Scholar] [CrossRef]
- Wasylenko, D.J.; Ganesamoorthy, C.; Henderson, M.A.; Berlinguette, C.P. Unraveling the Roles of the Acid Medium, Experimental Probes, and Terminal Oxidant, (NH4)2[Ce(NO3)6], in the Study of a Homogeneous Water Oxidation Catalyst. Inorg. Chem. 2011, 50, 3662–3672. [Google Scholar] [CrossRef]
- Chen, Z.; Concepcion, J.J.; Hu, X.; Yang, W.; Hoertz, P.G.; Meyer, T.J. Concerted O atom–proton transfer in the O–O bond forming step in water oxidation. Proc. Natl. Acad. Sci. USA 2010, 107, 7225–7229. [Google Scholar] [CrossRef]
- Wadsworth, E.; Duke, F.R.; Goetz, C. Present status of cerium (IV)-cerium (III) potentials. Anal. Chem. 1957, 29, 1824–1825. [Google Scholar] [CrossRef]
- Yagi, M.; Tomita, E.; Sakita, S.; Kuwabara, T.; Nagai, K. Self-Assembly of Active IrO2 Colloid Catalyst on an ITO Electrode for Efficient Electrochemical Water Oxidation. J. Phys. Chem. B 2005, 109, 21489–21491. [Google Scholar] [CrossRef]
- Nakagawa, T.; Bjorge, N.S.; Murray, R.W. Electrogenerated IrOx Nanoparticles as Dissolved Redox Catalysts for Water Oxidation. J. Am. Chem. Soc. 2009, 131, 15578–15579. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Schechter, A.; Feng, L. Iridium-based catalysts for oxygen evolution reaction in acidic media: Mechanism, catalytic promotion effects and recent progress. Nano Res. Energy 2023, 2, e9120056. [Google Scholar] [CrossRef]
- Eid, K.; Soliman, K.A.; Abdulmalik, D.; Mitoraj, D.; Sleim, M.H.; Liedke, M.O.; El-Sayed, H.A.; AlJaber, A.S.; Al-Qaradawi, I.Y.; Oliver Mendoza Reyes, O.M.; et al. Tailored fabrication of iridium nanoparticle sensitized titanium oxynitride nanotubes for solar driven water splitting: Experimental insights on the photocatalytic–activity–defects relationship. Catal. Sci. Technol. 2020, 10, 801–809. [Google Scholar] [CrossRef]
- Ooka, H.; Yamaguchi, A.; Takashima, T.; Hashimoto, K.; Nakamura, R. Efficiency of oxygen evolution on iridium oxide determined from the pH dependence of charge accumulation. J. Phys. Chem. C 2017, 121, 17873–17881. [Google Scholar] [CrossRef]
- Thomsen, J.M.; Huang, D.L.; Crabtree, R.H.; Brudvig, G.W. Iridium-based complexes for water oxidation. Dalton Trans. 2015, 44, 12452–12472. [Google Scholar] [CrossRef]
- Hintermair, U.; Hashmi, S.M.; Elimelech, M.; Crabtree, R.H. Particle formation during oxidation catalysis with Cp* iridium complexes. J. Am. Chem. Soc. 2012, 134, 9785–9795. [Google Scholar] [CrossRef]
- Jovic, V.; Chen, W.-T.; Blackford, M.G.; Idriss, H.; Waterhouse, G.I.N. Effect of Gold Loading and TiO2 Support Composition on the Activity of Au/TiO2 Photocatalysts for H2 Production from Ethanol-Water Mixtures. J. Catalysis 2013, 305, 307–317. [Google Scholar] [CrossRef]
- Fu, X.; Clark, L.A.; Yang, Q.; Anderson, M.A. Enhanced Photocatalytic Performance of Titania-Based Binary Metal Oxides: TiO2/SiO2 and TiO2/ZrO2. Environ. Sci. Technol. 1996, 30, 647–653. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | Area CPS. eV | Sensitivity Factor | Corrected Area | Atomic% | Mol. Wt. | Mol. Wt. × at.% | wt. % | |
|---|---|---|---|---|---|---|---|---|
| 1.96 wt. % Au–1.06 wt. % Pd | Au 4f | 63.80 | 5.24 | 12.18 | 0.33 | 107.86 | 35.53 | 1.64 |
| Pd 3d | 36.60 | 4.60 | 7.96 | 0.22 | 106.42 | 22.91 | 1.06 | |
| Ti 2p | 1318.90 | 1.80 | 733.54 | 19.85 | 47.87 | 950.02 | 43.94 | |
| O 1s | 1304.10 | 0.71 | 1834.18 | 49.63 | 16.00 | 794.03 | 36.72 | |
| C 1s | 328.00 | 0.30 | 1108.11 | 29.98 | 12.00 | 359.78 | 16.64 | |
| S1 | Total | 3695.95 | 100.00 | 2162.27 | 100.00 | |||
| 2.38 wt. % Au–0.65 wt. % Pd | Au 4f | 57.80 | 5.24 | 11.03 | 0.31 | 107.86 | 33.57 | 1.66 |
| Pd 3d | 2.96 | 4.60 | 0.64 | 0.02 | 106.42 | 1.93 | 0.10 | |
| Ti 2p | 1083.30 | 1.80 | 602.50 | 17.00 | 47.87 | 813.70 | 40.16 | |
| O 1s | 1164.80 | 0.71 | 1638.26 | 46.22 | 16.00 | 739.55 | 36.50 | |
| C 1s | 382.40 | 0.30 | 1291.89 | 36.45 | 12.00 | 437.40 | 21.59 | |
| S2 | Total | 3544.32 | 100.00 | 2026.14 | 100.00 | |||
| 1.45 wt. % Au–1.575 wt. % Pd | Au 4f | 13.70 | 5.24 | 2.61 | 0.08 | 196.97 | 15.33 | 0.68 |
| Pd 3d | 67.50 | 4.60 | 14.67 | 0.44 | 106.42 | 46.49 | 2.07 | |
| Ti 2p | 1292.70 | 1.80 | 718.97 | 21.41 | 47.87 | 1024.61 | 45.65 | |
| O 1s | 1318.90 | 0.71 | 1854.99 | 55.23 | 16.00 | 883.64 | 39.37 | |
| C 1s | 227.20 | 0.30 | 767.57 | 22.85 | 12.00 | 274.23 | 12.22 | |
| S3 | Total | 3358.81 | 100.00 | 2244.30 | 100.00 | |||
| 2.54 wt. % Au–0.46 wt. % Pd | Au 4f | 44.60 | 5.24 | 8.51 | 0.24 | 107.86 | 25.74 | 1.27 |
| Pd 3d | 1.80 | 4.60 | 0.39 | 0.01 | 106.42 | 1.17 | 0.06 | |
| Ti 2p | 1102.00 | 1.80 | 612.90 | 17.18 | 47.87 | 822.41 | 40.55 | |
| O 1s | 1193.00 | 0.71 | 1677.92 | 47.04 | 16.00 | 752.58 | 37.10 | |
| C 1s | 375.20 | 0.30 | 1267.57 | 35.53 | 12.00 | 426.40 | 21.02 | |
| S4 | Total | 3567.29 | 100.00 | 2028.29 | 100.00 | |||
| 1.15 wt. % Au–1.86 wt. % Pd | Au 4f | 11.70 | 5.24 | 2.23 | 0.06 | 107.86 | 7.01 | 0.34 |
| Pd 3d | 95.00 | 4.60 | 20.65 | 0.60 | 106.42 | 63.97 | 3.09 | |
| Ti 2p | 1049.20 | 1.80 | 583.54 | 16.99 | 47.87 | 813.03 | 39.33 | |
| O 1s | 1190.50 | 0.71 | 1674.40 | 48.74 | 16.00 | 779.80 | 37.72 | |
| C 1s | 341.80 | 0.30 | 1154.73 | 33.61 | 12.00 | 403.33 | 19.51 | |
| S5 | Total | 3435.55 | 100.00 | 2067.15 | 100.00 | |||
| 2.29 wt. % Au–3.72 wt. % Pd | Au 4f | 21.10 | 5.24 | 4.03 | 0.12 | 107.86 | 13.18 | 0.58 |
| Pd 3d | 155.00 | 4.60 | 33.70 | 1.02 | 106.42 | 108.85 | 4.81 | |
| Ti 2p | 1210.20 | 1.80 | 673.08 | 20.43 | 47.87 | 978.03 | 43.24 | |
| O 1s | 1293.70 | 0.71 | 1819.55 | 55.23 | 16.00 | 883.76 | 39.07 | |
| C 1s | 226.10 | 0.30 | 763.85 | 23.19 | 12.00 | 278.25 | 12.30 | |
| S6 | Total | 3294.20 | 100.00 | 2262.08 | 100.00 | |||
| 0.40 wt. % Au–0.65 wt. % Pd | Au 4f | 2.74 | 5.24 | 0.52 | 0.01 | 107.86 | 1.59 | 0.08 |
| Pd 3d | 1.68 | 4.60 | 0.36 | 0.01 | 106.42 | 1.09 | 0.06 | |
| Ti 2p | 1005.70 | 1.80 | 559.34 | 15.79 | 47.87 | 755.67 | 38.80 | |
| O 1s | 1128.30 | 0.71 | 1586.92 | 44.79 | 16.00 | 716.63 | 36.79 | |
| C 1s | 413.20 | 0.30 | 1395.95 | 39.40 | 12.00 | 472.79 | 24.27 | |
| S7 | Total | 3543.10 | 100.00 | 1947.77 | 100.00 | |||
| 3 wt. % Pd | Au 4f | 5.24 | 0.00 | 0.00 | 107.86 | 0.00 | 0.00 | |
| Pd 3d | 20.40 | 4.60 | 4.43 | 0.12 | 106.42 | 13.13 | 0.59 | |
| Ti 2p | 1453.30 | 1.80 | 808.29 | 22.49 | 47.87 | 1076.44 | 48.03 | |
| O 1s | 1424.50 | 0.71 | 2003.52 | 55.74 | 16.00 | 891.87 | 39.79 | |
| C 1s | 230.30 | 0.30 | 778.04 | 21.65 | 12.00 | 259.76 | 11.59 | |
| S8 | Total | 3594.28 | 100.00 | 2241.20 | 100.00 | |||
| 3 wt. % Au | Au 4f | 64.80 | 5.24 | 12.37 | 0.37 | 107.86 | 40.04 | 1.71 |
| Pd 3d | 4.60 | 0.00 | 0.00 | 106.42 | 0.00 | 0.00 | ||
| Ti 2p | 1459.20 | 1.80 | 811.57 | 24.36 | 47.87 | 1166.01 | 49.83 | |
| O 1s | 1366.00 | 0.71 | 1921.24 | 57.67 | 16.00 | 922.66 | 39.43 | |
| C 1s | 173.60 | 0.30 | 586.49 | 17.60 | 12.00 | 211.24 | 9.03 | |
| S9 | Total | 3331.66 | 100.00 | 2339.94 | 100.00 |
| Concentration of CAN (M) | [Ce4+]/[Au + Pd] | TON | TOF (min−1) | Moles/min |
|---|---|---|---|---|
| 0.023 | 105.31 | 13.02 | 0.07 | 2.89 × 10−7 |
| 0.046 | 210.63 | 21.20 | 0.14 | 6.32 × 10−7 |
| 0.068 | 315.94 | 37.75 | 0.16 | 6.96 × 10−7 |
| 0.091 | 421.26 | 71.77 | 0.17 | 7.32 × 10−7 |
| 0.137 | 631.89 | 107.81 | 0.34 | 1.49 × 10−6 |
| 0.182 | 842.52 | 121.33 | 0.35 | 1.57 × 10−6 |
| Catalyst Weight (g) | TON ([O2]/[Au + Pd]) | TOF (min−1) | Moles/min | Moles/(min/gcatal.) |
|---|---|---|---|---|
| 0.005 | 43.70 | 0.15 | 1.80 × 10−7 | 3.60 × 10−5 |
| 0.01 | 42.15 | 0.21 | 4.87 × 10−7 | 4.87 × 10−5 |
| 0.02 | 42.87 | 0.17 | 8.04 × 10−7 | 4.02 × 10−5 |
| 0.04 | 41.19 | 0.15 | 1.39 × 10−6 | 3.48 × 10−5 |
| Au wt. % | Pd wt. % | Nominal Atomic Ratio of Au/Pd | Moles/min |
|---|---|---|---|
| 1.45 | 1.57 | 1:2 | 1.00 × 10−6 |
| 1.15 | 1.86 | 1:3 | 1.39 × 10−6 |
| 2.29 | 3.72 | 1:3 | 2.30 × 10−6 |
| 1.96 | 1.06 | 1:1 | No reaction |
| 2.38 | 0.65 | 2:1 | No reaction |
| 2.54 | 0.46 | 3:1 | No reaction |
| Concentration of CAN (M) | Ce4+/Ir (Atomic Ratio) | TON | TOF (min−1) | Moles/min |
|---|---|---|---|---|
| 0.023 | 437.7 | 81.08 | 0.57 | 5.98 × 10−7 |
| 0.046 | 875.5 | 170.75 | 1.05 | 1.09 × 10−6 |
| 0.068 | 1313.2 | 243.25 | 1.54 | 1.60 × 10−6 |
| 0.091 | 1750.9 | 411.00 | 2.11 | 2.20 × 10−6 |
| 0.137 | 2626.4 | 629.80 | 4.12 | 4.29 × 10−6 |
| 0.182 | 3501.9 | 847.74 | 5.30 | 5.52 × 10−6 |
| Concentration of CAN (M) | Ce4+/Ir (Atomic Ratio) | TON | TOF (min−1) | Moles/min |
|---|---|---|---|---|
| 0.023 | 437.7 | 93.95 | 0.40 | 4.14 × 10−7 |
| 0.046 | 875.5 | 170.75 | 1.08 | 1.12 × 10−6 |
| 0.068 | 1313.2 | 253.98 | 1.37 | 1.43 × 10−6 |
| 0.091 | 1750.9 | 343.64 | 1.68 | 1.75 × 10−6 |
| 0.137 | 2626.4 | 450.04 | 1.81 | 1.88 × 10−6 |
| 0.182 | 3501.9 | 565.02 | 2.17 | 2.26 × 10−6 |
| Concentration of CAN (M) | Ce4+/Ir (Atomic Ratio) | TON | TOF (min−1) | Moles/min |
|---|---|---|---|---|
| 0.023 | 437.7 | 74.65 | 0.35 | 3.61 × 10−7 |
| 0.046 | 875.5 | 196.49 | 1.11 | 1.16 × 10−6 |
| 0.068 | 1313.2 | 226.52 | 1.19 | 1.24 × 10−6 |
| 0.091 | 1750.9 | 498.52 | 1.39 | 1.45 × 10−6 |
| 0.137 | 2626.4 | 506.67 | 1.49 | 1.55 × 10−6 |
| 0.182 | 3501.9 | 619.50 | 1.60 | 1.67 × 10−6 |
| Catalyst | Remarks |
|---|---|
| 1 wt. % Pt/TiO2 | No activity |
| 3 wt. % Au/TiO2 | No activity |
| 3 wt. % Pd/TiO2 | No activity |
| 1 wt. % Ru/SrTiO3 | No activity |
| 1 wt. % Rh–1 wt. % Ru/SrTiO3 | No activity |
| Fe2O3 | No activity |
| 0.1 wt. % Ag–0.3 wt. % Pd/TiO2 | No activity |
| Catalyst | Reaction Order | BET Surface Area m2/gcatal. | k (Rate Low) L/(min. gcatal.) | k (Rate Law) L/(min. m2) | k (L–H) mol/(min. gcatal.). | k (L–H) mol/(min. m2) | K (g/mol) |
|---|---|---|---|---|---|---|---|
| Au-Pd/TiO2 (A/R) | 0.8 | 40 | 3.07 × 10−4 | 7.68 × 10−6 | 1.7 × 10−4 | 4.25 × 10−6 | 4.12 |
| IrO2/TiO2 Anatase | 1.14 | 40 | 1.69 × 10−3 | 4.23 × 10−5 | 8.4 × 10−3 | 2.11 × 10−4 | 0.15 |
| IrO2/TiO2 (A/R) | 0.77 | 40.5 | 4.81 × 10−4 | 1.19 × 10−5 | 1.4 × 10−3 | 4.02 × 10−6 | 0.71 |
| IrO2/CeO2 | 0.67 | 30.6 | 1.34 × 10−4 | 4.36 × 10−6 | 6.0 × 10−4 | 3.27 × 10−6 | 1.48 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wahab, K.; Idriss, H. Water Oxidation over Au-Pd/TiO2 as a Substitute for Iridium-Based Catalysts. Catalysts 2024, 14, 87. https://doi.org/10.3390/catal14010087
Wahab K, Idriss H. Water Oxidation over Au-Pd/TiO2 as a Substitute for Iridium-Based Catalysts. Catalysts. 2024; 14(1):87. https://doi.org/10.3390/catal14010087
Chicago/Turabian StyleWahab, Khaja, and Hicham Idriss. 2024. "Water Oxidation over Au-Pd/TiO2 as a Substitute for Iridium-Based Catalysts" Catalysts 14, no. 1: 87. https://doi.org/10.3390/catal14010087
APA StyleWahab, K., & Idriss, H. (2024). Water Oxidation over Au-Pd/TiO2 as a Substitute for Iridium-Based Catalysts. Catalysts, 14(1), 87. https://doi.org/10.3390/catal14010087