Abstract
CO2 photoreduction into valuable hydrocarbons, such as CO, CH4, and C2H4, delivers a promising approach to address both environmental and energy challenges. Transition metal chalcogenides, particularly cadmium sulfide (CdS), have emerged as prominent candidates due to their tunable electronic properties and availability. This study delves into a comprehensive investigation of how CdS crystalline facets and sulfur-deficient surfaces modulate the product selectivity. Through employing density functional theory (DFT), we unravel the catalytic performance of various CdS crystal orientations and sulfur vacancy configurations. The results have shown that different CdS facets exhibit unique electronic characteristics and surface energetics, which influence the adsorption dynamics and reaction pathways. The introduction of sulfur vacancies further modulates the nature of active sites, leading to substantial shifts in product selectivity. A detailed investigation on the reaction mechanisms unveils that specific facets preferentially facilitate the formation of CO, while others are more conducive to the generation of hydrocarbons such as CH4 and C2H4, due to the variations in activation barriers and intermediate stabilities. These findings underscore the importance of crystal facet engineering and defect manipulation in tailoring catalyst performance thus providing valuable insights for the rational design of efficient and selective CO2 reduction metal catalysts.
1. Introduction
Given the ever-increasing global energy demand and the escalating environmental impact of fossil fuel consumption, the development of clean energy and carbon-neutral technologies has become a critical area of research. The transition to sustainable energy sources is not merely optional but essential for mitigating the adverse effects of climate change [1]. Among the various strategies explored, the catalytic conversion of carbon dioxide (CO2) into valuable chemical feedstocks has garnered significant attention since this approach could mitigate greenhouse gas (GHG) emissions while simultaneously promoting the production of renewable energy carriers and industrial materials. By capitalizing on the abundance and sustainability of solar power, the photocatalytic conversion of CO2 into valuable hydrocarbons offers a sustainable solution to the dual challenges of energy crisis and environmental pollution [2] and has emerged as a ‘hot’ topic in this research field.
Generally, CO2 photoreduction involves multi-electron reactions, and thus the effectiveness of CO2 photoreduction is primarily constrained by the optical properties, photoelectrochemical characteristics of the catalyst, and the geometric and electronic structures of active sites [3]. More specifically, the optical absorption properties determine the total amount of photogenerated electron–hole pairs during light irradiation, while the charge separation efficiency of these pairs affects their lifetime and the likelihood of electrons participating in the photocatalytic CO2 reduction reactions. In order to tailor the selectivity of reduction products, a rational design of the geometric and electronic structures of active sites is essential [4]. That being said, the feature of active sites is a key factor in the selective yield of required products. For gas–solid phase reactions, the CO2 reduction produced usually includes CO [5], CH4 [6], and C2H4 [7,8]. The CO2 reduction process is comprised of CO2 activation, adsorption, and hydrogeneration of intermediates. Of these, the selectivity is highly dependent on the CO adsorption ability of the active site [4,9]. A weak CO adsorption capacity of the active site promotes CO production, whereas an enhanced CO adsorption ability can significantly boost the potential for CO hydrogenation, ultimately leading to the formation of CH4 and/or C2 products. Notably, the formation of multi-carbon products (C2 and above) is usually associated with a unique active site structure. In such a system, adjacent active sites are capable of simultaneously activating and coupling at least two CO or CHO groups, thereby facilitating C−C bond formation [10,11,12]. For instance, Zhao et al. [13] found that the homonuclear dimer Fe2, anchored on C2N, exhibits significant selectivity for ethanol. Similarly, Duan et al. [14] conducted DFT studies on 21 bimetallic combinations and identified that heteronuclear bimetallic CrCo and MnCo@NG dual-atom catalysts (DACs) preferentially facilitate C−C coupling, leading to ethanol formation. Consequently, the strategic selection of active sites with tailored CO adsorption properties is crucial.
It is commonly acknowledged that different crystal facets of materials exhibit distinct active sites and variations in geometric and electronic structures due to differing atomic arrangements and surface energies. These variations result in unique active site coordination environments for each facet, which in turn influence CO adsorption characteristics and ultimately impact product selectivity in CO2 photocatalytic reduction [15]. Moreover, surface defect states and doping atoms are commonly introduced to enhance charge separation and reduce activation energy, thereby significantly influencing catalytic performance [16]. Therefore, co-investigating the properties of crystal facets and the effects of defects could provide valuable insights for optimizing CO2 reduction product selectivity.
Cadmium sulfide (CdS) serves as a prototypical photocatalyst, with its moderate band gap facilitating effective visible light absorption thus rendering it suitable for solar-driven catalytic processes. Additionally, CdS demonstrates outstanding photoelectrochemical properties, including efficient charge carrier generation and separation efficiency [17]. These attributes underscore its prominence as a promising material in photocatalysis, especially for CO2 reduction. CdS predominantly exists in the following two distinct structural phases: cubic and hexagonal. Extensive studies have focused on the (001) facets of hexagonal CdS [18,19,20,21]. For instance, Yuan et al. [22] synthesized hexagonal CdS with varying concentrations of S vacancies, leveraging electron diffraction analysis (EDA) to visible light-driven photocatalytic syngas formation rates of 115.6 and 959.4 μmol g−1 h−1 for CO and H2, respectively. DFT studies revealed that the (001) facet with S vacancies presents excellent CO2 reduction activity. Xiang et al. [18] introduced Cd vacancies into hexagonal CdS and incorporated Cu single atoms (SACs) and clusters, leading to a substantial enhancement in the selectivity of photocatalytic CO2 reduction to CO. Similarly, Zhou et al. [19] embedded Au atoms into defect-laden CdS (001) facets, implying that the redistribution of electron density significantly facilitated CO2 conversion. In contrast to systems with solely sulfur vacancies, which predominantly yielded CO, the introduction of Au SACs remarkably increased CO production and the formation of CH4. To the best of our knowledge, the majority of studies on cubic CdS have predominantly investigated the (111) facet, with carbon monoxide (CO) being the principal reduction product, akin to the (001) facet of hexagonal CdS [23,24,25,26]. However, our previous work [27] demonstrated the (110) facet of cubic CdS and suggested that the generation of S vacancies and introduction of ZIF-8 could result in a local Cd-Cd bimetallic structure on the surface capable of C2H4 formation. Although extensive efforts have been devoted to the photocatalytic reduction of CO2 using CdS, investigations have predominantly focused on specific crystal facets, and a comprehensive understanding of the mechanisms governing CO2 reduction product selectivity across different CdS facets remains insufficient. In addition to crystal facets, surface defects play a crucial role in modulating the catalytic performance of CdS. These defects could not only alter the geometric and electronic structures but also act as active sites for the adsorption and activation of CO2 molecules. Despite the recognized importance of sulfur vacancies, the interactions between sulfur vacancies and intact crystal facets during key stages of the CO2 reduction process, such as the adsorption of COOH and CO intermediates and the potential for C−C coupling, have not been comprehensively explored.
Additionally, in recent studies, density functional theory (DFT) calculations are frequently used to simulate complex reaction mechanisms, especially in the photocatalytic CO2 reduction reaction [22,28,29]. DFT calculations are often employed to obtain information such as the adsorption free energy of reaction intermediates, C−C coupling barriers, and surface charge distribution patterns, with the aim of predicting catalyst performance and understanding the mechanism of CO2 conversion processes on catalyst surfaces. Therefore, using DFT calculations to explore the effects of different crystal facets of CdS and S-vacancy modifications on CO2 reduction reactions is highly appropriate.
In order to address the gaps in the previous research, this study employs first-principles calculations to systematically investigate the photocatalytic CO2 reduction performance, product selectivity, and reaction mechanisms across different crystal facets of various CdS configurations. Additionally, we will explore the role of sulfur vacancies as dynamic catalytic sites and their interactions with the surrounding crystal structure throughout the photocatalytic process. This approach aims to elucidate the fundamental mechanisms governing CO2 reduction selectivity and efficiency on CdS, and to propose potential strategies for optimizing its performance.
2. Results and Discussion
2.1. Stability Analysis of Different Facet Configurations and Sulfur Defects
2.1.1. Stability of Different Crystalline Facets
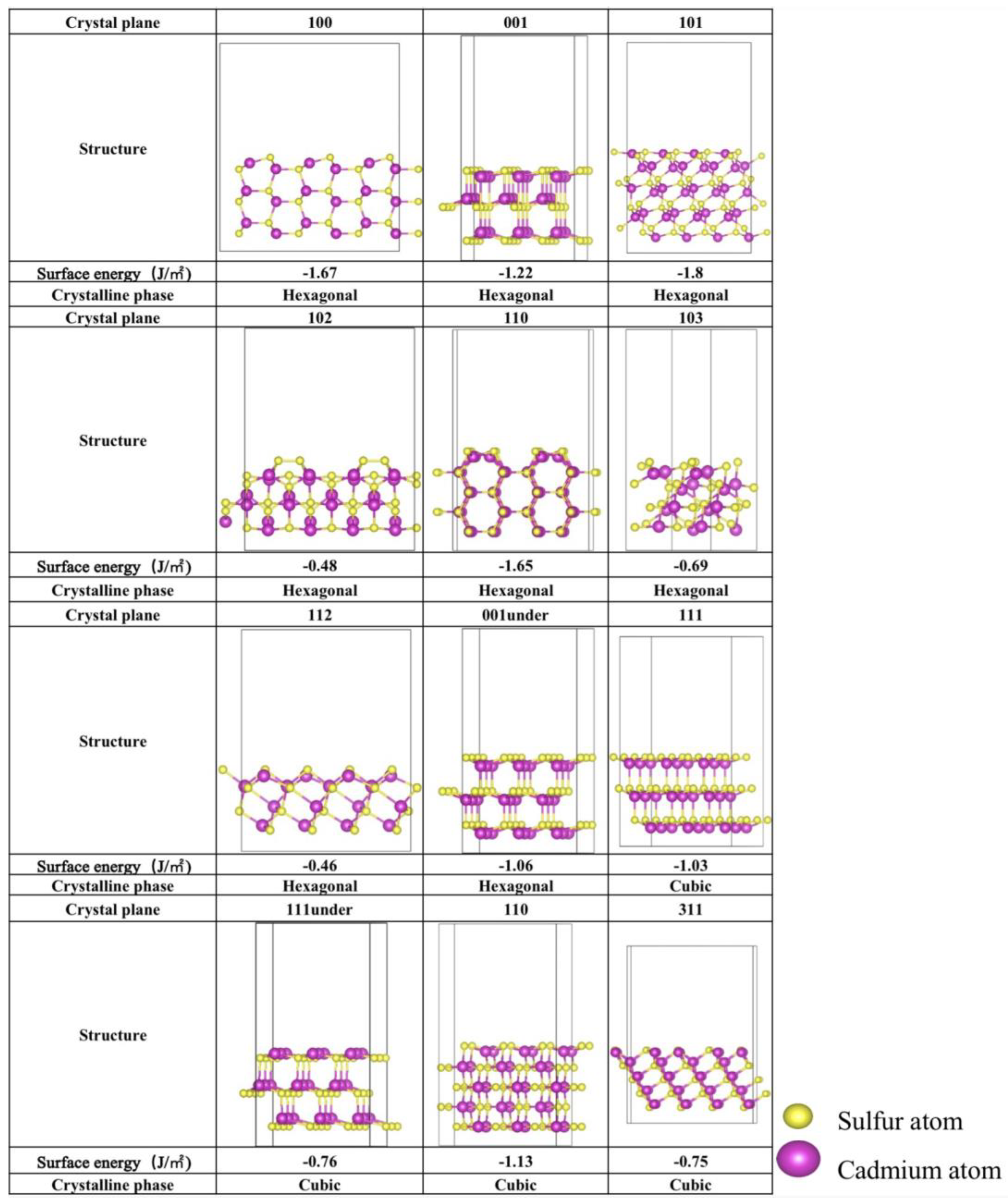
The stability of various crystal planes is closely tied to their surface energies. To quantify this stability, the surface energy of each crystalline facet was calculated and is presented in Figure 1. Both S-top and Cd-top surfaces were considered, with the configurations exhibiting the lowest surface energies also shown in Figure 2. As illustrated, all surface energies are negative, indicating that the crystalline facets are stable and experimentally feasible, consistent with XRD observations reported in previous studies [19,22,25,26,30]. In the hexagonal phase, the (101) and (100) planes show the lowest surface energies of −1.80 J/m2 and −1.65 J/m2, respectively. On the other hand, the (110) and (111) planes of cubic CdS have the lowest surface energies of −1.13 J/m2 and −1.03 J/m2, respectively, suggesting that these planes are more stable. Additionally, the surface structures of (100) and (001) from hexagonal CdS and (111), (111)-under, and (110) from cubic CdS align well with those most studied in prior research [19,22,25,26,30], validating the reliability of this study. Moreover, the (001) and (001)-under surfaces of the hexagonal CdS are similar to the (111)-under and (111) surfaces of cubic CdS, respectively, which may lead to similar performance. Consequently, the photocatalytic CO2 reduction performance of all the selected crystalline facets will be explored.
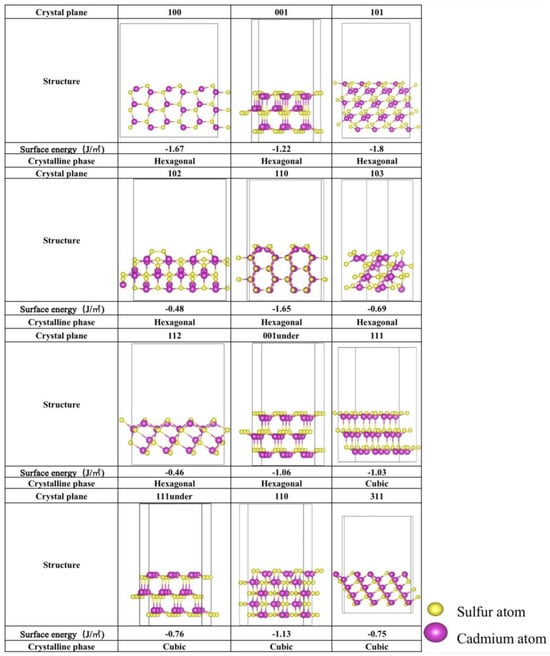
Figure 1.
The optimized crystalline facet configurations of hexagonal and cubic CdS. The corresponding surface energies γ (J/m2) are marked below the structure.
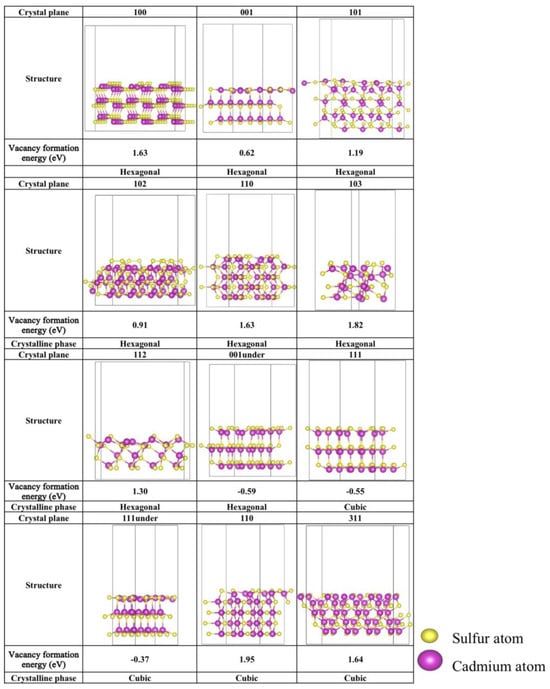
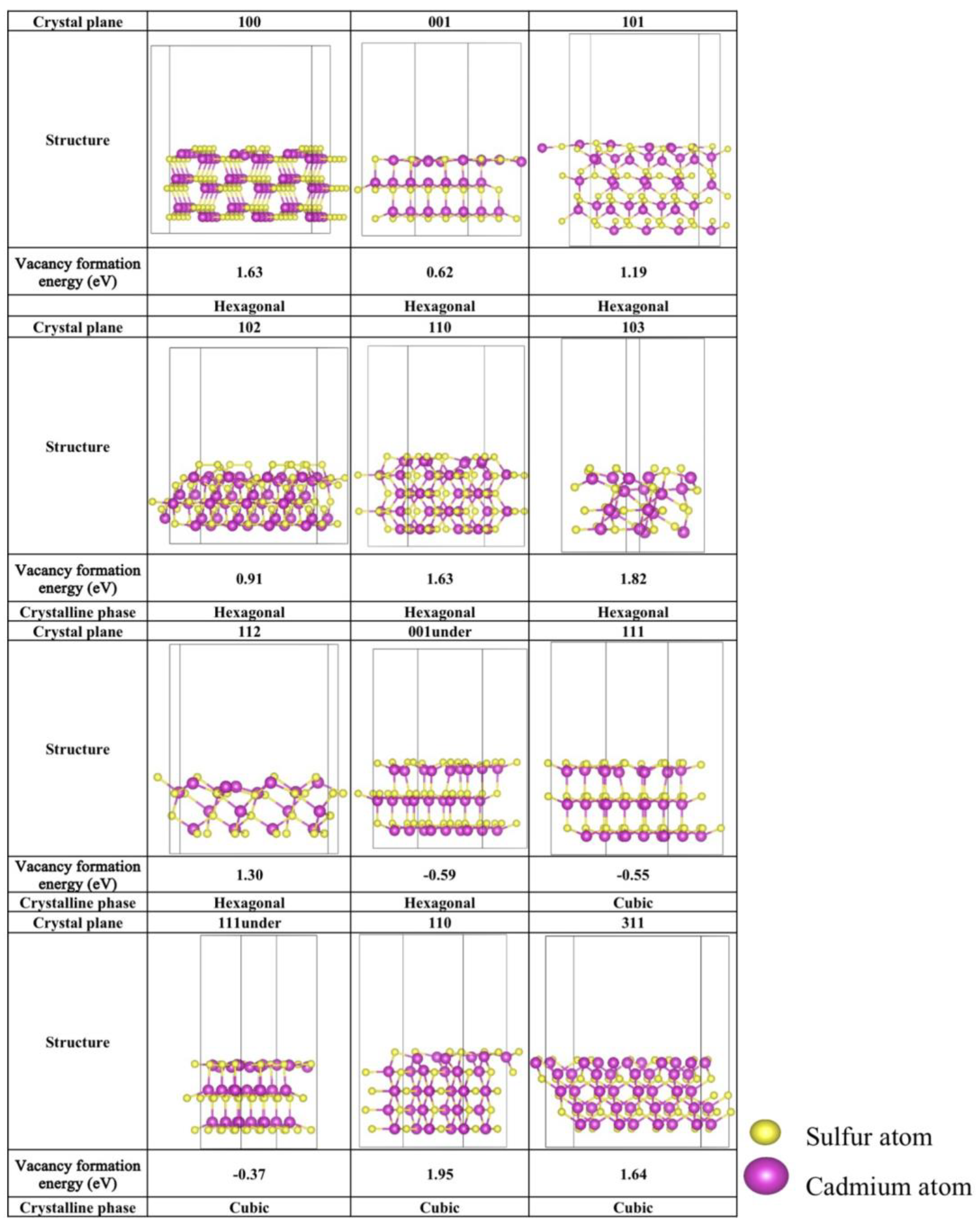
Figure 2.
The optimized S-vacancy crystalline facets configurations of hexagonal and cubic CdS. The corresponding S-vacancy formation energies (eV) are marked below the structure.
2.1.2. Stability of S-Vacancy in Various Crystalline Facets
To elucidate the stability of the S-vacancy crystalline facets, S-vacancy formation energies were first calculated. These energies, along with the optimized configurations of the S-vacancy crystalline facets, are summarized in Figure 2. Lower vacancy formation energies indicate a greater propensity for defect formation, whereas higher formation energies suggest higher stability against the formation of defects. For hexagonal CdS crystal planes, the (001)-under plane exhibits the lowest defect formation energy at −0.59 eV, followed by the (001) and (102) planes, which show relatively low formation energies of 0.62 eV and 0.91 eV, respectively. In the cubic phase, the (111) and (111)-under planes demonstrate comparatively stable surface structures, with defect formation energies of −0.55 eV and −0.37 eV, respectively. The (110) plane exhibits a defect formation energy of 0.92 eV. In contrast, other crystal planes display defect formation energies exceeding 1.0 eV, suggesting a diminished propensity for S-vacancy formation. This result elucidates the rationale behind the focus of previous studies on these particular planes. Therefore, for the upcoming photocatalytic CO2 reduction studies, the highlight would be placed on the (001) and (102) planes with S-vacancy of the hexagonal phase, as well as the (111), (111)-under, and (110) planes with S-vacancy of the cubic phase.
2.2. Analysis of the CO2 Reduction Reaction Pathways to CO, CH4, and C2H4
Generally, CO, CH4, and C2H4 are typically the predominant CO2 photoreduction products for gas–solid phase reactions. Initially, CO is formed via the following pathway: CO2 → COOH → CO. Methane production involves the further hydrogenation of CO through the following sequence of steps: CO → CHO/COH → … → CH4. The C2H4 formation necessitates a crucial C−C coupling step. Hence, this section first examines the formation process of CO and the free energy changes of its intermediates, followed by the investigation of CH4 and C2H4.
2.2.1. Reaction Pathway of CO2 Reduction to CO
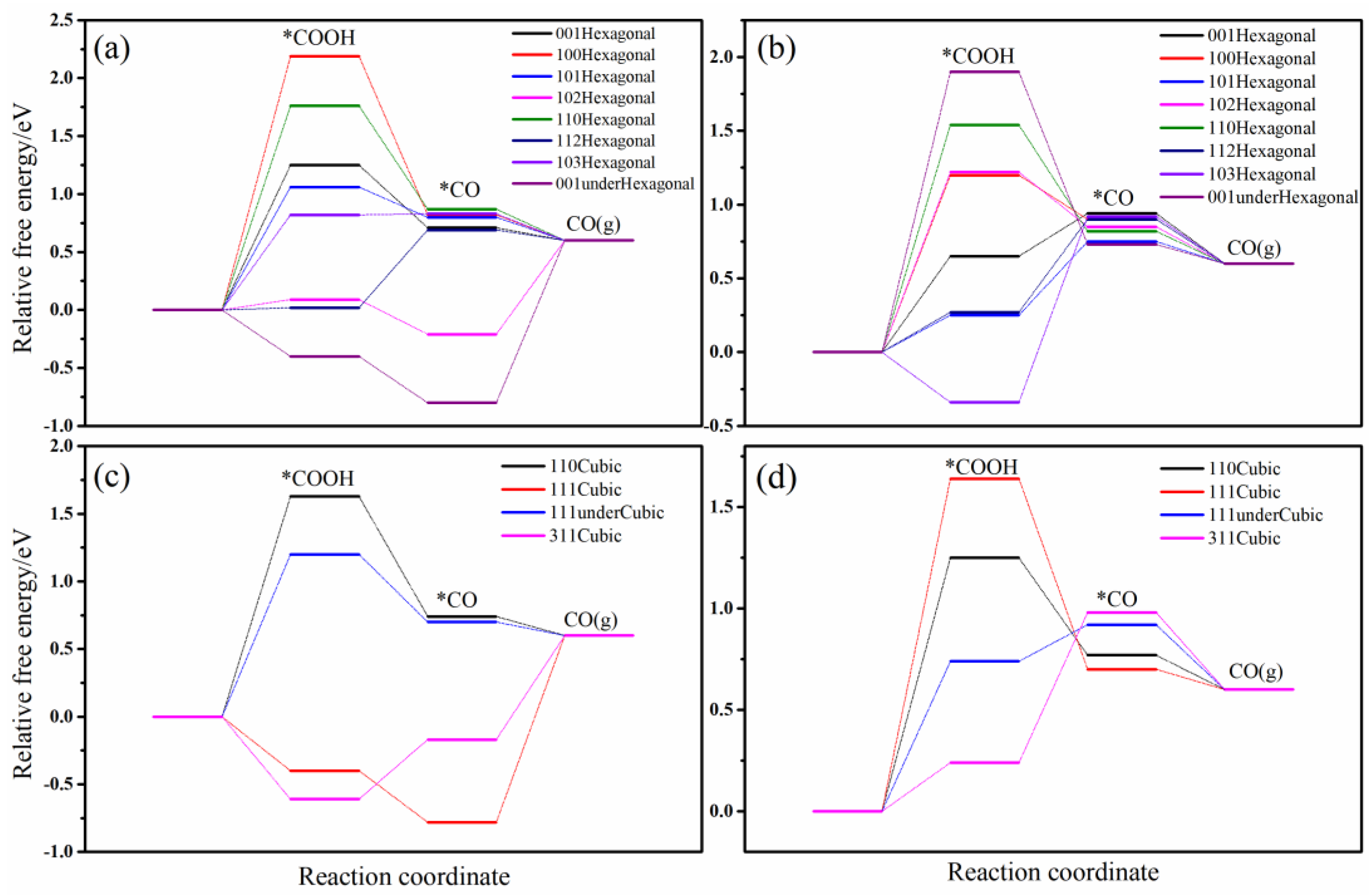
Figure 3 depicts the variations in free energy associated with the conversion of CO2 to CO across various crystallographic planes and configurations, both with and without sulfur vacancies. As presented in Figure 3a, the pristine hexagonal CdS surfaces exhibit considerable variability in the free energy barriers for CO2 activation to form COOH. Specifically, the (100), (110), (001), and (101) surfaces present high energy barriers exceeding 1.0 eV. The (103) surface shows a marginally lower barrier of 0.76 eV, while the (102) and (112) surfaces exhibit barriers near 0 eV, and the (001)-under surface even displays a negative barrier, signifying enhanced activity for CO2 hydrogenation to COOH. Upon conversion to CO on (001)-under, the adsorption free energy of CO is −0.79 eV, indicating a substantial detachment barrier. Consequently, the (102), (103), and (112) surfaces demonstrate superior overall activity for the CO2−to−CO reaction, with rate-determining steps being CO desorption, CO2 → COOH, and COOH → CO, and the corresponding energy barriers of 0.85, 0.76, and 0.73 eV, respectively. The lower CO adsorption energies (<0 eV) on the (102) and (111)-under surfaces suggest prolonged CO retention, which may facilitate further hydrogenation to methane or ethylene. Following the introduction of sulfur vacancies, the (103), (001), (112), and (101) surfaces show improved CO2 activation to COOH, with the free energy barriers of −0.32, 0.22, 0.26, and 0.62 eV, respectively. In contrast, the (001)-under, (110), (102), and (100) surfaces exhibit barriers exceeding 1 eV, reflecting reduced activity. Overall, the most active surfaces for CO2−to−CO conversion are the (001), (112), and (101) surfaces, with rate-determining steps of CO2 → COOH and COOH → CO. The significant COOH adsorption on the (103) surface results in a notable barrier for CO formation. Additionally, sulfur vacancies generally elevate CO adsorption energies on hexagonal CdS surfaces, promoting CO escape and thereby enhancing CO selectivity. In summary, sulfur vacancies enhance CO2−to−CO activity on the (001), (100), (101), (110), and (112) surfaces, underscoring the beneficial role of sulfur vacancies in CO production during CO2 reduction. Most pristine hexagonal CdS surfaces and all surfaces with sulfur vacancies predominantly produce CO from CO2 reduction, with the exception of the (102) and (001)-under surfaces, which exhibit the potential for alternative selectivity, consistent with the experimental observations that hexagonal CdS primarily yields CO [19,25].
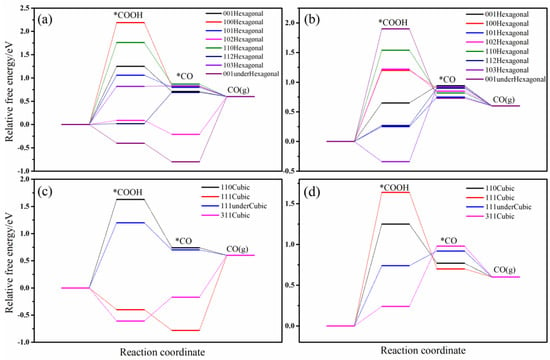
Figure 3.
The relative free energy images of the intermediates in CO2−to−CO process on various crystal planes. (a) Hexagonal surface, (b) Hexagonal surface with S-vacancy, (c) Cubic surface, (d) Cubic surface with S-vacancy. * represents adsorption state of reactive intermediates.
In the case of pristine cubic CdS surfaces, the (111) and (311) planes exhibit significant activity for CO2 activation to COOH, with free energy barriers of −0.42 and −0.60 eV, respectively. In contrast, the (111)-under and (110) surfaces show lower activity, characterized by energy barriers exceeding 1.2 eV. Among the surfaces examined, the rate-determining step for the (311) surface is CO desorption, which presents the lowest total energy barrier of 0.80 eV. Conversely, the (111) surface displays such a low CO adsorption energy that CO desorption is impeded. Notably, both the (311) and (111) surfaces exhibit potential for further hydrogenation based on their CO adsorption free energies. Comparing with other works, for example, Zhang et al. [25], Wu et al. [24], Li et al. [26], and Peng et al. [23] constructed (111)-under surface of cubic CdS, the energy barriers for CO2−to−CO are 0.84, 1.77, 2.34, and 0.80 eV, respectively, which are accordant with our result of 1.22 eV, further validating our calculation model and method.
Upon introducing sulfur vacancies, there is a significant reduction in the COOH adsorption free energies on the (111)-under and (110) surfaces, decreasing from 1.22 and 1.72 eV to 0.72 and 1.24 eV, respectively, thereby markedly enhancing their activity. However, the presence of sulfur vacancies on the (111) surface results in a COOH adsorption free energy exceeding 1.6 eV, indicating weakened activity. Although there is a slight increase in COOH adsorption energy on the (311) surface, its overall CO production activity remains relatively stable.
To summarize, the cubic (311) surface, the (111)-under surface, and hexagonal (001) surface with sulfur vacancies exhibit the highest CO2−to−CO activity among cubic CdS surfaces. The presence of sulfur vacancies also significantly enhances activity on the (110) surface. Moreover, the introduction of sulfur vacancies generally leads to elevated CO adsorption energies, indicating that CO remains the primary product of CO2 reduction, in alignment with prevailing experimental findings. Nevertheless, the exceptionally low CO adsorption energies observed on the pristine (311) and (111) surfaces suggest a potential for further hydrogenation, thereby identifying these surfaces as promising candidates for CO−to−CH4/C2H4 conversion. Additionally, the performance of hexagonal (001) with or without S-vacancy is similar to cubic (111)-under surface. Cubic (111) surface and hexagonal (001)-under show the same phenomenon.
2.2.2. Reaction Pathway of CO2 Reduction to CH4
From the perspective of CO adsorption free energy, the hexagonal (102) and (001)-under surfaces, as well as the cubic (311) and (111) surfaces, exhibit low CO adsorption free energies, below 0 eV. This result suggests that these surfaces have substantial potential for the deep hydrogenation of CO to selectively produce CH4. Recent experimental studies have demonstrated that the (110) surface of CuI with the F-43 m space group can produce C2 chemicals [31], and the sulfur vacancy (110) surface of cubic CdS with the same space group can also produce ethylene [27]. The CH4 selectivity of the sulfur vacancy (110) surface warrants further investigation.
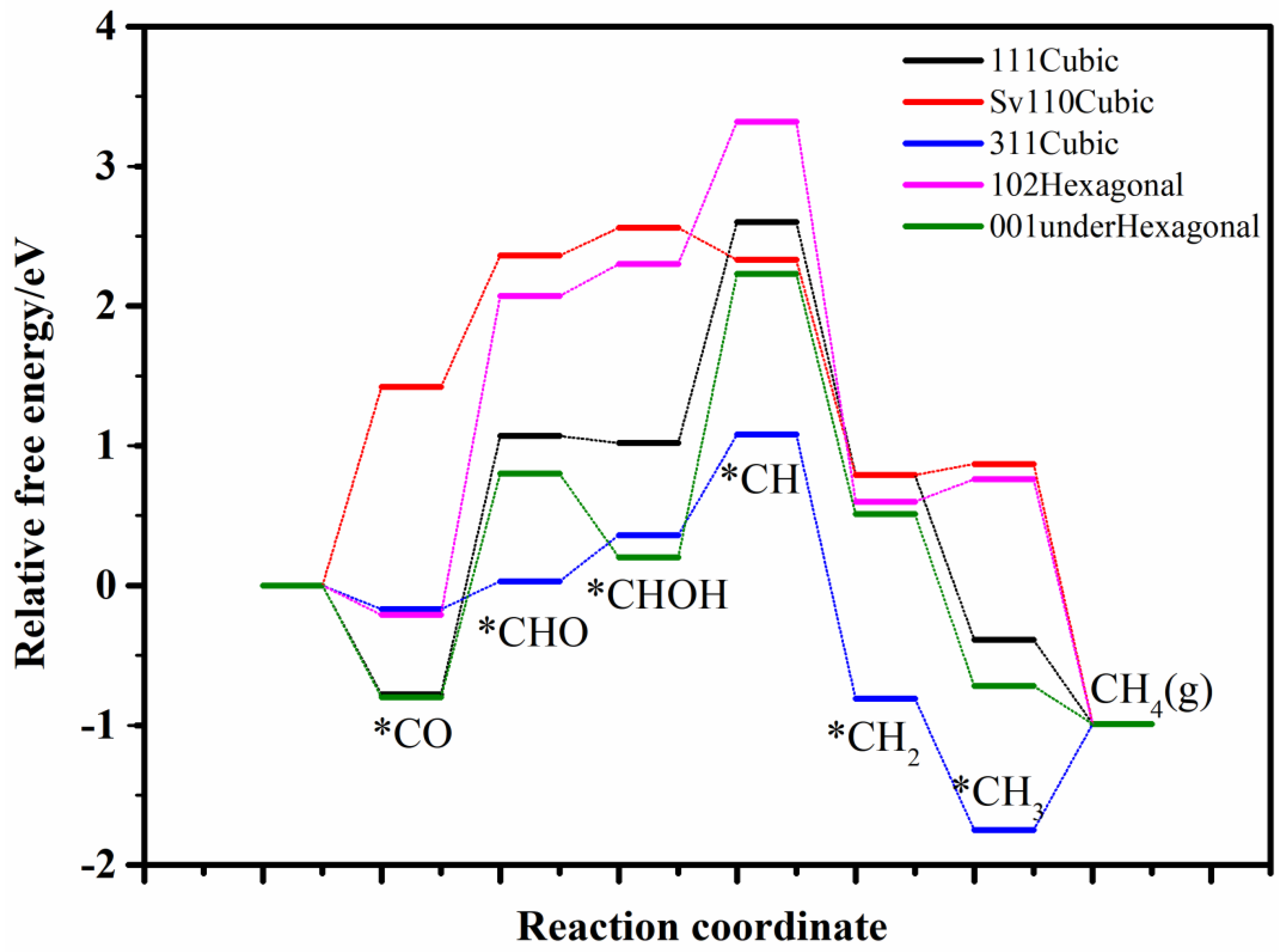
Given that, the potential for CO2 reduction to CH4 across these five surfaces was systematically explored, with the free energy profiles of intermediates in the CO−to−CH4 reaction illustrated in Figure 4. As illustrated, each surface exhibits a distinct rate-limiting step in the whole reaction process. For the cubic CdS (111) and hexagonal (001)-under surfaces, the rate-determining step is the conversion from CHOH to CH, with free energy barriers of 1.52 and 1.95 eV, respectively. On the cubic CdS (110) and hexagonal (102) surfaces, the rate-limiting step occurs at the CO to CHO transition, with barriers of 1.64 eV and 2.34 eV, respectively. For the cubic (311) surface, the rate-determining step involves the conversion from *CH3 to CH4 (g), with a free energy barrier of 1.40 eV.
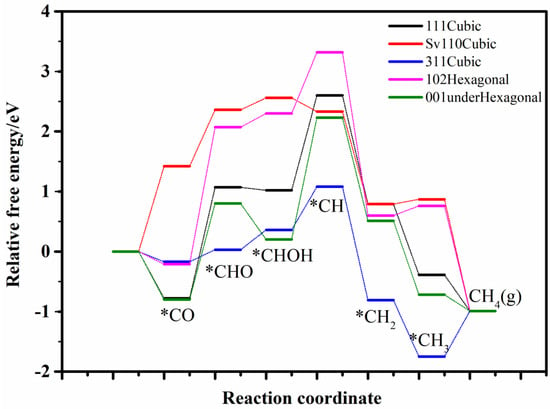
Figure 4.
The relative free energy profile of the intermediates in CO−to−CH4 process on selected various crystal planes. * represents adsorption state of reactive intermediates.
It could be concluded that all surfaces evaluated for CH4 production face significant free energy barriers at their respective rate-determining steps, resulting in low methane selectivity. This finding is consistent with experimental observations, which report minimal methane production [18,19,20,21,23,24,25,26].
2.2.3. Reaction Pathway of CO2 Reduction to C2H4
Previous studies [10,11,12] have demonstrated that the selective formation of C2 products is normally linked to the local structure of active sites, with bimetallic atomic arrangements showing promise for C–C coupling. Among the optimized perfect and S-vacancy surfaces of hexagonal and cubic CdS, only the (311) surface exhibits both significant CO adsorption capabilities and the presence of a local bimetallic atomic structure. In contrast, the cubic S-vacancy CdS (110) surface also features a local bimetallic structure, its CO adsorption is less robust. The introduction of S vacancies on the hexagonal (001) surface, which features a tri-cadmium atomic arrangement, indicates the potential for C−C coupling. However, there is no research work demonstrating the ethylene production from this surface.
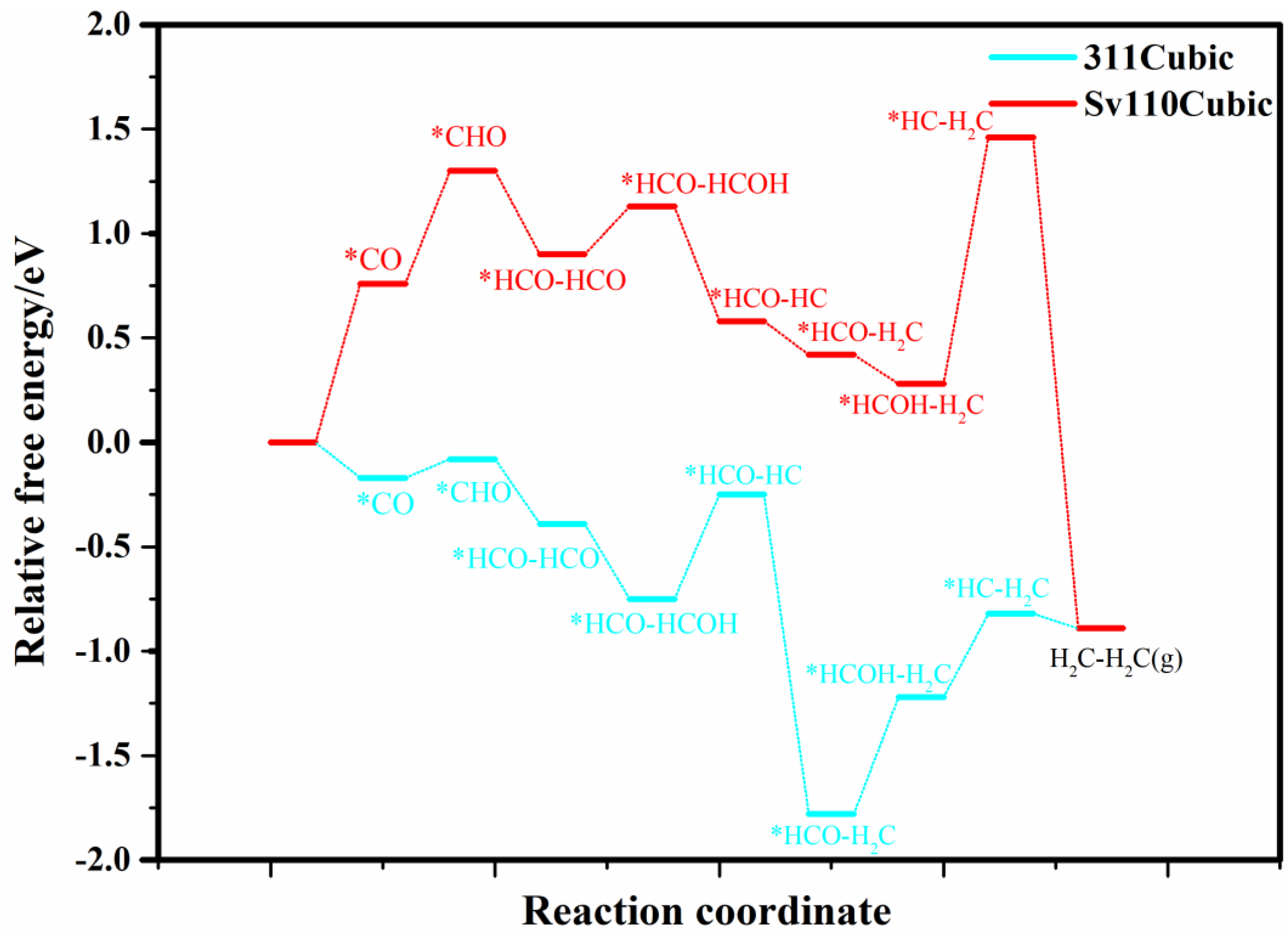
Therefore, the following section focuses on the cubic (311) perfect surface and the S-vacancy (110) surface. The relative free energies of intermediates for CO−to−C2H4 conversion are presented in Figure 5. As illustrated, both the (311) and S-vacancy (110) surfaces demonstrate potential for ethylene production. For the (311) surface, the rate-limiting step is HCO−HCOH to HCO−HC, with a free energy barrier of 0.55 eV. For the S-vacancy (110) surface, the rate-limiting step is CO to CHO, with a barrier of 0.65 eV. It indicates that both the (311) and S-vacancy (110) surfaces of cubic CdS have the potential for C−C coupling and selective C2H4 production.
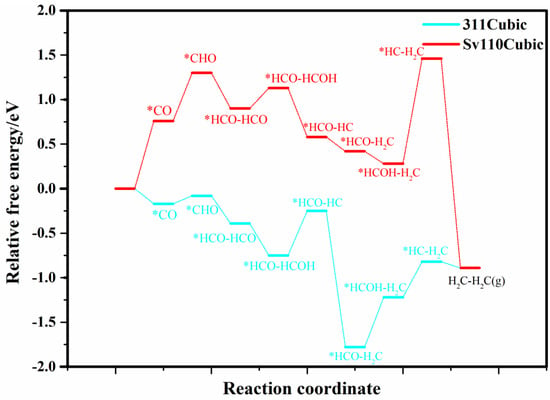
Figure 5.
The relative free energy profile of the intermediates in the CO−to−C2H4 process on selected cubic (311) and S-vacancy (110) planes. * represents adsorption state of reactive intermediates.
2.3. Theoretical Analysis of CO Adsorption and C−C Coupling Mechanism
2.3.1. Investigation of CO Adsorption against Different Active Surface
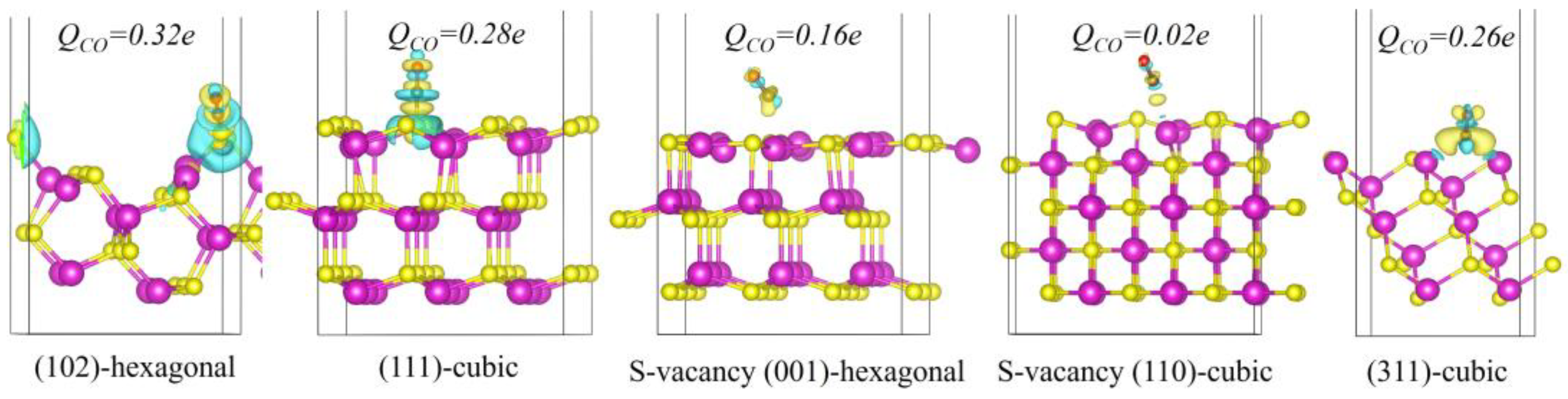
The adsorption properties of CO play a pivotal role in determining the selectivity of CO−reduction products. To elucidate the mechanisms governing the selectivity of CO2 reduction on hexagonal and cubic phases, we systematically investigated the adsorption characteristics of CO on several prominent crystal facets, namely the hexagonal phase S-vacancy (001), the hexagonal phase (102) with lower CO adsorption free energy, the cubic phase (311), the cubic phase (111), and the cubic phase CdS (110) with S-vacancy that exhibits potential for C−C coupling. Figure 6 presents the CO adsorption structures and charge density difference maps for these facets. The results imply that facets with higher CO adsorption free energies, such as the hexagonal phase (102) and the cubic phase (111), show CO is absorbed via top-site interactions with S atoms. It is evidenced by substantial charge transfer in the electron density difference maps. Bader charge analysis further reveals that the number of charge transfers for these facets are 0.32 and 0.28 e, respectively, highlighting the significant confinement effect of surface sulfur atoms on CO.
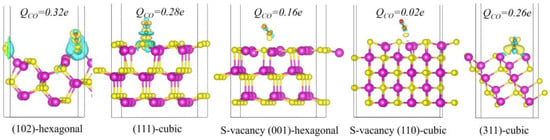
Figure 6.
The electron density difference of CO adsorbed on (102)—hexagonal, (111)—cubic, S-vacancy (001)—hexagonal, S-vacancy (110)—cubic and (311)—cubic surfaces. The transferred bader charge is marked above the CO adsorption structures. The isosurface level is set at 0.003 eV/Å. The purple and yellow spheres represent cadmium atom and sulfur atom, respectively.
In contrast, the hexagonal phase S-vacancy (001) and cubic phase S-vacancy (110) exhibit high CO adsorption free energy and weaker adsorption interactions. On these facets, CO also adsorbs in a top-site manner over Cd atoms. The electron density difference maps for these facets indicate a decrease in electron density between the surface Cd atoms and CO, indicating limited charge transfer. Bader charge calculations for these facets yield values of 0.16 eV and 0.02 eV, respectively, suggesting that the interactions between CO and the surface Cd atoms are predominantly governed by weak van der Waals forces. This behavior is attributed to the inert nature of Cd atoms, which is due to their filled d orbitals. On the cubic phase (311), the electron density is slightly higher compared to the S-vacancy (110) facet, and CO adsorbs via a bridge–site interaction between two Cd atoms. Quantitative Bader charge analysis for the (311) facet shows a charge transfer of 0.26 eV, indicating that the two surface Cd atoms work synergistically to adsorb the CO molecule. Differing from the two Cd atoms on the S-vacancy (110) facet, the Cd atoms on the cubic phase (311) are positioned closer together, which enhances CO adsorption and facilitates further hydrogenation of CO on the (311) facets.
2.3.2. Investigation of C–C Coupling Mechanism against Different Active Surfaces
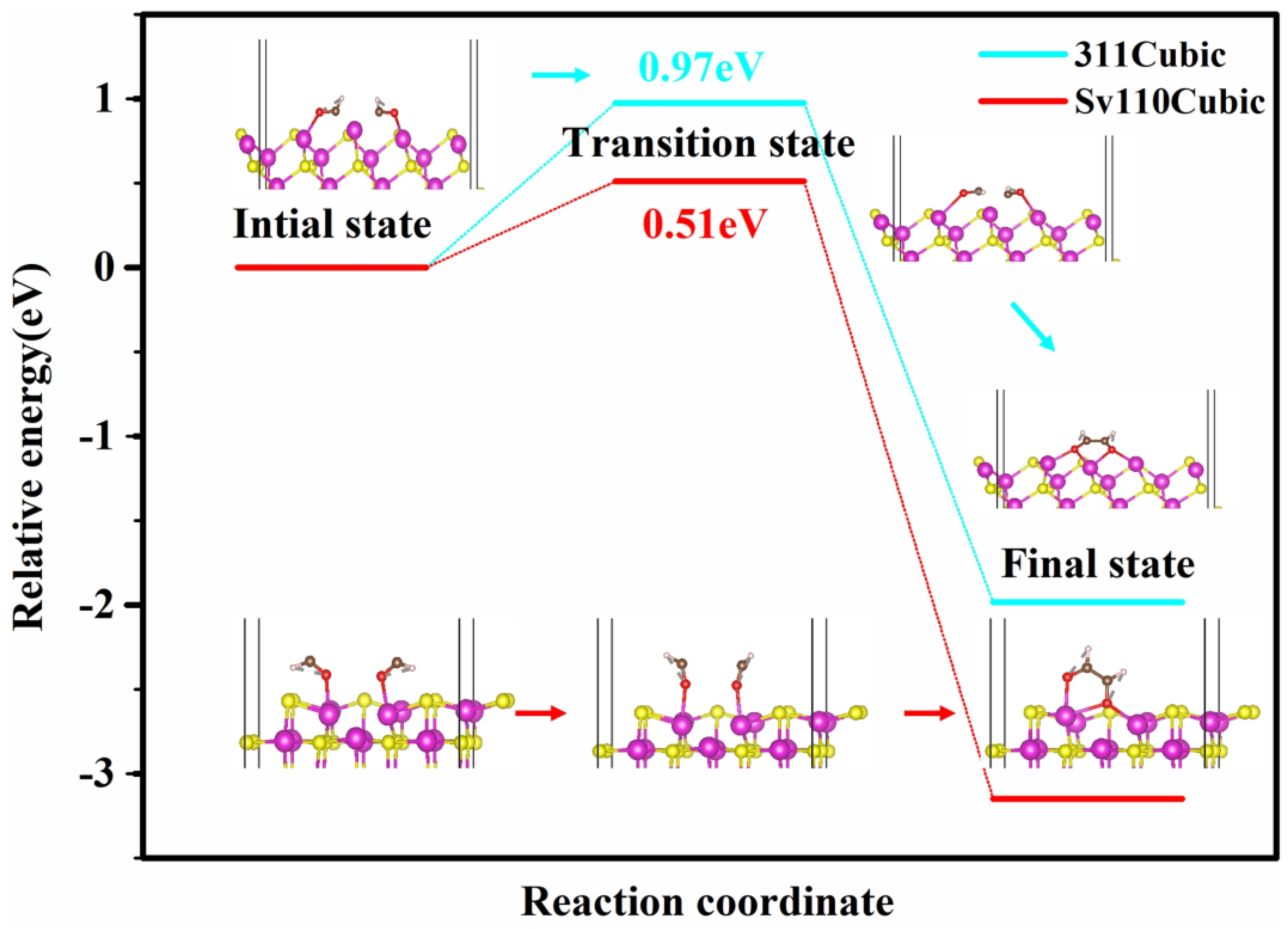
The C−C coupling barrier serves as a crucial indicator of the formation of multi-carbon products during CO2 reduction. To elucidate the C−C coupling mechanisms on the cubic phase (311) and S-vacancy (110) crystal facets, we employed the CI−NEB method to determine the transition states between the initial and final states of C−C coupling. The results are presented in Figure 7. The energy barriers for CHO–CHO coupling reactions on the cubic phase (311) and S-vacancy (110) crystal facets are 0.97 eV and 0.49 eV, respectively. On the (311) facet, Cd atoms are arranged linearly with a Cd–Cd distance of 3.74 Å. As demonstrated in Figure 6, the low-coordinated Cd atom (with only two S atom-coordination) would lead to CO–bridge adsorption mode, thereby facilitating C–C coupling within the localized structure formed by three Cd atoms. In contrast, on the S-vacancy (110) surface, C–C coupling reactions are facilitated by two Cd atoms adjacent to the S-vacancy. The CO adsorption characteristics reveal that a single Cd atom is insufficient to effectively fix CO, leading to an inadequate amount of CO for C–C coupling. This explains why the S-vacancy (110) surface exhibits a lower C–C coupling energy barrier but a lower yield of multi-carbon products. To address this challenge, Peng et al. [27] proposed the addition of organic ligands on the surface to restrict CO dissociation, thereby enhancing C2H4 selectivity. Alternative strategies, such as the introduction of heteroatoms to synergistically adsorb CO, may also prove effective. Given that CO adsorption on the current S-vacancy CdS (110) surface occurs in a top-site manner, introducing heteroatoms could promote a co-bridge adsorption mode for CO, thereby enhancing CO-surface interactions, reducing the activation energy barrier for CO hydrogenation, and consequently providing a greater feedstock for C−C coupling.
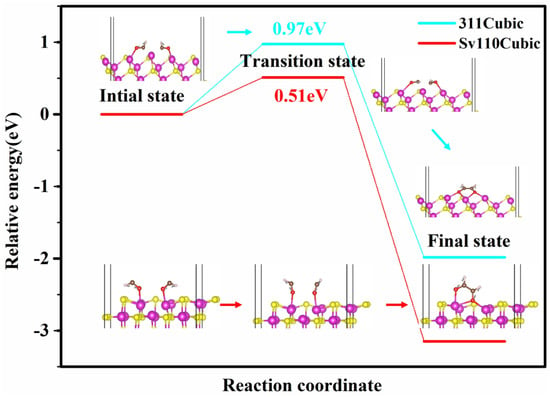
Figure 7.
The relative free energy profile of the intermediates in CO−to−C2H4 process on selected cubic (311) and S-vacancy (110) planes. The purple spheres, yellow spheres, red spheres, brown spheres and white spheres represent cadmium, sulfur, oxygen, carbon and hydrogen atoms, respectively.
3. Materials and Methods
Density functional theory (DFT) was employed through the Vienna Ab initio Simulation Package (VASP 5.4.4) to investigate the influence of crystalline facets and sulfur defects in cadmium sulfide (CdS) during the photocatalytic CO2 reduction process. The PBE exchange–correlation functional in the generalized gradient approximation (GGA) [32,33] with Grimme’s DFT−D3 dispersion correction [34] was used to better describe the weak interactions within the system. The electronic structure calculations were conducted based on the plane–wave method [35], with an energy cutoff set above 400 eV, and Brillouin zone integrations were performed using a Gamma-centered k-point mesh. For geometric structure optimization, the force convergence criterion was set at 0.02 eV/Å, and the energy convergence criterion at 10−5 eV, ensuring the accuracy of the computations. The CI−NEB method [36] was employed to explore key reaction pathways(C–C coupling) and transition states to determine reaction barriers. The electronic structure was analyzed using charge density difference and Bader charge analysis to elucidate the electronic characteristics and charge transfer behaviors associated with the facets and defects during the reaction process.
Surface energy is the energy necessary to create a surface from a bulk crystal and is defined as follows:
In this equation, and are the energies of the relaxed and unrelaxed surfaces, respectively. is the number of atoms in the slab, and represents the bulk energy per atom, which is calculated by dividing the total energy of the bulk structure by the number of atoms within that structure. The in the formula refers to the surface area of the slab layer used in the calculation. When calculating the surface area, the first step is to determine the basis vectors parallel to the surface of the slab, which are assumed to be and , The surface area of slab is usually the area of the parallelogram enclosed by these two basis vectors and , which can be calculated by vector cross product:
By viewing the structural file, and can be expressed in coordinate form and Therefore, the surface can be calculated using the following formula:
The formation energy of S-vacancy in each crystal plane was calculated by the following formula:
Here, and represent the relaxed S-vacancy and perfect surface without S-vacancy, respectively. is the energy of one S atom in its bulk structure.
The computational hydrogen electrode (CHE) model, as introduced by Nørskov et al. [37,38], is employed to determine the Gibbs free energy changes for each step of the CO2 reduction reactions. In this model, the electrode potential, U, is measured against the reversible hydrogen electrode (RHE). At U = 0 V, the change in Gibbs free energy, , is given by the following equation:
Here, represents the energy difference between the products and reactants at the active sites and is directly calculated using DFT. The terms , , , and are corrections for zero-point energy, entropy, corrections for pH, and enthalpy at a temperature of 298.15 K, respectively, all derived from vibrational frequency data. The relative free energy of each intermediate CxHyOz in the whole CO2 reduction process is determined through the following equation:
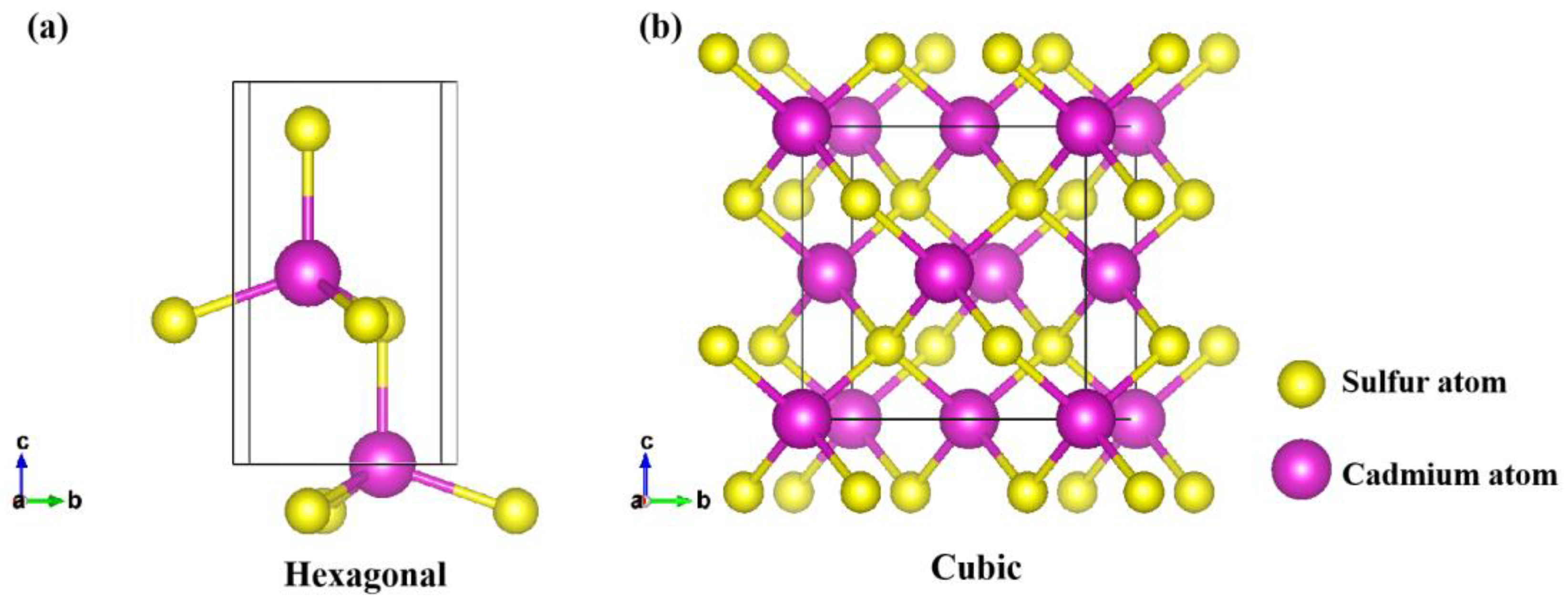
In addition, all the crystal planes investigated were cleaved from bulk cubic and hexagonal CdS, as shown in Figure 8. The planes (001), (101), (100), (110), (102), (103), (112), and (001)-under were derived from hexagonal CdS, while (111), (110), (311), and (111)-under were cleaved from bulk cubic CdS. Each slab supercell was constructed with a specific number of unit cells, depending on the surface size. To prevent unphysical interactions between periodic images, vacuum layers of 15 Å were included above each plane. The S-vacancy surfaces were created by removing a sulfur atom from the corresponding surfaces.
Figure 8.
The bulk structures of (a) hexagonal and (b) cubic CdS.
4. Conclusions
This study provides a detailed theoretical study of how different CdS crystalline facets and sulfur-deficient surfaces influence CO2 photoreduction selectivity. Through DFT calculation, we examined the catalytic performance of various CdS crystal orientations and sulfur vacancy configurations. The findings reveal that hexagonal (001) and cubic (111) facets exhibit similar performance and stability, while sulfur vacancies induce instability in certain facets. The hexagonal (001) and (102) planes, along with cubic (111), (111)-under, and (110) planes, show better stability. Sulfur vacancies generally increase CO adsorption free energy, enhancing CO selectivity, with the highest CO2−to−CO activity over cubic (311) and hexagonal (001) facets. In terms of CO−to−CH4 conversion, all facets display high energy barriers, leading to low CH4 selectivity. For CO−to−C2H4 conversion, cubic (311) and S-vacancy (110) facets exhibit lower energy barriers and better activity. CO adsorption analysis indicates strong interactions on hexagonal (102) and cubic (111) facets due to charge transfer, while hexagonal (001) and cubic (110) facets show weaker interactions. More importantly, the cubic (311) facet, with low-coordinated Cd–Cd bridge sites, facilitates CO adsorption and exhibits low C−C coupling barriers (0.97 eV for (311) and 0.51 eV for S-vacancy (110)). We propose that heteroatom doping near sulfur vacancies could enhance CO adsorption without weakening the C–C coupling ability. Overall, perfect CdS facets demonstrate high CO selectivity, with the sulfur vacancies potentially improving this selectivity, while CH4 selectivity remains low. Hexagonal phases are less promising for C–C coupling, whereas cubic (311) and S-vacancy (110) facets show potential for CO2 reduction to C2H4.
Author Contributions
S.L. and Y.M.: conceptualization, methodology, and writing—original draft preparation; Y.Z. and L.W.: software and investigation; D.X., L.T. and H.Z.: writing—review and editing; T.W., G.Y. and H.X.: conceptualization, writing—review and editing, and supervision. All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by Ningbo International Science & Technology Cooperation Program (2023H005), Ningbo Natural Science Foundation (2023J366), and Central Public−interest Scientific Institution Basal Research Fund (GYZX240406).
Data Availability Statement
Data are contained within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Arunkumar, K.; Kumar, A.; Hassan, M.M.; Chauhan, S.; Lovely James, A.; Thethi, H.P.; Helena Raj, V.; Reddy, Y.M.; Ramesh Kumar, D.; Swaminathan, P.; et al. Advances in CCU Methods: Handling Release of Carbon for the Impact of Climate Change. E3S Web Conf. 2024, 529, 03018. [Google Scholar] [CrossRef]
- Gong, E.; Ali, S.; Hiragond, C.B.; Kim, H.S.; Powar, N.S.; Kim, D.; Kim, H.; In, S.-I. Solar fuels: Research and development strategies to accelerate photocatalytic CO2 conversion into hydrocarbon fuels. Energy Environ. Sci. 2022, 15, 880–937. [Google Scholar] [CrossRef]
- Jiao, X.; Zheng, K.; Liang, L.; Li, X.; Sun, Y.; Xie, Y. Fundamentals and challenges of ultrathin 2D photocatalysts in boosting CO2 photoreduction. Chem. Soc. Rev. 2020, 49, 6592–6604. [Google Scholar] [CrossRef]
- Fu, J.; Jiang, K.; Qiu, X.; Yu, J.; Liu, M. Product selectivity of photocatalytic CO2 reduction reactions. Mater. Today 2020, 32, 222–243. [Google Scholar] [CrossRef]
- Zhang, Y.; Johannessen, B.; Zhang, P.; Gong, J.; Ran, J.; Qiao, S.Z. Reversed electron transfer in dual single atom catalyst for boosted photoreduction of CO2. Adv. Mater. 2023, 35, 2306923. [Google Scholar] [CrossRef]
- Ban, C.; Wang, Y.; Feng, Y.; Zhu, Z.; Duan, Y.; Ma, J.; Zhang, X.; Liu, X.; Zhou, K.; Zou, H. Photochromic single atom Ag/TiO2 catalysts for selective CO2 reduction to CH4. Energy Environ. Sci. 2024, 17, 518–530. [Google Scholar] [CrossRef]
- Chang, X.; Wang, T.; Gong, J. CO2 photo-reduction: Insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environ. Sci. 2016, 9, 2177–2196. [Google Scholar] [CrossRef]
- Behera, A.; Kar, A.K.; Srivastava, R. Challenges and prospects in the selective photoreduction of CO2 to C1 and C2 products with nanostructured materials: A review. Mater. Horiz. 2022, 9, 607–639. [Google Scholar] [CrossRef]
- Wang, H.-N.; Zou, Y.-H.; Sun, H.-X.; Chen, Y.; Li, S.-L.; Lan, Y.-Q. Recent progress and perspectives in heterogeneous photocatalytic CO2 reduction through a solid–gas mode. Coord. Chem. Rev. 2021, 438, 213906. [Google Scholar] [CrossRef]
- Sun, F.-L.; Lin, C.-B.; Zhang, W.; Chen, Q.; Chen, W.-X.; Li, X.-N.; Zhuang, G.-L. Dual activation and C−C coupling on single atom catalyst for CO2 photoreduction. NPJ Comput. Mater. 2023, 9, 220. [Google Scholar] [CrossRef]
- Albero, J.; Peng, Y.; García, H. Photocatalytic CO2 reduction to C2+ products. Acs Catal. 2020, 10, 5734–5749. [Google Scholar] [CrossRef]
- Cohen, K.Y.; Evans, R.; Dulovic, S.; Bocarsly, A.B. Using light and electrons to bend carbon dioxide: Developing and understanding catalysts for CO2 conversion to fuels and feedstocks. Acc. Chem. Res. 2022, 55, 944–954. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, S.; Zhao, J. Selective C−C Coupling by Spatially Confined Dimeric Metal Centers. iScience 2020, 23, 101051. [Google Scholar] [CrossRef]
- Li, H.; Wu, D.; Wu, J.; Song, Y.; Lv, W.; Duan, Z.; Ma, D. Mechanistic understanding of the electrocatalytic conversion of CO into C2+ products by double-atom catalysts. Mater. Today Phys. 2023, 37, 101203. [Google Scholar] [CrossRef]
- Zhu, C.; Wei, X.; Li, W.; Pu, Y.; Sun, J.; Tang, K.; Wan, H.; Ge, C.; Zou, W.; Dong, L. Crystal-plane effects of CeO2 {110} and CeO2 {100} on photocatalytic CO2 reduction: Synergistic interactions of oxygen defects and hydroxyl groups. ACS Sustain. Chem. Eng. 2020, 8, 14397–14406. [Google Scholar] [CrossRef]
- Liu, L.; Wang, S.; Huang, H.; Zhang, Y.; Ma, T. Surface sites engineering on semiconductors to boost photocatalytic CO2 reduction. Nano Energy 2020, 75, 104959. [Google Scholar] [CrossRef]
- Sun, K.; Qian, Y.; Jiang, H.L. Metal-Organic Frameworks for Photocatalytic Water Splitting and CO2 Reduction. Angew. Chem. Int. Ed. 2023, 62, e202217565. [Google Scholar] [CrossRef]
- Cheng, L.; Li, B.; Yin, H.; Fan, J.; Xiang, Q. Cu clusters immobilized on Cd-defective cadmium sulfide nano-rods towards photocatalytic CO2 reduction. J. Mater. Sci. Technol. 2022, 118, 54–63. [Google Scholar] [CrossRef]
- Cao, Y.; Guo, L.; Dan, M.; Doronkin, D.E.; Han, C.; Rao, Z.; Liu, Y.; Meng, J.; Huang, Z.; Zheng, K.; et al. Modulating electron density of vacancy site by single Au atom for effective CO2 photoreduction. Nat. Commun. 2021, 12, 1675. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, L.; Sheng, J.; Tang, F.; Zeng, K.; Wang, L.; Liang, K.; Hu, H.; Liu, Y.-N. Photostable core-shell CdS/ZIF-8 composite for enhanced photocatalytic reduction of CO2. Appl. Surf. Sci. 2019, 498, 143899. [Google Scholar] [CrossRef]
- Cai, S.; Zhang, M.; Li, J.; Chen, J.; Jia, H. Anchoring Single-Atom Ru on CdS with Enhanced CO2 Capture and Charge Accumulation for High Selectivity of Photothermocatalytic CO2 Reduction to Solar Fuels. Sol. RRL 2021, 5, 2000313. [Google Scholar] [CrossRef]
- Tian, T.; Jin, X.; Guo, N.; Li, H.; Han, Y.; Yuan, Y. CdS/ethylenediamine nanowires 3D photocatalyst with rich sulfur vacancies for efficient syngas production from CO2 photoreduction. Appl. Catal. B Environ. 2022, 308, 121227. [Google Scholar] [CrossRef]
- Qin, B.; Li, Y.; Wang, H.; Yang, G.; Cao, Y.; Yu, H.; Zhang, Q.; Liang, H.; Peng, F. Efficient electrochemical reduction of CO2 into CO promoted by sulfur vacancies. Nano Energy 2019, 60, 43–51. [Google Scholar] [CrossRef]
- Wang, N.; Cheong, S.; Yoon, D.-E.; Lu, P.; Lee, H.; Lee, Y.K.; Park, Y.-S.; Lee, D.C. Efficient, Selective CO2 Photoreduction Enabled by Facet-Resolved Redox-Active Sites on Colloidal CdS Nanosheets. J. Am. Chem. Soc. 2022, 144, 16974–16983. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, Y.; Xie, G.; Huang, Z.; Peng, L.; Yu, C.; Xie, X.; Qu, S.; Zhang, N. Isolated Cu Sites in CdS Hollow Nanocubes with Doping-Location-Dependent Performance for Photocatalytic CO2 Reduction. ACS Catal. 2024, 14, 1468–1479. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, Z.; Wang, J.; Chen, Z.; Jiang, G.; Zhang, Q.; Li, Z. Generating Long-Lived Charge Carriers in CdS Quantum Dots by Cu-Doping for Photocatalytic CO2 Reduction. Inorg. Chem. 2024, 63, 2234–2240. [Google Scholar] [CrossRef]
- Tian, F.; Zhang, H.; Liu, S.; Wu, T.; Yu, J.; Wang, D.; Jin, X.; Peng, C. Visible-light-driven CO2 reduction to ethylene on CdS: Enabled by structural relaxation-induced intermediate dimerization and enhanced by ZIF-8 coating. Appl. Catal. B Environ. 2021, 285, 119834. [Google Scholar] [CrossRef]
- Wang, T.; Chen, L.; Chen, C.; Huang, M.; Huang, Y.; Liu, S.; Li, B. Engineering catalytic interfaces in Cuδ+/CeO2-TiO2 photocatalysts for synergistically boosting CO2 reduction to ethylene. ACS Nano 2022, 16, 2306–2318. [Google Scholar] [CrossRef]
- Shao, W.; Li, X.; Zhu, J.; Zu, X.; Liang, L.; Hu, J.; Pan, Y.; Zhu, J.; Yan, W.; Sun, Y.; et al. Metaln+−Metalδ+ pair sites steer C−C coupling for selective CO2 photoreduction to C2 hydrocarbons. Nano Res. 2022, 15, 1882–1891. [Google Scholar] [CrossRef]
- Zeng, R.; Wang, W.; Cai, G.; Huang, Z.; Tao, J.; Tang, D.; Zhu, C. Single-atom platinum nanocatalyst-improved catalytic efficiency with enzyme-DNA supermolecular architectures. Nano Energy 2020, 74, 104931. [Google Scholar] [CrossRef]
- Xue, W.; Liu, H.; Chen, X.; Yang, X.; Yang, R.; Liu, Y.; Li, M.; Yang, X.; Xia, B.Y.; You, B. Operando reconstruction towards stable CuI nanodots with favorable facets for selective CO2 electroreduction to C2H4. Sci. China Chem. 2023, 66, 1834–1843. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT−D) for the 94 elements H−Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).