


Insights into the Effect of Crystal Facets and Sulfur Defects on the Product Selectivity of Various CdS Configurations for CO2 Photoreduction: A DFT Study

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
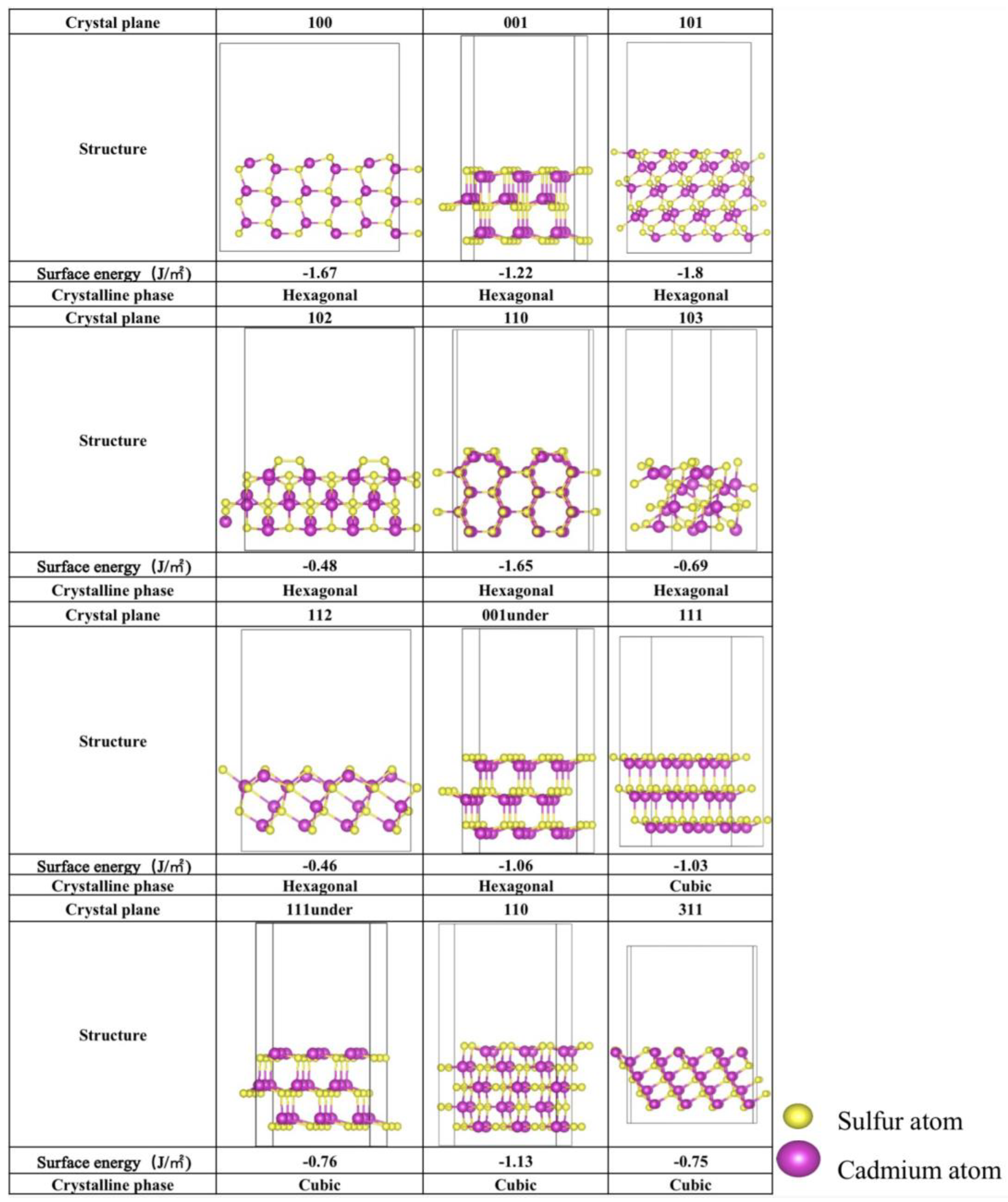
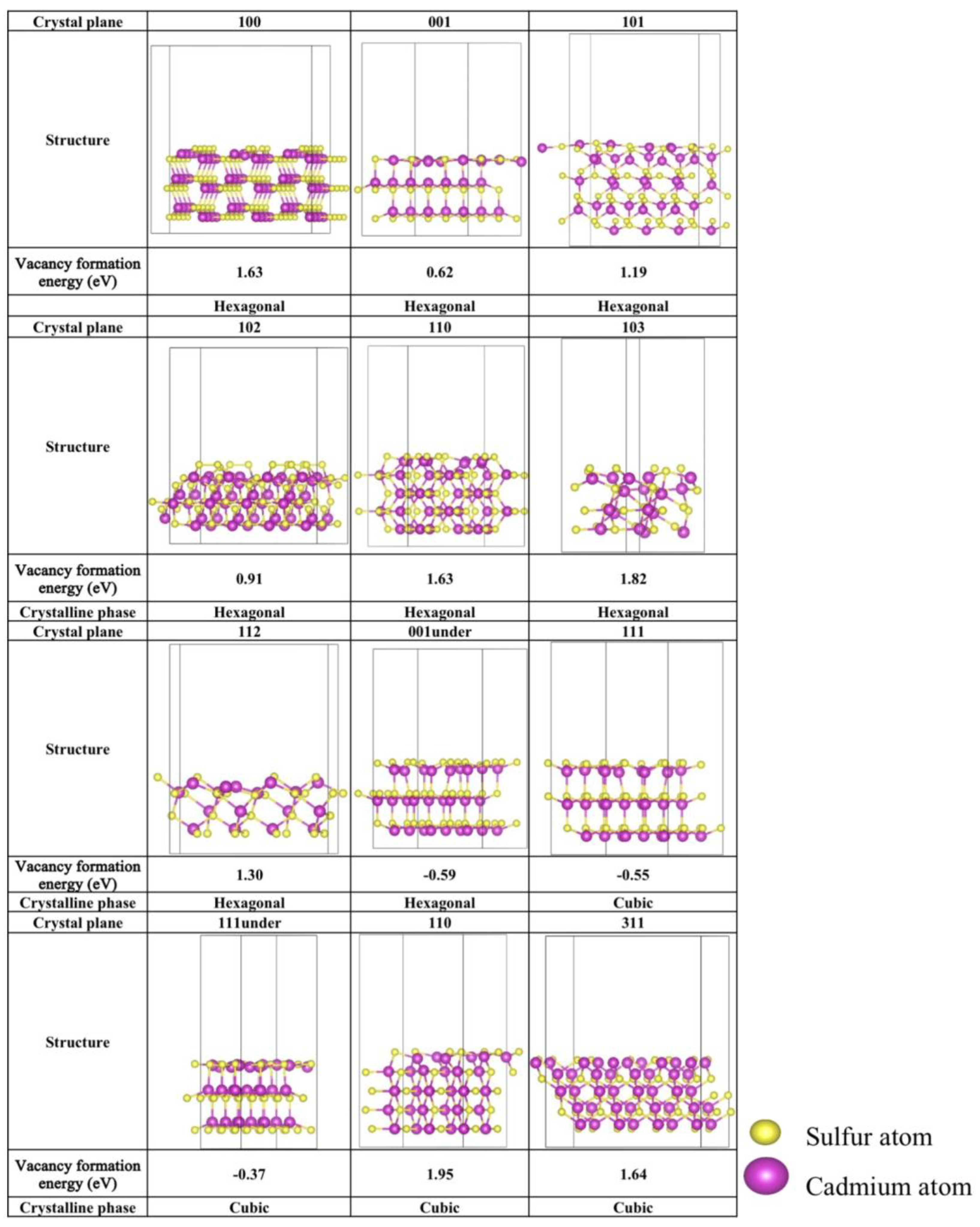
2.1. Stability Analysis of Different Facet Configurations and Sulfur Defects
2.1.1. Stability of Different Crystalline Facets
2.1.2. Stability of S-Vacancy in Various Crystalline Facets
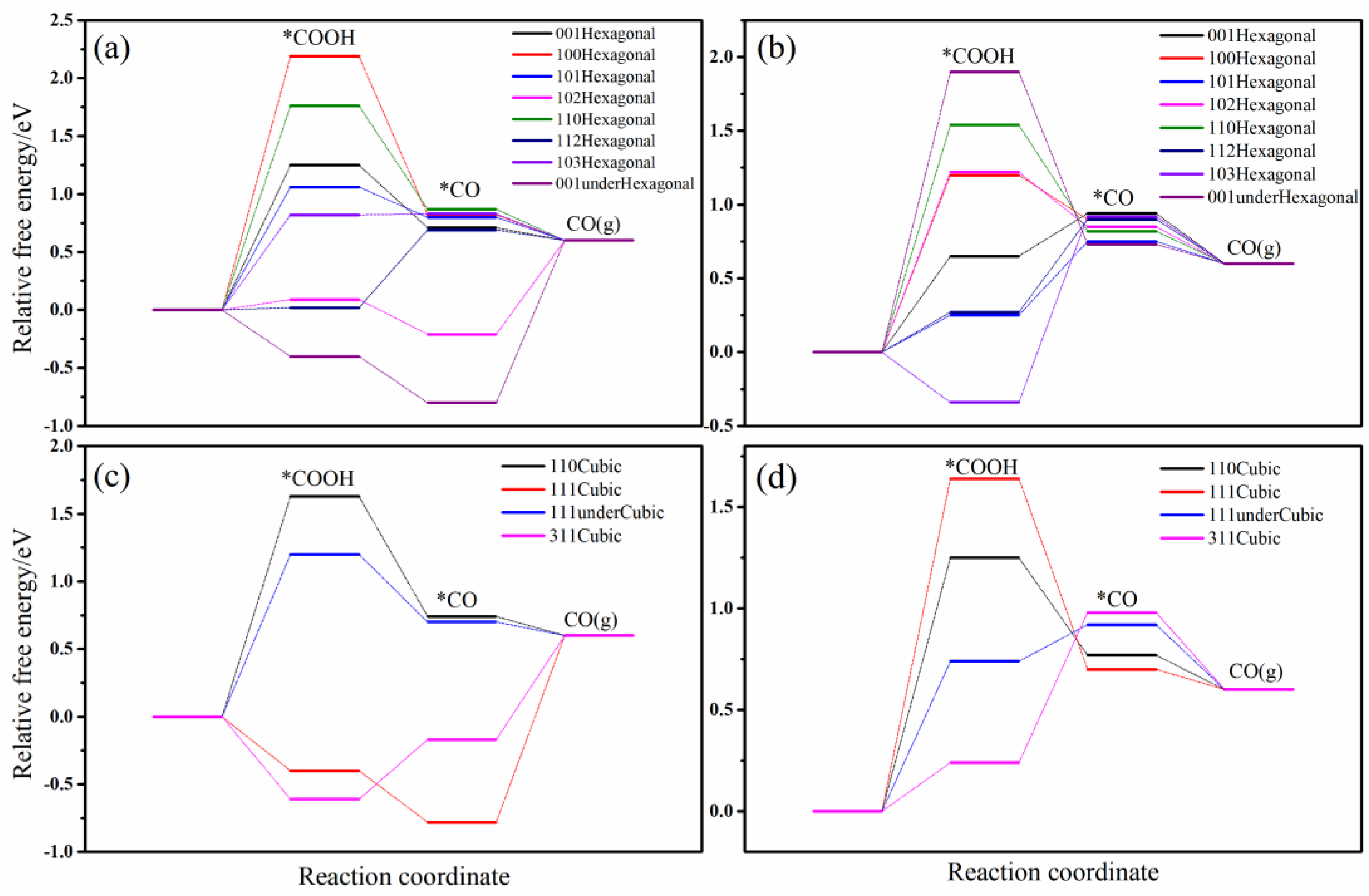
2.2. Analysis of the CO2 Reduction Reaction Pathways to CO, CH4, and C2H4
2.2.1. Reaction Pathway of CO2 Reduction to CO
2.2.2. Reaction Pathway of CO2 Reduction to CH4
2.2.3. Reaction Pathway of CO2 Reduction to C2H4
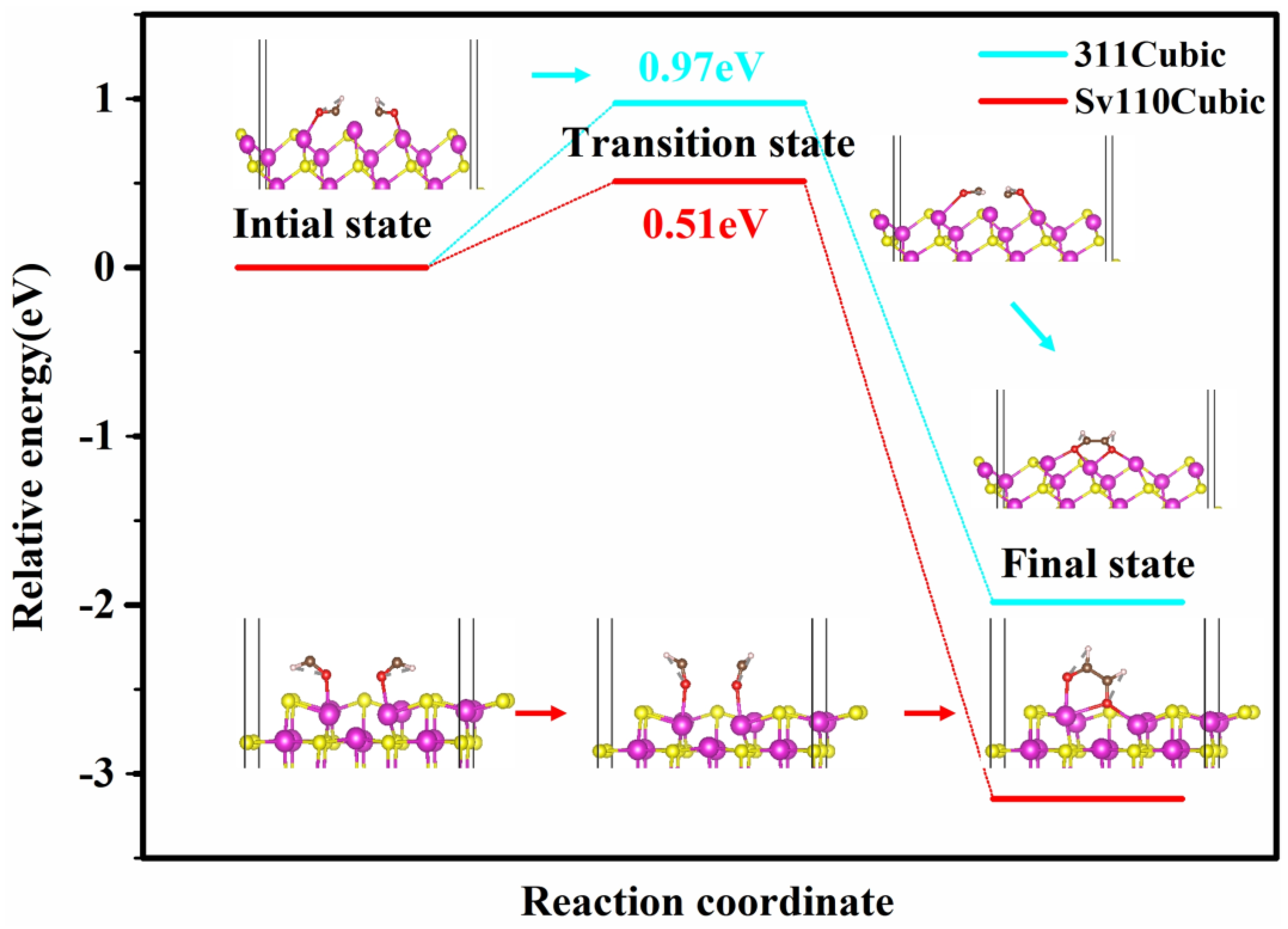
2.3. Theoretical Analysis of CO Adsorption and C−C Coupling Mechanism
2.3.1. Investigation of CO Adsorption against Different Active Surface
2.3.2. Investigation of C–C Coupling Mechanism against Different Active Surfaces
3. Materials and Methods
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Arunkumar, K.; Kumar, A.; Hassan, M.M.; Chauhan, S.; Lovely James, A.; Thethi, H.P.; Helena Raj, V.; Reddy, Y.M.; Ramesh Kumar, D.; Swaminathan, P.; et al. Advances in CCU Methods: Handling Release of Carbon for the Impact of Climate Change. E3S Web Conf. 2024, 529, 03018. [Google Scholar] [CrossRef]
- Gong, E.; Ali, S.; Hiragond, C.B.; Kim, H.S.; Powar, N.S.; Kim, D.; Kim, H.; In, S.-I. Solar fuels: Research and development strategies to accelerate photocatalytic CO2 conversion into hydrocarbon fuels. Energy Environ. Sci. 2022, 15, 880–937. [Google Scholar] [CrossRef]
- Jiao, X.; Zheng, K.; Liang, L.; Li, X.; Sun, Y.; Xie, Y. Fundamentals and challenges of ultrathin 2D photocatalysts in boosting CO2 photoreduction. Chem. Soc. Rev. 2020, 49, 6592–6604. [Google Scholar] [CrossRef]
- Fu, J.; Jiang, K.; Qiu, X.; Yu, J.; Liu, M. Product selectivity of photocatalytic CO2 reduction reactions. Mater. Today 2020, 32, 222–243. [Google Scholar] [CrossRef]
- Zhang, Y.; Johannessen, B.; Zhang, P.; Gong, J.; Ran, J.; Qiao, S.Z. Reversed electron transfer in dual single atom catalyst for boosted photoreduction of CO2. Adv. Mater. 2023, 35, 2306923. [Google Scholar] [CrossRef]
- Ban, C.; Wang, Y.; Feng, Y.; Zhu, Z.; Duan, Y.; Ma, J.; Zhang, X.; Liu, X.; Zhou, K.; Zou, H. Photochromic single atom Ag/TiO2 catalysts for selective CO2 reduction to CH4. Energy Environ. Sci. 2024, 17, 518–530. [Google Scholar] [CrossRef]
- Chang, X.; Wang, T.; Gong, J. CO2 photo-reduction: Insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environ. Sci. 2016, 9, 2177–2196. [Google Scholar] [CrossRef]
- Behera, A.; Kar, A.K.; Srivastava, R. Challenges and prospects in the selective photoreduction of CO2 to C1 and C2 products with nanostructured materials: A review. Mater. Horiz. 2022, 9, 607–639. [Google Scholar] [CrossRef]
- Wang, H.-N.; Zou, Y.-H.; Sun, H.-X.; Chen, Y.; Li, S.-L.; Lan, Y.-Q. Recent progress and perspectives in heterogeneous photocatalytic CO2 reduction through a solid–gas mode. Coord. Chem. Rev. 2021, 438, 213906. [Google Scholar] [CrossRef]
- Sun, F.-L.; Lin, C.-B.; Zhang, W.; Chen, Q.; Chen, W.-X.; Li, X.-N.; Zhuang, G.-L. Dual activation and C−C coupling on single atom catalyst for CO2 photoreduction. NPJ Comput. Mater. 2023, 9, 220. [Google Scholar] [CrossRef]
- Albero, J.; Peng, Y.; García, H. Photocatalytic CO2 reduction to C2+ products. Acs Catal. 2020, 10, 5734–5749. [Google Scholar] [CrossRef]
- Cohen, K.Y.; Evans, R.; Dulovic, S.; Bocarsly, A.B. Using light and electrons to bend carbon dioxide: Developing and understanding catalysts for CO2 conversion to fuels and feedstocks. Acc. Chem. Res. 2022, 55, 944–954. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, S.; Zhao, J. Selective C−C Coupling by Spatially Confined Dimeric Metal Centers. iScience 2020, 23, 101051. [Google Scholar] [CrossRef]
- Li, H.; Wu, D.; Wu, J.; Song, Y.; Lv, W.; Duan, Z.; Ma, D. Mechanistic understanding of the electrocatalytic conversion of CO into C2+ products by double-atom catalysts. Mater. Today Phys. 2023, 37, 101203. [Google Scholar] [CrossRef]
- Zhu, C.; Wei, X.; Li, W.; Pu, Y.; Sun, J.; Tang, K.; Wan, H.; Ge, C.; Zou, W.; Dong, L. Crystal-plane effects of CeO2 {110} and CeO2 {100} on photocatalytic CO2 reduction: Synergistic interactions of oxygen defects and hydroxyl groups. ACS Sustain. Chem. Eng. 2020, 8, 14397–14406. [Google Scholar] [CrossRef]
- Liu, L.; Wang, S.; Huang, H.; Zhang, Y.; Ma, T. Surface sites engineering on semiconductors to boost photocatalytic CO2 reduction. Nano Energy 2020, 75, 104959. [Google Scholar] [CrossRef]
- Sun, K.; Qian, Y.; Jiang, H.L. Metal-Organic Frameworks for Photocatalytic Water Splitting and CO2 Reduction. Angew. Chem. Int. Ed. 2023, 62, e202217565. [Google Scholar] [CrossRef]
- Cheng, L.; Li, B.; Yin, H.; Fan, J.; Xiang, Q. Cu clusters immobilized on Cd-defective cadmium sulfide nano-rods towards photocatalytic CO2 reduction. J. Mater. Sci. Technol. 2022, 118, 54–63. [Google Scholar] [CrossRef]
- Cao, Y.; Guo, L.; Dan, M.; Doronkin, D.E.; Han, C.; Rao, Z.; Liu, Y.; Meng, J.; Huang, Z.; Zheng, K.; et al. Modulating electron density of vacancy site by single Au atom for effective CO2 photoreduction. Nat. Commun. 2021, 12, 1675. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, L.; Sheng, J.; Tang, F.; Zeng, K.; Wang, L.; Liang, K.; Hu, H.; Liu, Y.-N. Photostable core-shell CdS/ZIF-8 composite for enhanced photocatalytic reduction of CO2. Appl. Surf. Sci. 2019, 498, 143899. [Google Scholar] [CrossRef]
- Cai, S.; Zhang, M.; Li, J.; Chen, J.; Jia, H. Anchoring Single-Atom Ru on CdS with Enhanced CO2 Capture and Charge Accumulation for High Selectivity of Photothermocatalytic CO2 Reduction to Solar Fuels. Sol. RRL 2021, 5, 2000313. [Google Scholar] [CrossRef]
- Tian, T.; Jin, X.; Guo, N.; Li, H.; Han, Y.; Yuan, Y. CdS/ethylenediamine nanowires 3D photocatalyst with rich sulfur vacancies for efficient syngas production from CO2 photoreduction. Appl. Catal. B Environ. 2022, 308, 121227. [Google Scholar] [CrossRef]
- Qin, B.; Li, Y.; Wang, H.; Yang, G.; Cao, Y.; Yu, H.; Zhang, Q.; Liang, H.; Peng, F. Efficient electrochemical reduction of CO2 into CO promoted by sulfur vacancies. Nano Energy 2019, 60, 43–51. [Google Scholar] [CrossRef]
- Wang, N.; Cheong, S.; Yoon, D.-E.; Lu, P.; Lee, H.; Lee, Y.K.; Park, Y.-S.; Lee, D.C. Efficient, Selective CO2 Photoreduction Enabled by Facet-Resolved Redox-Active Sites on Colloidal CdS Nanosheets. J. Am. Chem. Soc. 2022, 144, 16974–16983. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, Y.; Xie, G.; Huang, Z.; Peng, L.; Yu, C.; Xie, X.; Qu, S.; Zhang, N. Isolated Cu Sites in CdS Hollow Nanocubes with Doping-Location-Dependent Performance for Photocatalytic CO2 Reduction. ACS Catal. 2024, 14, 1468–1479. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, Z.; Wang, J.; Chen, Z.; Jiang, G.; Zhang, Q.; Li, Z. Generating Long-Lived Charge Carriers in CdS Quantum Dots by Cu-Doping for Photocatalytic CO2 Reduction. Inorg. Chem. 2024, 63, 2234–2240. [Google Scholar] [CrossRef]
- Tian, F.; Zhang, H.; Liu, S.; Wu, T.; Yu, J.; Wang, D.; Jin, X.; Peng, C. Visible-light-driven CO2 reduction to ethylene on CdS: Enabled by structural relaxation-induced intermediate dimerization and enhanced by ZIF-8 coating. Appl. Catal. B Environ. 2021, 285, 119834. [Google Scholar] [CrossRef]
- Wang, T.; Chen, L.; Chen, C.; Huang, M.; Huang, Y.; Liu, S.; Li, B. Engineering catalytic interfaces in Cuδ+/CeO2-TiO2 photocatalysts for synergistically boosting CO2 reduction to ethylene. ACS Nano 2022, 16, 2306–2318. [Google Scholar] [CrossRef]
- Shao, W.; Li, X.; Zhu, J.; Zu, X.; Liang, L.; Hu, J.; Pan, Y.; Zhu, J.; Yan, W.; Sun, Y.; et al. Metaln+−Metalδ+ pair sites steer C−C coupling for selective CO2 photoreduction to C2 hydrocarbons. Nano Res. 2022, 15, 1882–1891. [Google Scholar] [CrossRef]
- Zeng, R.; Wang, W.; Cai, G.; Huang, Z.; Tao, J.; Tang, D.; Zhu, C. Single-atom platinum nanocatalyst-improved catalytic efficiency with enzyme-DNA supermolecular architectures. Nano Energy 2020, 74, 104931. [Google Scholar] [CrossRef]
- Xue, W.; Liu, H.; Chen, X.; Yang, X.; Yang, R.; Liu, Y.; Li, M.; Yang, X.; Xia, B.Y.; You, B. Operando reconstruction towards stable CuI nanodots with favorable facets for selective CO2 electroreduction to C2H4. Sci. China Chem. 2023, 66, 1834–1843. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Wang, Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system. Phys. Rev. B 1996, 54, 16533. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT−D) for the 94 elements H−Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Meng, Y.; Zhong, Y.; Wang, L.; Xue, D.; Tong, L.; Zhang, H.; Wu, T.; Yu, G.; Xiao, H. Insights into the Effect of Crystal Facets and Sulfur Defects on the Product Selectivity of Various CdS Configurations for CO2 Photoreduction: A DFT Study. Catalysts 2024, 14, 688. https://doi.org/10.3390/catal14100688
Liu S, Meng Y, Zhong Y, Wang L, Xue D, Tong L, Zhang H, Wu T, Yu G, Xiao H. Insights into the Effect of Crystal Facets and Sulfur Defects on the Product Selectivity of Various CdS Configurations for CO2 Photoreduction: A DFT Study. Catalysts. 2024; 14(10):688. https://doi.org/10.3390/catal14100688
Chicago/Turabian StyleLiu, Shuai, Yang Meng, Yidong Zhong, Leiping Wang, Dingming Xue, Lei Tong, Honglei Zhang, Tao Wu, Guangsuo Yu, and Hang Xiao. 2024. "Insights into the Effect of Crystal Facets and Sulfur Defects on the Product Selectivity of Various CdS Configurations for CO2 Photoreduction: A DFT Study" Catalysts 14, no. 10: 688. https://doi.org/10.3390/catal14100688