
Advancements in Serine Protease Inhibitors: From Mechanistic Insights to Clinical Applications



Abstract
:
1. Introduction
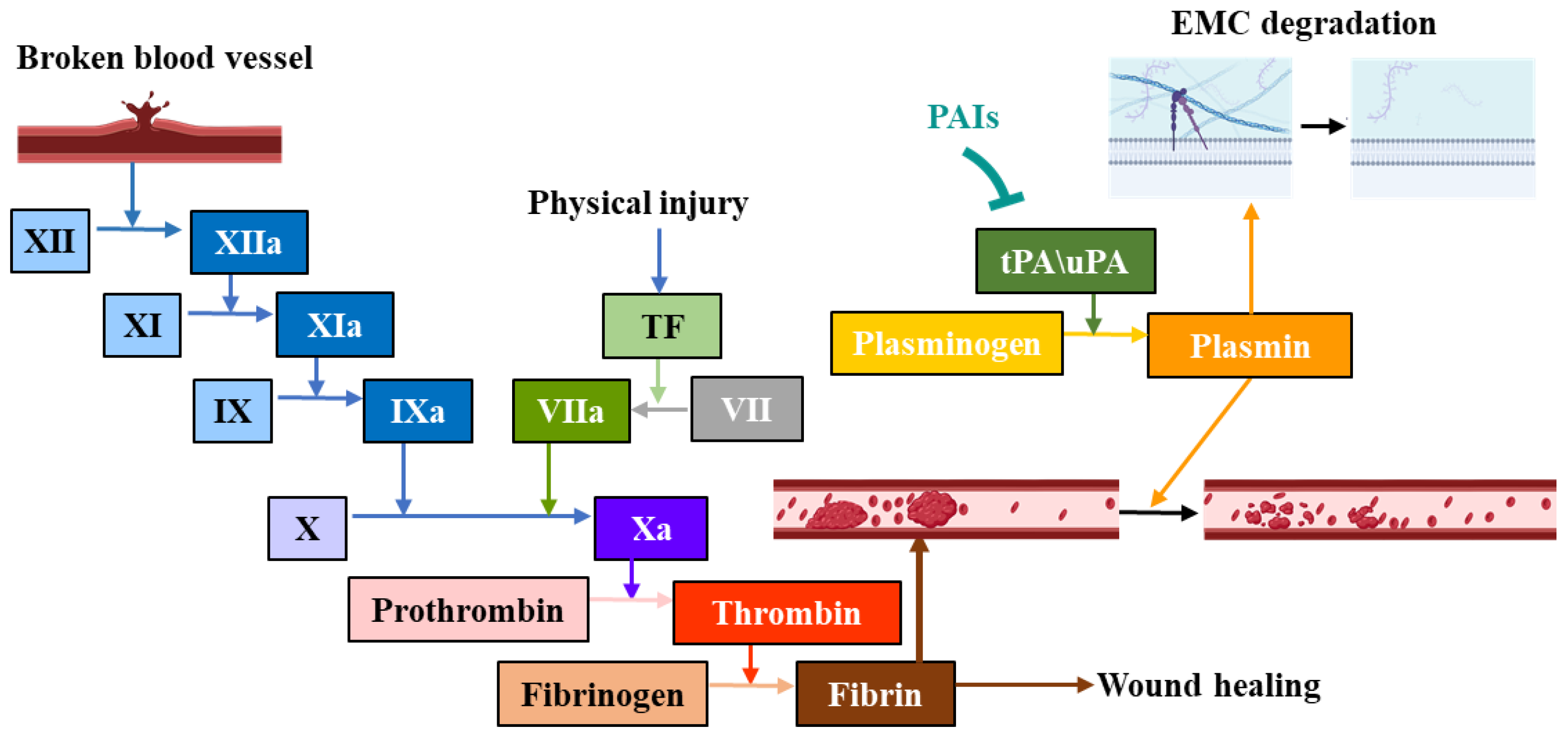
2. Mechanisms and Classification of Serine Protease Inhibitors
3. Different Types of Serine Protease Inhibitors
3.1. Small-Molecule Inhibitors
3.2. Nucleotide Drugs
3.2.1. Antisense Oligonucleotides (ASOs)
3.2.2. siRNA
4. Biomacromolecular Inhibitors
4.1. Peptides and Peptide Mimetics
4.2. Antibody
5. Challenges of Serine Protease Inhibitors in Clinical Applications
6. General Strategies for Overcoming Challenges in the Development of Serine Protease Inhibitors
6.1. Structure-Based Drug Design Remains the Primary Approach for Inhibitor Development
6.2. Drug Delivery Systems Enhance the Clinical Utilization of Poorly Soluble Inhibitors and Broad-Spectrum Inhibitors
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Rawlings, N.D.; Barrett, A.J. MEROPS: The peptidase database. Nucleic Acids Res. 2000, 28, 323–325. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.B.; Chou, K.C. Identification of proteases and their types. Anal. Biochem. 2009, 385, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Burchacka, E.; Pięta, P.; Łupicka-Słowik, A. Recent advances in fungal serine protease inhibitors. Biomed. Pharmacother. 2022, 146, 112523. [Google Scholar] [CrossRef]
- Soreide, K.; Janssen, E.A.; Körner, H.; Baak, J.P. Trypsin in colorectal cancer: Molecular biological mechanisms of proliferation, invasion, and metastasis. J. Pathol. 2006, 209, 147–156. [Google Scholar] [CrossRef]
- Andriulli, A.; Masoero, G.; Felder, M.; Vantini, I.; Petrillo, M.; Cavallini, G.; Bianchi Porro, G.; Dobrilla, G.; Verme, G. Circulating trypsin-like immunoreactivity in chronic pancreatitis. Dig. Dis. Sci. 1981, 26, 532–537. [Google Scholar] [CrossRef]
- Yoshida, N.; Yoshikawa, T. Basic and translational research on proteinase-activated receptors: Implication of proteinase/proteinase-activated receptor in gastrointestinal inflammation. J. Pharmacol. Sci. 2008, 108, 415–421. [Google Scholar] [CrossRef]
- Keragala, C.B.; Medcalf, R.L. Plasminogen: An enigmatic zymogen. Blood 2021, 137, 2881–2889. [Google Scholar] [CrossRef]
- Ali, A.E.; Becker, R.C. The foundation for investigating factor XI as a target for inhibition in human cardiovascular disease. J. Thromb. Thrombolysis 2024, 1–14, online ahead of print. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Degn, S.E.; Thiel, S. Humoral pattern recognition and the complement system. Scand. J. Immunol. 2013, 78, 181–193. [Google Scholar] [CrossRef]
- Carroll, M.C. Complement and humoral immunity. Vaccine 2008, 26 (Suppl. 8), I28–I33. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Remuzzi, G. Overview of complement activation and regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.M.; Heinlein, C.; Kim, T.; Hernandez, S.A.; Malik, M.S.; True, L.D.; Morrissey, C.; Corey, E.; Montgomery, B.; Mostaghel, E.; et al. The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis. Cancer Discov. 2014, 4, 1310–1325. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.J.; Huang, C.C.; Lin, H.Y.; Juan, C.P.; Lan, S.W.; Shyu, H.Y.; Wu, S.R.; Hsiao, P.W.; Huang, H.P.; Shun, C.T.; et al. Androgen-Induced TMPRSS2 Activates Matriptase and Promotes Extracellular Matrix Degradation, Prostate Cancer Cell Invasion, Tumor Growth, and Metastasis. Cancer Res. 2015, 75, 2949–2960. [Google Scholar] [CrossRef]
- Cheng, J.; Zhou, J.; Fu, S.; Fu, J.; Zhou, B.; Chen, H.; Fu, J.; Wei, C. Prostate adenocarcinoma and COVID-19: The possible impacts of TMPRSS2 expressions in susceptibility to SARS-CoV-2. J. Cell Mol. Med. 2021, 25, 4157–4165. [Google Scholar] [CrossRef]
- Sharony, R.; Yu, P.J.; Park, J.; Galloway, A.C.; Mignatti, P.; Pintucci, G. Protein targets of inflammatory serine proteases and cardiovascular disease. J. Inflamm. 2010, 7, 45. [Google Scholar] [CrossRef]
- Heutinck, K.M.; ten Berge, I.J.; Hack, C.E.; Hamann, J.; Rowshani, A.T. Serine proteases of the human immune system in health and disease. Mol. Immunol. 2010, 47, 1943–1955. [Google Scholar] [CrossRef]
- Meyer-Hoffert, U. Reddish, scaly, and itchy: How proteases and their inhibitors contribute to inflammatory skin diseases. Arch. Immunol. Ther. Exp. 2009, 57, 345–354. [Google Scholar] [CrossRef]
- Moldoveanu, B.; Otmishi, P.; Jani, P.; Walker, J.; Sarmiento, X.; Guardiola, J.; Saad, M.; Yu, J. Inflammatory mechanisms in the lung. J. Inflamm. Res. 2009, 2, 1–11. [Google Scholar]
- Soualmia, F.; El Amri, C. Serine protease inhibitors to treat inflammation: A patent review (2011–2016). Expert. Opin. Ther. Pat. 2018, 28, 93–110. [Google Scholar] [CrossRef]
- Micalet, A.; Tappouni, L.J.; Peszko, K.; Karagianni, D.; Lam, A.; Counsell, J.R.; Quezada, S.A.; Moeendarbary, E.; Cheema, U. Urokinase-type plasminogen activator (uPA) regulates invasion and matrix remodelling in colorectal cancer. Matrix Biol. Plus 2023, 19–20, 100137. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R., Jr.; Nunneley, J.W.; Barnard, D.; Pöhlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015, 116, 76–84. [Google Scholar] [CrossRef]
- Mykytyn, A.Z.; Breugem, T.I.; Riesebosch, S.; Schipper, D.; van den Doel, P.B.; Rottier, R.J.; Lamers, M.M.; Haagmans, B.L. SARS-CoV-2 entry into human airway organoids is serine protease-mediated and facilitated by the multibasic cleavage site. Elife 2021, 10, e64508. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, W.; Reiser, G. Trypsin and trypsin-like proteases in the brain: Proteolysis and cellular functions. Cell Mol. Life Sci. 2008, 65, 237–252. [Google Scholar] [CrossRef]
- Kruithof, E.K. Plasminogen activator inhibitors—A review. Enzyme 1988, 40, 113–121. [Google Scholar] [CrossRef]
- Bode, W.; Huber, R. Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem. 1992, 204, 433–451. [Google Scholar] [CrossRef]
- Krowarsch, D.; Cierpicki, T.; Jelen, F.; Otlewski, J. Canonical protein inhibitors of serine proteases. Cell Mol. Life Sci. 2003, 60, 2427–2444. [Google Scholar] [CrossRef]
- Laskowski, M., Jr.; Kato, I. Protein inhibitors of proteinases. Annu. Rev. Biochem. 1980, 49, 593–626. [Google Scholar] [CrossRef]
- Zakharova, E.; Horvath, M.P.; Goldenberg, D.P. Structure of a serine protease poised to resynthesize a peptide bond. Proc. Natl. Acad. Sci. USA 2009, 106, 11034–11039. [Google Scholar] [CrossRef]
- Micheelsen, P.O.; Vévodová, J.; De Maria, L.; Ostergaard, P.R.; Friis, E.P.; Wilson, K.; Skjøt, M. Structural and mutational analyses of the interaction between the barley alpha-amylase/subtilisin inhibitor and the subtilisin savinase reveal a novel mode of inhibition. J. Mol. Biol. 2008, 380, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.L.; Fuentes-Prior, P.; Sadler, J.E.; Huber, R.; Bode, W. Characterization of the residues involved in the human alpha-thrombin-haemadin complex: An exosite II-binding inhibitor. Biochemistry 2002, 41, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Rydel, T.J.; Tulinsky, A.; Bode, W.; Huber, R. Refined structure of the hirudin-thrombin complex. J. Mol. Biol. 1991, 221, 583–601. [Google Scholar] [CrossRef]
- Jiang, L.; Yuan, C.; Huang, M. A general strategy to inhibit serine protease by targeting its autolysis loop. FASEB J. 2021, 35, e21259. [Google Scholar] [CrossRef]
- Redondo, S.; Martínez, M.P.; Ramajo, M.; Navarro-Dorado, J.; Barez, A.; Tejerina, T. Pharmacological basis and clinical evidence of dabigatran therapy. J. Hematol. Oncol. 2011, 4, 53. [Google Scholar] [CrossRef]
- Deftereos, S.; Anatoliotakis, N.; Giannopoulos, G.; Kaoukis, A.; Mavri, M.; Pyrgakis, V.; Stefanadis, C. Novel direct factor IIa and Xa inhibitors: Mechanisms of action and preclinical studies. Curr. Clin. Pharmacol. 2012, 7, 149–165. [Google Scholar] [CrossRef]
- Bennett, C.F.; Swayze, E.E. RNA targeting therapeutics: Molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 259–293. [Google Scholar] [CrossRef]
- Blair, H.A.; Keating, G.M. Dabigatran Etexilate: A Review in Nonvalvular Atrial Fibrillation. Drugs 2017, 77, 331–344. [Google Scholar] [CrossRef]
- Antonijevic, N.M.; Zivkovic, I.D.; Jovanovic, L.M.; Matic, D.M.; Kocica, M.J.; Mrdovic, I.B.; Kanjuh, V.I.; Culafic, M.D. Dabigatran—Metabolism, Pharmacologic Properties and Drug Interactions. Curr. Drug Metab. 2017, 18, 622–635. [Google Scholar] [CrossRef]
- Evans, H.C.; Perry, C.M.; Faulds, D. Ximelagatran/Melagatran: A review of its use in the prevention of venous thromboembolism in orthopaedic surgery. Drugs 2004, 64, 649–678. [Google Scholar] [CrossRef]
- Brighton, T.A. The direct thrombin inhibitor melagatran/ximelagatran. Med. J. Aust. 2004, 181, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, R.; Tran, M.; Gore, J.M.; Spencer, F.A. A review of the oral direct thrombin inhibitor ximelagatran: Not yet the end of the warfarin era. Am. Heart J. 2005, 150, 19–26. [Google Scholar] [CrossRef]
- Berry, C.N.; Girardot, C.; Lecoffre, C.; Lunven, C. Effects of the synthetic thrombin inhibitor argatroban on fibrin- or clot-incorporated thrombin: Comparison with heparin and recombinant Hirudin. Thromb. Haemost. 1994, 72, 381–386. [Google Scholar] [CrossRef]
- O’Brien, P.J.; Mureebe, L. Direct thrombin inhibitors. J. Cardiovasc. Pharmacol. Ther. 2012, 17, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Lip, G.Y.; Rasmussen, L.H.; Olsson, S.B.; Jensen, E.C.; Persson, A.L.; Eriksson, U.; Wåhlander, K.F. Oral direct thrombin inhibitor AZD0837 for the prevention of stroke and systemic embolism in patients with non-valvular atrial fibrillation: A randomized dose-guiding, safety, and tolerability study of four doses of AZD0837 vs. vitamin K antagonists. Eur. Heart J. 2009, 30, 2897–2907. [Google Scholar] [CrossRef]
- Squizzato, A.; Dentali, F.; Steidl, L.; Ageno, W. New direct thrombin inhibitors. Intern. Emerg. Med. 2009, 4, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Stone, G.W.; Witzenbichler, B.; Guagliumi, G.; Peruga, J.Z.; Brodie, B.R.; Dudek, D.; Kornowski, R.; Hartmann, F.; Gersh, B.J.; Pocock, S.J.; et al. Bivalirudin during primary PCI in acute myocardial infarction. N. Engl. J. Med. 2008, 358, 2218–2230. [Google Scholar] [CrossRef]
- Parry, M.A.; Maraganore, J.M.; Stone, S.R. Kinetic mechanism for the interaction of Hirulog with thrombin. Biochemistry 1994, 33, 14807–14814. [Google Scholar] [CrossRef]
- Siddiqui, F.; Hoppensteadt, D.; Jeske, W.; Iqbal, O.; Tafur, A.; Fareed, J. Factor Xa Inhibitory Profile of Apixaban, Betrixaban, Edoxaban, and Rivaroxaban Does Not Fully Reflect Their Biologic Spectrum. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029619847524. [Google Scholar] [CrossRef]
- Mandernach, M.W.; Beyth, R.J.; Rajasekhar, A. Apixaban for the prophylaxis and treatment of deep vein thrombosis and pulmonary embolism: An evidence-based review. Ther. Clin. Risk Manag. 2015, 11, 1273–1282. [Google Scholar] [CrossRef]
- Mekaj, Y.H.; Mekaj, A.Y.; Duci, S.B.; Miftari, E.I. New oral anticoagulants: Their advantages and disadvantages compared with vitamin K antagonists in the prevention and treatment of patients with thromboembolic events. Ther. Clin. Risk Manag. 2015, 11, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Bauer, K.A. Recent progress in anticoagulant therapy: Oral direct inhibitors of thrombin and factor Xa. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 12–19. [Google Scholar] [CrossRef] [PubMed]
- Bonar, R.; Favaloro, E.J.; Mohammed, S.; Ahuja, M.; Pasalic, L.; Sioufi, J.; Marsden, K. The effect of the direct factor Xa inhibitors apixaban and rivaroxaban on haemostasis tests: A comprehensive assessment using in vitro and ex vivo samples. Pathology 2016, 48, 60–71. [Google Scholar] [CrossRef]
- Byon, W.; Garonzik, S.; Boyd, R.A.; Frost, C.E. Apixaban: A Clinical Pharmacokinetic and Pharmacodynamic Review. Clin. Pharmacokinet. 2019, 58, 1265–1279. [Google Scholar] [CrossRef]
- Chan, N.C.; Bhagirath, V.; Eikelboom, J.W. Profile of betrixaban and its potential in the prevention and treatment of venous thromboembolism. Vasc. Health Risk Manag. 2015, 11, 343–351. [Google Scholar] [CrossRef]
- Connolly, S.J.; Eikelboom, J.; Dorian, P.; Hohnloser, S.H.; Gretler, D.D.; Sinha, U.; Ezekowitz, M.D. Betrixaban compared with warfarin in patients with atrial fibrillation: Results of a phase 2, randomized, dose-ranging study (Explore-Xa). Eur. Heart J. 2013, 34, 1498–1505. [Google Scholar] [CrossRef]
- Feng, W.; Wu, K.; Liu, Z.; Kong, G.; Deng, Z.; Chen, S.; Wu, Y.; Chen, M.; Liu, S.; Wang, H. Oral direct factor Xa inhibitor versus enoxaparin for thromboprophylaxis after hip or knee arthroplasty: Systemic review, traditional meta-analysis, dose-response meta-analysis and network meta-analysis. Thromb. Res. 2015, 136, 1133–1144. [Google Scholar] [CrossRef]
- Srinivasan, S.; Ajmal, M.; Pecci, C.; Lassar, T. Edoxaban in cardiovascular disease management: Review. Br. J. Clin. Pharmacol. 2022, 88, 535–540. [Google Scholar] [CrossRef]
- Perzborn, E.; Roehrig, S.; Straub, A.; Kubitza, D.; Mueck, W.; Laux, V. Rivaroxaban: A new oral factor Xa inhibitor. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 376–381. [Google Scholar] [CrossRef]
- Perzborn, E.; Strassburger, J.; Wilmen, A.; Pohlmann, J.; Roehrig, S.; Schlemmer, K.H.; Straub, A. In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939—An oral, direct Factor Xa inhibitor. J. Thromb. Haemost. 2005, 3, 514–521. [Google Scholar] [CrossRef]
- Bondarenko, M.; Curti, C.; Montana, M.; Rathelot, P.; Vanelle, P. Efficacy and toxicity of factor Xa inhibitors. J. Pharm. Pharm. Sci. 2013, 16, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Birchansky, J.; Frishman, W.H. Milvexian: A Focus on a New Oral Anticoagulant that Targets Factor XIa for Thromboembolism Prophylaxis. Cardiol. Rev. 2024, online ahead of print. [CrossRef]
- Perera, V.; Wang, Z.; Luettgen, J.; Li, D.; DeSouza, M.; Cerra, M.; Seiffert, D. First-in-human study of milvexian, an oral, direct, small molecule factor XIa inhibitor. Clin. Transl. Sci. 2022, 15, 330–342. [Google Scholar] [CrossRef]
- Schubart, A.; Anderson, K.; Mainolfi, N.; Sellner, H.; Ehara, T.; Adams, C.M.; Mac Sweeney, A.; Liao, S.M.; Crowley, M.; Littlewood-Evans, A.; et al. Small-molecule factor B inhibitor for the treatment of complement-mediated diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 7926–7931. [Google Scholar] [CrossRef]
- Hernández-Mitre, M.P.; Tong, S.Y.C.; Denholm, J.T.; Dore, G.J.; Bowen, A.C.; Lewin, S.R.; Venkatesh, B.; Hills, T.E.; McQuilten, Z.; Paterson, D.L.; et al. Nafamostat Mesylate for Treatment of COVID-19 in Hospitalised Patients: A Structured, Narrative Review. Clin. Pharmacokinet. 2022, 61, 1331–1343. [Google Scholar] [CrossRef]
- Ohshio, G.; Saluja, A.K.; Leli, U.; Sengupta, A.; Steer, M.L. Esterase inhibitors prevent lysosomal enzyme redistribution in two noninvasive models of experimental pancreatitis. Gastroenterology 1989, 96, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Talukdar, R.; Saikia, N.; Singal, D.K.; Tandon, R. Chronic pancreatitis: Evolving paradigms. Pancreatology 2006, 6, 440–449. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Smith, J.C.; Krüger, N.; Sørensen, L.K.; Søgaard, O.S.; Hasselstrøm, J.B.; Winkler, M.; Hempel, T.; Raich, L.; et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. bioRxiv 2020, 65, 103255. [Google Scholar] [CrossRef]
- Mack, G.S.; Marshall, A. Lost in migration. Nat. Biotechnol. 2010, 28, 214–229. [Google Scholar] [CrossRef]
- Setyono-Han, B.; Stürzebecher, J.; Schmalix, W.A.; Muehlenweg, B.; Sieuwerts, A.M.; Timmermans, M.; Magdolen, V.; Schmitt, M.; Klijn, J.G.; Foekens, J.A. Suppression of rat breast cancer metastasis and reduction of primary tumour growth by the small synthetic urokinase inhibitor WX-UK1. Thromb. Haemost. 2005, 93, 779–786. [Google Scholar] [CrossRef]
- Plasse, T.F.; Fathi, R.; Fehrmann, C.; McComsey, G.A. Upamostat: A serine protease inhibitor for antiviral, gastrointestinal, and anticancer indications. Expert Opin. Investig. Drugs 2023, 32, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Ekici, O.D.; Paetzel, M.; Dalbey, R.E. Unconventional serine proteases: Variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 2008, 17, 2023–2037. [Google Scholar] [CrossRef] [PubMed]
- Franco, F.M.; Jones, D.E.; Harris, P.K.; Han, Z.; Wildman, S.A.; Jarvis, C.M.; Janetka, J.W. Structure-based discovery of small molecule hepsin and HGFA protease inhibitors: Evaluation of potency and selectivity derived from distinct binding pockets. Bioorg. Med. Chem. 2015, 23, 2328–2343. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Gong, L.; Yuan, C.; Xu, M.; Wang, X.; Jiang, L.; Huang, M. A structural mechanism of flavonoids in inhibiting serine proteases. Food Funct. 2017, 8, 2437–2443. [Google Scholar] [CrossRef]
- Jiang, L.; Zhang, X.; Zhou, Y.; Chen, Y.; Luo, Z.; Li, J.; Yuan, C.; Huang, M. Halogen bonding for the design of inhibitors by targeting the S1 pocket of serine proteases. RSC Adv. 2018, 8, 28189–28197. [Google Scholar] [CrossRef]
- Visser, M.E.; Witztum, J.L.; Stroes, E.S.; Kastelein, J.J. Antisense oligonucleotides for the treatment of dyslipidaemia. Eur. Heart J. 2012, 33, 1451–1458. [Google Scholar] [CrossRef]
- Lippi, G.; Harenberg, J.; Mattiuzzi, C.; Favaloro, E.J. Next generation antithrombotic therapy: Focus on antisense therapy against coagulation factor XI. Semin. Thromb. Hemost. 2015, 41, 255–262. [Google Scholar] [CrossRef]
- Liu, C.; Chen, J.; Chen, H.; Zhang, T.; He, D.; Luo, Q.; Chi, J.; Hong, Z.; Liao, Y.; Zhang, S.; et al. PCSK9 Inhibition: From Current Advances to Evolving Future. Cells 2022, 11, 2972. [Google Scholar] [CrossRef]
- Bethune, C.; Walsh, M.; Jung, B.; Yu, R.; Geary, R.S.; Bhanot, S. Pharmacokinetics and Pharmacodynamics of Ionis-FXIRx, an Antisense Inhibitor of Factor XI, in Patients with End-Stage Renal Disease on Hemodialysis. Blood 2017, 130, 1116. [Google Scholar] [CrossRef]
- Fredenburgh, J.C.; Weitz, J.I. News at XI: Moving beyond factor Xa inhibitors. J. Thromb. Haemost. 2023, 21, 1692–1702. [Google Scholar] [CrossRef]
- Frampton, J.E. Inclisiran: A Review in Hypercholesterolemia. Am. J. Cardiovasc. Drugs 2023, 23, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.S. Small-interfering RNA targeting proprotein convertase subtilisin/kexin type 9 might promote fatty liver disease and hepatocellular carcinoma through upregulation of CD36. Tumor. Biol. 2023, 45, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, J.; Wang, X.; Yan, S.; Xu, Y.; San, M.; Tang, W.; Yang, F.; Cao, Z.; Li, W.; et al. Functional characterization of a new non-Kunitz serine protease inhibitor from the scorpion Lychas mucronatus. Int. J. Biol. Macromol. 2015, 72, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Jouiaei, M.; Casewell, N.R.; Yanagihara, A.A.; Nouwens, A.; Cribb, B.W.; Whitehead, D.; Jackson, T.N.; Ali, S.A.; Wagstaff, S.C.; Koludarov, I.; et al. Firing the sting: Chemically induced discharge of cnidae reveals novel proteins and peptides from box jellyfish (Chironex fleckeri) venom. Toxins 2015, 7, 936–950. [Google Scholar] [CrossRef]
- Pomin, V.H.; Mourão, P.A. Specific sulfation and glycosylation-a structural combination for the anticoagulation of marine carbohydrates. Front. Cell. Infect. Microbiol. 2014, 4, 33. [Google Scholar] [CrossRef]
- Sabotič, J.; Bleuler-Martinez, S.; Renko, M.; Avanzo Caglič, P.; Kallert, S.; Štrukelj, B.; Turk, D.; Aebi, M.; Kos, J.; Künzler, M. Structural basis of trypsin inhibition and entomotoxicity of cospin, serine protease inhibitor involved in defense of Coprinopsis cinerea fruiting bodies. J. Biol. Chem. 2012, 287, 3898–3907. [Google Scholar] [CrossRef]
- Harish, B.S.; Uppuluri, K.B. Microbial serine protease inhibitors and their therapeutic applications. Int. J. Biol. Macromol. 2018, 107, 1373–1387. [Google Scholar] [CrossRef]
- Mosolov, V.V.; Valueva, T.A. Proteinase inhibitors and their function in plants: A review. Appl. Biochem. Microbiol. 2005, 41, 227–246. [Google Scholar] [CrossRef]
- Speranskaya, A.S.; Krinitsina, A.A.; Kudryavtseva, A.V.; Poltronieri, P.; Santino, A.; Oparina, N.Y.; Dmitriev, A.A.; Belenikin, M.S.; Guseva, M.A.; Shevelev, A.B. Impact of recombination on polymorphism of genes encoding Kunitz-type protease inhibitors in the genus Solanum. Biochimie 2012, 94, 1687–1696. [Google Scholar] [CrossRef]
- Cid-Gallegos, M.S.; Corzo-Ríos, L.J.; Jiménez-Martínez, C.; Sánchez-Chino, X.M. Protease Inhibitors from Plants as Therapeutic Agents- A Review. Plant Foods Hum. Nutr. 2022, 77, 20–29. [Google Scholar] [CrossRef]
- Kanost, M.R. Serine proteinase inhibitors in arthropod immunity. Dev. Comp. Immunol. 1999, 23, 291–301. [Google Scholar] [CrossRef]
- Mishra, M. Evolutionary Aspects of the Structural Convergence and Functional Diversification of Kunitz-Domain Inhibitors. J. Mol. Evol. 2020, 88, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Royston, D. The current place of aprotinin in the management of bleeding. Anaesthesia 2015, 70 (Suppl. 1), 46-e17. [Google Scholar] [CrossRef] [PubMed]
- Himbert, S.; D’Alessandro, A.; Qadri, S.M.; Majcher, M.J.; Hoare, T.; Sheffield, W.P.; Nagao, M.; Nagle, J.F.; Rheinstädter, M.C. The bending rigidity of the red blood cell cytoplasmic membrane. PLoS ONE 2022, 17, e0269619. [Google Scholar] [CrossRef] [PubMed]
- Ivachtchenko, A.V.; Ivashchenko, A.A.; Shkil, D.O.; Ivashchenko, I.A. Aprotinin-Drug against Respiratory Diseases. Int. J. Mol. Sci. 2023, 24, 11173. [Google Scholar] [CrossRef]
- Padín, J.F.; Pérez-Ortiz, J.M.; Redondo-Calvo, F.J. Aprotinin (I): Understanding the Role of Host Proteases in COVID-19 and the Importance of Pharmacologically Regulating Their Function. Int. J. Mol. Sci. 2024, 25, 7553. [Google Scholar] [CrossRef]
- Padín, J.F.; Pérez-Ortiz, J.M.; Redondo-Calvo, F.J. Aprotinin (II): Inhalational Administration for the Treatment of COVID-19 and Other Viral Conditions. Int. J. Mol. Sci. 2024, 25, 7209. [Google Scholar] [CrossRef]
- Ma, D.; Mizurini, D.M.; Assumpção, T.C.; Li, Y.; Qi, Y.; Kotsyfakis, M.; Ribeiro, J.M.; Monteiro, R.Q.; Francischetti, I.M. Desmolaris, a novel factor XIa anticoagulant from the salivary gland of the vampire bat (Desmodus rotundus) inhibits inflammation and thrombosis in vivo. Blood 2013, 122, 4094–4106. [Google Scholar] [CrossRef]
- Decrem, Y.; Rath, G.; Blasioli, V.; Cauchie, P.; Robert, S.; Beaufays, J.; Frère, J.M.; Feron, O.; Dogné, J.M.; Dessy, C.; et al. Ir-CPI, a coagulation contact phase inhibitor from the tick Ixodes ricinus, inhibits thrombus formation without impairing hemostasis. J. Exp. Med. 2009, 206, 2381–2395. [Google Scholar] [CrossRef]
- Kuhar, K.; Kansal, R.; Subrahmanyam, B.; Koundal, K.R.; Miglani, K.; Gupta, V.K. A Bowman–Birk protease inhibitor with antifeedant and antifungal activity from Dolichos biflorus. Acta Physiol. Plant. 2013, 35, 1887–1903. [Google Scholar] [CrossRef]
- Clemente, A.; Arques Mdel, C. Bowman-Birk inhibitors from legumes as colorectal chemopreventive agents. World J. Gastroenterol. 2014, 20, 10305–10315. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.R. Cancer prevention by protease inhibitors. Prev. Med. 1993, 22, 796–811. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.R.; Wan, X.S. Effects of the Bowman-Birk inhibitor on growth, invasion, and clonogenic survival of human prostate epithelial cells and prostate cancer cells. Prostate 2002, 50, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.R. The Bowman-Birk inhibitor from soybeans as an anticarcinogenic agent. Am. J. Clin. Nutr. 1998, 68, 1406s–1412s. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, T.; Kröger, B.; Bialojan, S.; Lemaire, H.G.; Höffken, H.W.; Reuschenbach, P.; Otte, M.; Dodt, J. A Kazal-type inhibitor with thrombin specificity from Rhodnius prolixus. J. Biol. Chem. 1993, 268, 16216–16222. [Google Scholar] [CrossRef]
- Mägert, H.J.; Ständker, L.; Kreutzmann, P.; Zucht, H.D.; Reinecke, M.; Sommerhoff, C.P.; Fritz, H.; Forssmann, W.G. LEKTI, a novel 15-domain type of human serine proteinase inhibitor. J. Biol. Chem. 1999, 274, 21499–21502. [Google Scholar] [CrossRef]
- Augustin, R.; Siebert, S.; Bosch, T.C. Identification of a kazal-type serine protease inhibitor with potent anti-staphylococcal activity as part of Hydra’s innate immune system. Dev. Comp. Immunol. 2009, 33, 830–837. [Google Scholar] [CrossRef]
- Dietrich, M.A.; Słowińska, M.; Karol, H.; Adamek, M.; Steinhagen, D.; Hejmej, A.; Bilińska, B.; Ciereszko, A. Serine protease inhibitor Kazal-type 2 is expressed in the male reproductive tract of carp with a possible role in antimicrobial protection. Fish Shellfish Immunol. 2017, 60, 150–163. [Google Scholar] [CrossRef]
- Kim, B.Y.; Lee, K.S.; Zou, F.M.; Wan, H.; Choi, Y.S.; Yoon, H.J.; Kwon, H.W.; Je, Y.H.; Jin, B.R. Antimicrobial activity of a honeybee (Apis cerana) venom Kazal-type serine protease inhibitor. Toxicon 2013, 76, 110–117. [Google Scholar] [CrossRef]
- Mende, K.; Petoukhova, O.; Koulitchkova, V.; Schaub, G.A.; Lange, U.; Kaufmann, R.; Nowak, G. Dipetalogastin, a potent thrombin inhibitor from the blood-sucking insect. Dipetalogaster maximus cDNA cloning, expression and characterization. Eur. J. Biochem. 1999, 266, 583–590. [Google Scholar] [CrossRef]
- Campos, I.T.; Amino, R.; Sampaio, C.A.; Auerswald, E.A.; Friedrich, T.; Lemaire, H.G.; Schenkman, S.; Tanaka, A.S. Infestin, a thrombin inhibitor presents in Triatoma infestans midgut, a Chagas’ disease vector: Gene cloning, expression and characterization of the inhibitor. Insect. Biochem. Mol. Biol. 2002, 32, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Campos, I.T.; Tanaka-Azevedo, A.M.; Tanaka, A.S. Identification and characterization of a novel factor XIIa inhibitor in the hematophagous insect, Triatoma infestans (Hemiptera: Reduviidae). FEBS Lett. 2004, 577, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.M.; Tanaka-Azevedo, A.M.; Araujo, M.S.; Juliano, M.A.; Tanaka, A.S. Characterization of thrombin inhibitory mechanism of rAaTI, a Kazal-type inhibitor from Aedes aegypti with anticoagulant activity. Biochimie 2011, 93, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, D.C.; Shapanka, R.; Greene, L.J. The primary structure of the human pancreatic secretory trypsin inhibitor. Amino acid sequence of the reduced S-aminoethylated protein. Arch. Biochem. Biophys. 1977, 179, 189–199. [Google Scholar] [CrossRef]
- Horii, A.; Kobayashi, T.-A.; Tomita, N.; Yamamoto, T.; Fukushige, S.; Murotsu, T.; Ogawa, M.; Mori, T.; Matsubara, K.i. Primary structure of human pancreatic secretory trypsin inhibitor (PSTI) gene. Biochem. Biophys. Res. Commun. 1987, 149, 635–641. [Google Scholar] [CrossRef]
- Mehner, C.; Radisky, E.S. Bad Tumors Made Worse: SPINK1. Front. Cell. Dev. Biol. 2019, 7, 10. [Google Scholar] [CrossRef]
- Lin, T.C. Functional Roles of SPINK1 in Cancers. Int. J. Mol. Sci. 2021, 22, 3814. [Google Scholar] [CrossRef]
- Guo, M.; Zhou, X.; Han, X.; Zhang, Y.; Jiang, L. SPINK1 is a prognosis predicting factor of non-small cell lung cancer and regulates redox homeostasis. Oncol. Lett. 2019, 18, 6899–6908. [Google Scholar] [CrossRef]
- Lu, F.; Shah, P.A.; Rao, A.; Gifford-Hollingsworth, C.; Chen, A.; Trey, G.; Soryal, M.; Talat, A.; Aslam, A.; Nasir, B.; et al. Liver Cancer-Specific Serine Protease Inhibitor Kazal Is a Potentially Novel Biomarker for the Early Detection of Hepatocellular Carcinoma. Clin. Transl. Gastroenterol. 2020, 11, e00271. [Google Scholar] [CrossRef]
- Jia, J.; Ga, L.; Liu, Y.; Yang, Z.; Wang, Y.; Guo, X.; Ma, R.; Liu, R.; Li, T.; Tang, Z.; et al. Serine Protease Inhibitor Kazal Type 1, A Potential Biomarker for the Early Detection, Targeting, and Prediction of Response to Immune Checkpoint Blockade Therapies in Hepatocellular Carcinoma. Front. Immunol. 2022, 13, 923031. [Google Scholar] [CrossRef]
- Appleyard, G.; Tisdale, M. Inhibition of the growth of human coronavirus 229E by leupeptin. J. Gen. Virol. 1985, 66 Pt 2, 363–366. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, J.; Kandasamy, S.; Iruthayaraj, A.; Magudeeswaran, S.; Chinnasamy, K.; Poomani, K. Strong Binding of Leupeptin with TMPRSS2 Protease May Be an Alternative to Camostat and Nafamostat for SARS-CoV-2 Repurposed Drug: Evaluation from Molecular Docking and Molecular Dynamics Simulations. Appl. Biochem. Biotechnol. 2021, 193, 1909–1923. [Google Scholar] [CrossRef] [PubMed]
- Di Nisio, M.; Middeldorp, S.; Büller, H.R. Direct thrombin inhibitors. N. Engl. J. Med. 2005, 353, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Esslinger, H.U.; Haas, S.; Maurer, R.; Lassmann, A.; Dübbers, K.; Müller-Peltzer, H. Pharmacodynamic and safety results of PEG-Hirudin in healthy volunteers. Thromb. Haemost. 1997, 77, 911–919. [Google Scholar] [CrossRef]
- Bossavy, J.P.; Sakariassen, K.S.; Rübsamen, K.; Thalamas, C.; Boneu, B.; Cadroy, Y. Comparison of the antithrombotic effect of PEG-hirudin and heparin in a human ex vivo model of arterial thrombosis. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1348–1353. [Google Scholar] [CrossRef]
- Johns, D.G.; Campeau, L.C.; Banka, P.; Bautmans, A.; Bueters, T.; Bianchi, E.; Branca, D.; Bulger, P.G.; Crevecoeur, I.; Ding, F.X.; et al. Orally Bioavailable Macrocyclic Peptide That Inhibits Binding of PCSK9 to the Low Density Lipoprotein Receptor. Circulation 2023, 148, 144–158. [Google Scholar] [CrossRef]
- Zhang, Z.; Shen, C.; Fang, M.; Han, Y.; Long, C.; Liu, W.; Yang, M.; Liu, M.; Zhang, D.; Cao, Q.; et al. Novel contact-kinin inhibitor sylvestin targets thromboinflammation and ameliorates ischemic stroke. Cell Mol. Life Sci. 2022, 79, 240. [Google Scholar] [CrossRef]
- Lin, K.; Perni, R.B.; Kwong, A.D.; Lin, C. VX-950, a novel hepatitis C virus (HCV) NS3-4A protease inhibitor, exhibits potent antiviral activities in HCv replicon cells. Antimicrob. Agents Chemother. 2006, 50, 1813–1822. [Google Scholar] [CrossRef]
- Xu, P.; Andreasen, P.A.; Huang, M. Structural Principles in the Development of Cyclic Peptidic Enzyme Inhibitors. Int. J. Biol. Sci. 2017, 13, 1222–1233. [Google Scholar] [CrossRef]
- Huntington, J.A. Serpin structure, function and dysfunction. J. Thromb. Haemost. 2011, 9 (Suppl. 1), 26–34. [Google Scholar] [CrossRef]
- Kellici, T.F.; Pilka, E.S.; Bodkin, M.J. Small-molecule modulators of serine protease inhibitor proteins (serpins). Drug Discov. Today 2021, 26, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Matamala, N.; Martínez, M.T.; Lara, B.; Pérez, L.; Vázquez, I.; Jimenez, A.; Barquín, M.; Ferrarotti, I.; Blanco, I.; Janciauskiene, S.; et al. Alternative transcripts of the SERPINA1 gene in alpha-1 antitrypsin deficiency. J. Transl. Med. 2015, 13, 211. [Google Scholar] [CrossRef] [PubMed]
- Sharp, H.L. The current status of alpha-1-antityrpsin, a protease inhibitor, in gastrointestinal disease. Gastroenterology 1976, 70, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Jonigk, D.; Al-Omari, M.; Maegel, L.; Müller, M.; Izykowski, N.; Hong, J.; Hong, K.; Kim, S.H.; Dorsch, M.; Mahadeva, R.; et al. Anti-inflammatory and immunomodulatory properties of α1-antitrypsin without inhibition of elastase. Proc. Natl. Acad. Sci. USA 2013, 110, 15007–15012. [Google Scholar] [CrossRef]
- Connolly, B.; Isaacs, C.; Cheng, L.; Asrani, K.H.; Subramanian, R.R. SERPINA1 mRNA as a Treatment for Alpha-1 Antitrypsin Deficiency. J. Nucleic Acids 2018, 2018, 8247935. [Google Scholar] [CrossRef]
- Herman, W.A.; Seńko, A.; Korczowska, I.; Lacka, K. Assessment of selected serum inflammatory markers of acute phase response and their correlations with adrenal androgens and metabolic syndrome in a population of men over the age of 40. Pol. Arch. Med. Wewn. 2009, 119, 704–711. [Google Scholar] [CrossRef]
- Kalsheker, N.A. Alpha 1-antichymotrypsin. Int. J. Biochem. Cell Biol. 1996, 28, 961–964. [Google Scholar] [CrossRef]
- Forsyth, S.; Horvath, A.; Coughlin, P. A review and comparison of the murine alpha1-antitrypsin and alpha1-antichymotrypsin multigene clusters with the human clade A serpins. Genomics 2003, 81, 336–345. [Google Scholar] [CrossRef]
- Duranton, J.; Boudier, C.; Belorgey, D.; Mellet, P.; Bieth, J.G. DNA strongly impairs the inhibition of cathepsin G by alpha(1)-antichymotrypsin and alpha(1)-proteinase inhibitor. J. Biol. Chem. 2000, 275, 3787–3792. [Google Scholar] [CrossRef]
- Heit, C.; Jackson, B.C.; McAndrews, M.; Wright, M.W.; Thompson, D.C.; Silverman, G.A.; Nebert, D.W.; Vasiliou, V. Update of the human and mouse SERPIN gene superfamily. Hum. Genom. 2013, 7, 22. [Google Scholar] [CrossRef]
- Björk, I.; Olson, S.T. Antithrombin. A bloody important serpin. Adv. Exp. Med. Biol. 1997, 425, 17–33. [Google Scholar] [PubMed]
- Horie, S.; Ishii, H.; Kazama, M. Heparin-like glycosaminoglycan is a receptor for antithrombin III-dependent but not for thrombin-dependent prostacyclin production in human endothelial cells. Thromb. Res. 1990, 59, 895–904. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.H.; Sniecinski, R.M.; Welsby, I.J.; Levi, M. Antithrombin: Anti-inflammatory properties and clinical applications. Thromb. Haemost. 2016, 115, 712–728. [Google Scholar] [CrossRef] [PubMed]
- García Merino, A. Monoclonal antibodies. Basic features. Neurologia 2011, 26, 301–306. [Google Scholar] [CrossRef]
- Sounni, N.E.; Noel, A. Targeting the tumor microenvironment for cancer therapy. Clin. Chem. 2013, 59, 85–93. [Google Scholar] [CrossRef]
- Gouni-Berthold, I.; Berthold, H.K. PCSK9 antibodies for the treatment of hypercholesterolemia. Nutrients 2014, 6, 5517–5533. [Google Scholar] [CrossRef]
- Blom, D.J.; Hala, T.; Bolognese, M.; Lillestol, M.J.; Toth, P.D.; Burgess, L.; Ceska, R.; Roth, E.; Koren, M.J.; Ballantyne, C.M.; et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N. Engl. J. Med. 2014, 370, 1809–1819. [Google Scholar] [CrossRef]
- Lorentz, C.U.; Verbout, N.G.; Wallisch, M.; Hagen, M.W.; Shatzel, J.J.; Olson, S.R.; Puy, C.; Hinds, M.T.; McCarty, O.J.T.; Gailani, D.; et al. Contact Activation Inhibitor and Factor XI Antibody, AB023, Produces Safe, Dose-Dependent Anticoagulation in a Phase 1 First-In-Human Trial. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 799–809. [Google Scholar] [CrossRef]
- Thomas, D.; Thelen, K.; Kraff, S.; Schwers, S.; Schiffer, S.; Unger, S.; Yassen, A.; Boxnick, S. BAY 1213790, a fully human IgG1 antibody targeting coagulation factor XIa: First evaluation of safety, pharmacodynamics, and pharmacokinetics. Res. Pract. Thromb. Haemost. 2019, 3, 242–253. [Google Scholar] [CrossRef]
- Jiang, L.; Botkjaer, K.A.; Andersen, L.M.; Yuan, C.; Andreasen, P.A.; Huang, M. Rezymogenation of active urokinase induced by an inhibitory antibody. Biochem. J. 2013, 449, 161–166. [Google Scholar] [CrossRef]
- Kromann-Hansen, T.; Louise Lange, E.; Peter Sørensen, H.; Hassanzadeh-Ghassabeh, G.; Huang, M.; Jensen, J.K.; Muyldermans, S.; Declerck, P.J.; Komives, E.A.; Andreasen, P.A. Discovery of a novel conformational equilibrium in urokinase-type plasminogen activator. Sci. Rep. 2017, 7, 3385. [Google Scholar] [CrossRef]
- Lindner, T.; Loktev, A.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Jäger, D.; Mier, W.; Haberkorn, U. Development of Quinoline-Based Theranostic Ligands for the Targeting of Fibroblast Activation Protein. J. Nucl. Med. 2018, 59, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.A.; Fujimura, T.; Uno, T.; Terada, T.; Hirano, K.I.; Hosokawa, H.; Ohta, A.; Miyata, T.; Ando, K.; Yahata, T. Plasminogen activator inhibitor-1 promotes immune evasion in tumors by facilitating the expression of programmed cell death-ligand 1. Front. Immunol. 2024, 15, 1365894. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, Y.; Shao, D.; Pan, Y.; Gao, X.; Zhao, P.; Liu, Q.; Shang, G.; Shang, W.; Fu, Z.; et al. High expression of serine protease inhibitor kazal type 1 predicts poor prognosis and promotes the progression and invasion of oral tongue squamous cell carcinoma. Arch. Oral Biol. 2024, 164, 106003. [Google Scholar] [CrossRef]
- Enriquez, A.; Baranchuk, A.; Redfearn, D.; Simpson, C.; Abdollah, H.; Michael, K. Dabigatran for the prevention and treatment of thromboembolic disorders. Expert. Rev. Cardiovasc. Ther. 2015, 13, 529–540. [Google Scholar] [CrossRef]
- Ricca Gonçalves, L.; Robalo Nunes, A. Dabigatran Reversal: A Practical Approach. Acta Med. Port. 2022, 35, 394–398. [Google Scholar] [CrossRef]
- Paik, J. Dabigatran Etexilate: A Review in Pediatric Venous Thromboembolism. Paediatr. Drugs 2022, 24, 423–431. [Google Scholar] [CrossRef]
- Lin, S.; Wang, Y.; Zhang, L.; Guan, W. Dabigatran must be used carefully: Literature review and recommendations for management of adverse events. Drug Des. Devel. Ther. 2019, 13, 1527–1533. [Google Scholar] [CrossRef]
- Turpie, A.G.G.; Purdham, D.; Ciaccia, A. Nonvitamin K antagonist oral anticoagulant use in patients with renal impairment. Ther. Adv. Cardiovasc. Dis. 2017, 11, 243–256. [Google Scholar] [CrossRef]
- Cortese, F.; Calculli, G.; Gesualdo, M.; Cecere, A.; Zito, A.; De Vito, F.; Carbonara, R.; Carbonara, S.; Cortese, A.M.; Ciccone, M.M. Idarucizumab: What Should We Know? Curr. Drug Targets 2018, 19, 81–88. [Google Scholar] [CrossRef]
- Eriksson, B.I. Clinical experience of melagatran/ximelagatran in major orthopaedic surgery. Thromb. Res. 2003, 109 (Suppl. 1), S23–S29. [Google Scholar] [CrossRef] [PubMed]
- Francis, C.W. Ximelagatran: A new oral anticoagulant. Best Pract. Res. Clin. Haematol. 2004, 17, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Koehl, J.L.; Hayes, B.D.; Al-Samkari, H.; Rosovsky, R. A comprehensive evaluation of apixaban in the treatment of venous thromboembolism. Expert Rev. Hematol. 2020, 13, 155–173. [Google Scholar] [CrossRef]
- Murphy, G.; Grace, Y.; Chaudry, S.; Chamoun, R. Betrixaban: A Novel Oral Anticoagulant With a New Niche. J. Pharm. Technol. 2018, 34, 123–133. [Google Scholar] [CrossRef]
- Eisen, A.; Ruff, C.T. Edoxaban in patients with atrial fibrillation. Ther. Adv. Cardiovasc. Dis. 2017, 11, 81–90. [Google Scholar] [CrossRef]
- Raskob, G.E.; van Es, N.; Verhamme, P.; Carrier, M.; Di Nisio, M.; Garcia, D.; Grosso, M.A.; Kakkar, A.K.; Kovacs, M.J.; Mercuri, M.F.; et al. Edoxaban for the Treatment of Cancer-Associated Venous Thromboembolism. N. Engl. J. Med. 2018, 378, 615–624. [Google Scholar] [CrossRef]
- Kruger, P.C.; Eikelboom, J.W.; Douketis, J.D.; Hankey, G.J. Deep vein thrombosis: Update on diagnosis and management. Med. J. Aust. 2019, 210, 516–524. [Google Scholar] [CrossRef]
- Male, C.; Lensing, A.W.A.; Palumbo, J.S.; Kumar, R.; Nurmeev, I.; Hege, K.; Bonnet, D.; Connor, P.; Hooimeijer, H.L.; Torres, M.; et al. Rivaroxaban compared with standard anticoagulants for the treatment of acute venous thromboembolism in children: A randomised, controlled, phase 3 trial. Lancet Haematol. 2020, 7, e18–e27. [Google Scholar] [CrossRef]
- Ahrens, I.; Peter, K.; Lip, G.Y.; Bode, C. Development and clinical applications of novel oral anticoagulants. Part II. Drugs under clinical investigation. Discov. Med. 2012, 13, 445–450. [Google Scholar]
- Guertin, K.R.; Choi, Y.M. The discovery of the Factor Xa inhibitor otamixaban: From lead identification to clinical development. Curr. Med. Chem. 2007, 14, 2471–2481. [Google Scholar] [CrossRef]
- Goldstein, S.; Bates, E.R.; Bhatt, D.L.; Cao, C.; Holmes, D.; Kupfer, S.; Martinez, F.; Spaeder, J.; Weitz, J.I.; Ye, Z.; et al. Phase 2 study of TAK-442, an oral factor Xa inhibitor, in patients following acute coronary syndrome. Thromb. Haemost. 2014, 111, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Agnelli, G.; Haas, S.; Ginsberg, J.S.; Krueger, K.A.; Dmitrienko, A.; Brandt, J.T. A phase II study of the oral factor Xa inhibitor LY517717 for the prevention of venous thromboembolism after hip or knee replacement. J. Thromb. Haemost. 2007, 5, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Wong, L.; Ko, B.S.; Yoon, S.S.; Li, K.; Baltcheva, I.; Nidamarthy, P.K.; Chawla, R.; Junge, G.; Yap, E.S. Iptacopan monotherapy in patients with paroxysmal nocturnal hemoglobinuria: A 2-cohort open-label proof-of-concept study. Blood Adv. 2022, 6, 4450–4460. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Rizk, D.V.; Perkovic, V.; Maes, B.; Kashihara, N.; Rovin, B.; Trimarchi, H.; Sprangers, B.; Meier, M.; Kollins, D.; et al. Results of a randomized double-blind placebo-controlled Phase 2 study propose iptacopan as an alternative complement pathway inhibitor for IgA nephropathy. Kidney Int. 2024, 105, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Breining, P.; Frølund, A.L.; Højen, J.F.; Gunst, J.D.; Staerke, N.B.; Saedder, E.; Cases-Thomas, M.; Little, P.; Nielsen, L.P.; Søgaard, O.S.; et al. Camostat mesylate against SARS-CoV-2 and COVID-19-Rationale, dosing and safety. Basic Clin. Pharmacol. Toxicol. 2021, 128, 204–212. [Google Scholar] [CrossRef]
- Froriep, D.; Clement, B.; Bittner, F.; Mendel, R.R.; Reichmann, D.; Schmalix, W.; Havemeyer, A. Activation of the anti-cancer agent upamostat by the mARC enzyme system. Xenobiotica 2013, 43, 780–784. [Google Scholar] [CrossRef]
- Beierlein, W.; Scheule, A.M.; Dietrich, W.; Ziemer, G. Forty years of clinical aprotinin use: A review of 124 hypersensitivity reactions. Ann. Thorac. Surg. 2005, 79, 741–748. [Google Scholar] [CrossRef]
- Dietrich, W.; Späth, P.; Ebell, A.; Richter, J.A. Prevalence of anaphylactic reactions to aprotinin: Analysis of two hundred forty-eight reexposures to aprotinin in heart operations. J. Thorac. Cardiovasc. Surg. 1997, 113, 194–201. [Google Scholar] [CrossRef]
- Suda, H.; Aoyagi, T.; Hamada, M.; Takeuchi, T.; Umezawa, H. Antipain, a new protease inhibitor isolated from actinomycetes. J. Antibiot. 1972, 25, 263–266. [Google Scholar] [CrossRef]
- Raphaelis, S.; Frommlet, F.; Mayer, H.; Koller, A. Implementation of a nurse-led self-management support intervention for patients with cancer-related pain: A cluster randomized phase-IV study with a stepped wedge design (EvANtiPain). BMC Cancer 2020, 20, 559. [Google Scholar] [CrossRef]
- Buijs, N.; Vlaming, H.C.; van Haren, M.J.; Martin, N.I. Synthetic Studies with Bacitracin A and Preparation of Analogues Containing Alternative Zinc Binding Groups. Chembiochem 2022, 23, e202200547. [Google Scholar] [CrossRef] [PubMed]
- Gennemark, P.; Walter, K.; Clemmensen, N.; Rekić, D.; Nilsson, C.A.M.; Knöchel, J.; Hölttä, M.; Wernevik, L.; Rosengren, B.; Kakol-Palm, D.; et al. An oral antisense oligonucleotide for PCSK9 inhibition. Sci. Transl. Med. 2021, 13, eabe9117. [Google Scholar] [CrossRef] [PubMed]
- Graetz, T.J.; Tellor, B.R.; Smith, J.R.; Avidan, M.S. Desirudin: A review of the pharmacology and clinical application for the prevention of deep vein thrombosis. Expert Rev. Cardiovasc. Ther. 2011, 9, 1101–1109. [Google Scholar] [CrossRef]
- Ballantyne, C.M.; Banka, P.; Mendez, G.; Garcia, R.; Rosenstock, J.; Rodgers, A.; Mendizabal, G.; Mitchel, Y.; Catapano, A.L. Phase 2b Randomized Trial of the Oral PCSK9 Inhibitor MK-0616. J. Am. Coll. Cardiol. 2023, 81, 1553–1564. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Kwong, A.D.; Perni, R.B. Discovery and development of VX-950, a novel, covalent, and reversible inhibitor of hepatitis C virus NS3.4A serine protease. Infect. Disord. Drug Targets 2006, 6, 3–16. [Google Scholar] [CrossRef]
- Roujeau, J.C.; Mockenhaupt, M.; Tahan, S.R.; Henshaw, J.; Martin, E.C.; Harding, M.; van Baelen, B.; Bengtsson, L.; Singhal, P.; Kauffman, R.S.; et al. Telaprevir-related dermatitis. JAMA Dermatol. 2013, 149, 152–158. [Google Scholar] [CrossRef]
- Garcias-Ladaria, J.; Pérez-Ferriols, A.; Ortega-García, P.; Diago, M. Management of refractory telaprevir-induced dermatitis using oral corticosteroids. Actas Dermosifiliogr. 2014, 105, e55–e60. [Google Scholar] [CrossRef]
- Bentounes, N.K.; Melicine, S.; Martin, A.C.; Smadja, D.M.; Gendron, N. Development of new anticoagulant in 2023: Prime time for anti-factor XI and XIa inhibitors. J. Med. Vasc. 2023, 48, 69–80. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Scott, A.M.; Wiseman, G.; Welt, S.; Adjei, A.; Lee, F.T.; Hopkins, W.; Divgi, C.R.; Hanson, L.H.; Mitchell, P.; Gansen, D.N.; et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 2003, 9, 1639–1647. [Google Scholar]
- Hao, Y.; Yang, Y.L.; Wang, Y.C.; Li, J. Effect of the Early Application of Evolocumab on Blood Lipid Profile and Cardiovascular Prognosis in Patients with Extremely High-Risk Acute Coronary Syndrome. Int. Heart J. 2022, 63, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Pérez de Isla, L.; Díaz-Díaz, J.L.; Romero, M.J.; Muñiz-Grijalvo, O.; Mediavilla, J.D.; Argüeso, R.; Sánchez Muñoz-Torrero, J.F.; Rubio, P.; Álvarez-Baños, P.; Ponte, P.; et al. Alirocumab and Coronary Atherosclerosis in Asymptomatic Patients with Familial Hypercholesterolemia: The ARCHITECT Study. Circulation 2023, 147, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Lafayette, R.A.; Rovin, B.H.; Reich, H.N.; Tumlin, J.A.; Floege, J.; Barratt, J. Safety, Tolerability and Efficacy of Narsoplimab, a Novel MASP-2 Inhibitor for the Treatment of IgA Nephropathy. Kidney Int. Rep. 2020, 5, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I.; Bauersachs, R.; Becker, B.; Berkowitz, S.D.; Freitas, M.C.S.; Lassen, M.R.; Metzig, C.; Raskob, G.E. Effect of Osocimab in Preventing Venous Thromboembolism Among Patients Undergoing Knee Arthroplasty: The FOXTROT Randomized Clinical Trial. JAMA 2020, 323, 130–139. [Google Scholar] [CrossRef]
- Poulsen, A.; Kang, C.; Keller, T.H. Drug design for flavivirus proteases: What are we missing? Curr. Pharm. Des. 2014, 20, 3422–3427. [Google Scholar] [CrossRef]
- Kang, C.; Keller, T.H.; Luo, D. Zika Virus Protease: An Antiviral Drug Target. Trends Microbiol. 2017, 25, 797–808. [Google Scholar] [CrossRef]
- Saha, N.; Robev, D.; Himanen, J.P.; Nikolov, D.B. ADAM proteases: Emerging role and targeting of the non-catalytic domains. Cancer Lett. 2019, 467, 50–57. [Google Scholar] [CrossRef]
- Xie, X.; Guo, N.; Xue, G.; Xie, D.; Yuan, C.; Harrison, J.; Li, J.; Jiang, L.; Huang, M. Solution Structure of SpoIVB Reveals Mechanism of PDZ Domain-Regulated Protease Activity. Front. Microbiol. 2019, 10, 1232. [Google Scholar] [CrossRef]
- Oberst, M.D.; Williams, C.A.; Dickson, R.B.; Johnson, M.D.; Lin, C.Y. The activation of matriptase requires its noncatalytic domains, serine protease domain, and its cognate inhibitor. J. Biol. Chem. 2003, 278, 26773–26779. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Afosah, D.K. Recent advances in the discovery and development of factor XI/XIa inhibitors. Med. Res. Rev. 2018, 38, 1974–2023. [Google Scholar] [CrossRef]
- Fjällskog, M.L.; Frii, L.; Bergh, J. Is Cremophor EL, solvent for paclitaxel, cytotoxic? Lancet 1993, 342, 873. [Google Scholar] [CrossRef] [PubMed]
- Fader, A.N.; Rose, P.G. Abraxane for the treatment of gynecologic cancer patients with severe hypersensitivity reactions to paclitaxel. Int. J. Gynecol. Cancer 2009, 19, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Borensztajn, K.; Spek, C.A. Blood coagulation factor Xa as an emerging drug target. Expert Opin. Ther. Targets 2011, 15, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Polack, B. Thrombin: A multifunctional enzyme. Ann. Biol. Clin. 2003, 61, 23–31. [Google Scholar]
- Ding, L.; Shu, Z.; Hao, J.; Luo, X.; Ye, X.; Zhu, W.; Duan, W.; Chen, Z. Schixator, a new FXa inhibitor from Schistosoma japonicum with antithrombotic effect and low bleeding risk. Biochem. Biophys. Res. Commun. 2022, 603, 138–143. [Google Scholar] [CrossRef]
- Narayanan, S. Multifunctional roles of thrombin. Ann. Clin. Lab. Sci. 1999, 29, 275–280. [Google Scholar]
- Laboissière, M.C.; Young, M.M.; Pinho, R.G.; Todd, S.; Fletterick, R.J.; Kuntz, I.; Craik, C.S. Computer-assisted mutagenesis of ecotin to engineer its secondary binding site for urokinase inhibition. J. Biol. Chem. 2002, 277, 26623–26631. [Google Scholar] [CrossRef]
- Mackman, R.L.; Katz, B.A.; Breitenbucher, J.G.; Hui, H.C.; Verner, E.; Luong, C.; Liu, L.; Sprengeler, P.A. Exploiting subsite S1 of trypsin-like serine proteases for selectivity: Potent and selective inhibitors of urokinase-type plasminogen activator. J. Med. Chem. 2001, 44, 3856–3871. [Google Scholar] [CrossRef]
- Sperl, S.; Jacob, U.; Arroyo de Prada, N.; Stürzebecher, J.; Wilhelm, O.G.; Bode, W.; Magdolen, V.; Huber, R.; Moroder, L. (4-aminomethyl)phenylguanidine derivatives as nonpeptidic highly selective inhibitors of human urokinase. Proc. Natl. Acad. Sci. USA 2000, 97, 5113–5118. [Google Scholar] [CrossRef]
- Spencer, J.R.; McGee, D.; Allen, D.; Katz, B.A.; Luong, C.; Sendzik, M.; Squires, N.; Mackman, R.L. 4-Aminoarylguanidine and 4-aminobenzamidine derivatives as potent and selective urokinase-type plasminogen activator inhibitors. Bioorg. Med. Chem. Lett. 2002, 12, 2023–2026. [Google Scholar] [CrossRef]
- Duffy, F.J.; Devocelle, M.; Shields, D.C. Computational approaches to developing short cyclic peptide modulators of protein-protein interactions. Methods Mol. Biol. 2015, 1268, 241–271. [Google Scholar] [CrossRef] [PubMed]
- Guerra, Y.; Armijos-Jaramillo, V.; Pons, T.; Tejera, E.; Berry, C. Canonical or noncanonical? Structural plasticity of serine protease-binding loops in Kunitz-STI protease inhibitors. Protein Sci. 2023, 32, e4570. [Google Scholar] [CrossRef] [PubMed]
- Gryaznov, S.; Skorski, T.; Cucco, C.; Nieborowska-Skorska, M.; Chiu, C.Y.; Lloyd, D.; Chen, J.K.; Koziolkiewicz, M.; Calabretta, B. Oligonucleotide N3’-->P5’ phosphoramidates as antisense agents. Nucleic Acids Res. 1996, 24, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Summerton, J. Morpholino antisense oligomers: The case for an RNase H-independent structural type. Biochim. Biophys. Acta 1999, 1489, 141–158. [Google Scholar] [CrossRef]
- Addison, M.L.; Ranasinghe, P.; Webb, D.J. Novel Pharmacological Approaches in the Treatment of Hypertension: A Focus on RNA-Based Therapeutics. Hypertension 2023, 80, 2243–2254. [Google Scholar] [CrossRef]
- Brown, D.A.; Kang, S.H.; Gryaznov, S.M.; DeDionisio, L.; Heidenreich, O.; Sullivan, S.; Xu, X.; Nerenberg, M.I. Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J. Biol. Chem. 1994, 269, 26801–26805. [Google Scholar] [CrossRef]
- Teplova, M.; Minasov, G.; Tereshko, V.; Inamati, G.B.; Cook, P.D.; Manoharan, M.; Egli, M. Crystal structure and improved antisense properties of 2’-O-(2-methoxyethyl)-RNA. Nat. Struct. Biol. 1999, 6, 535–539. [Google Scholar] [CrossRef]
- Koshkin, A.A.; Singh, S.K.; Nielsen, P.; Rajwanshi, V.K.; Kumar, R.; Meldgaard, M.; Olsen, C.E.; Wengel, J. LNA (Locked Nucleic Acids): Synthesis of the adenine, cytosine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron 1998, 54, 3607–3630. [Google Scholar] [CrossRef]
- Swayze, E.E.; Siwkowski, A.M.; Wancewicz, E.V.; Migawa, M.T.; Wyrzykiewicz, T.K.; Hung, G.; Monia, B.P.; Bennett, C.F. Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res. 2007, 35, 687–700. [Google Scholar] [CrossRef]
- Koizumi, M. ENA oligonucleotides as therapeutics. Curr. Opin. Mol. Ther. 2006, 8, 144–149. [Google Scholar]
- Seth, P.P.; Siwkowski, A.; Allerson, C.R.; Vasquez, G.; Lee, S.; Prakash, T.P.; Wancewicz, E.V.; Witchell, D.; Swayze, E.E. Short antisense oligonucleotides with novel 2’-4’ conformationaly restricted nucleoside analogues show improved potency without increased toxicity in animals. J. Med. Chem. 2009, 52, 10–13. [Google Scholar] [CrossRef] [PubMed]
- Jason, T.L.; Koropatnick, J.; Berg, R.W. Toxicology of antisense therapeutics. Toxicol. Appl. Pharmacol. 2004, 201, 66–83. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.H.; Lim, S.; Wong, W.S. Antisense oligonucleotides: From design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 2006, 33, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Wurster, C.D.; Ludolph, A.C. Antisense oligonucleotides in neurological disorders. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418776932. [Google Scholar] [CrossRef]
- Bhalani, D.V.; Nutan, B.; Kumar, A.; Singh Chandel, A.K. Bioavailability Enhancement Techniques for Poorly Aqueous Soluble Drugs and Therapeutics. Biomedicines 2022, 10, 2055. [Google Scholar] [CrossRef]
- Li, H.; Wang, Z.; Yu, S.; Chen, S.; Zhou, Y.; Qu, Y.; Xu, P.; Jiang, L.; Yuan, C.; Huang, M. Albumin-based drug carrier targeting urokinase receptor for cancer therapy. Int. J. Pharm. 2023, 634, 122636. [Google Scholar] [CrossRef]
- Meng, W.; He, C.; Hao, Y.; Wang, L.; Li, L.; Zhu, G. Prospects and challenges of extracellular vesicle-based drug delivery system: Considering cell source. Drug Deliv. 2020, 27, 585–598. [Google Scholar] [CrossRef]
- Vandooren, J.; Opdenakker, G.; Loadman, P.M.; Edwards, D.R. Proteases in cancer drug delivery. Adv. Drug Deliv. Rev. 2016, 97, 144–155. [Google Scholar] [CrossRef]
- Liu, S.; Aaronson, H.; Mitola, D.J.; Leppla, S.H.; Bugge, T.H. Potent antitumor activity of a urokinase-activated engineered anthrax toxin. Proc. Natl. Acad. Sci. USA 2003, 100, 657–662. [Google Scholar] [CrossRef]
- Liu, S.; Netzel-Arnett, S.; Birkedal-Hansen, H.; Leppla, S.H. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res. 2000, 60, 6061–6067. [Google Scholar]
- Devy, L.; de Groot, F.M.; Blacher, S.; Hajitou, A.; Beusker, P.H.; Scheeren, H.W.; Foidart, J.M.; Noël, A. Plasmin-activated doxorubicin prodrugs containing a spacer reduce tumor growth and angiogenesis without systemic toxicity. FASEB J. 2004, 18, 565–567. [Google Scholar] [CrossRef] [PubMed]
- Fielding, A.K.; Maurice, M.; Morling, F.J.; Cosset, F.L.; Russell, S.J. Inverse targeting of retroviral vectors: Selective gene transfer in a mixed population of hematopoietic and nonhematopoietic cells. Blood 1998, 91, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Y.; Wang, F.; Gan, X.; Zheng, T.; Chen, M.; Wei, L.; Chen, J.; Yu, C. CES1-Triggered Liver-Specific Cargo Release of CRISPR/Cas9 Elements by Cationic Triadic Copolymeric Nanoparticles Targeting Gene Editing of PCSK9 for Hyperlipidemia Amelioration. Adv. Sci. 2023, 10, e2300502. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Graham, M.J.; Yu, J.; Carty, R.; Low, A.; Chappell, A.; Schmidt, K.; Zhao, C.; Aghajan, M.; Murray, H.F.; et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014, 42, 8796–8807. [Google Scholar] [CrossRef] [PubMed]
- Wohlfart, S.; Gelperina, S.; Kreuter, J. Transport of drugs across the blood-brain barrier by nanoparticles. J. Control Release 2012, 161, 264–273. [Google Scholar] [CrossRef]
- Jang, W.D.; Jeon, S.; Kim, S.; Lee, S.Y. Drugs repurposed for COVID-19 by virtual screening of 6,218 drugs and cell-based assay. Proc. Natl. Acad. Sci. USA 2021, 118, e2024302118. [Google Scholar] [CrossRef]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody-Drug Conjugates-A Tutorial Review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef]
- LeBeau, A.M.; Duriseti, S.; Murphy, S.T.; Pepin, F.; Hann, B.; Gray, J.W.; VanBrocklin, H.F.; Craik, C.S. Targeting uPAR with Antagonistic Recombinant Human Antibodies in Aggressive Breast Cancer. Cancer Res. 2013, 73, 2070–2081. [Google Scholar] [CrossRef]
- Li, R.; Zheng, K.; Hu, P.; Chen, Z.; Zhou, S.; Chen, J.; Yuan, C.; Chen, S.; Zheng, W.; Ma, E.; et al. A novel tumor targeting drug carrier for optical imaging and therapy. Theranostics 2014, 4, 642–659. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Li, R.; Zhou, X.; Hu, P.; Zhang, Y.; Huang, Y.; Chen, Z.; Huang, M. Dual actions of albumin packaging and tumor targeting enhance the antitumor efficacy and reduce the cardiotoxicity of doxorubicin in vivo. Int. J. Nanomed. 2015, 10, 5327–5342. [Google Scholar] [CrossRef]
- Zhou, Y.; Yu, S.; Chen, D.; Li, H.; Xu, P.; Yuan, C.; Jiang, L.; Huang, M. Nafamostat Mesylate in Combination with the Mouse Amino-Terminal Fragment of Urokinase-Human Serum Albumin Improves the Treatment Outcome of Triple-Negative Breast Cancer Therapy. Mol. Pharm. 2023, 20, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zheng, K.; Li, R.; Chen, Z.; Yuan, C.; Hu, P.; Chen, J.; Xue, J.; Huang, M. A drug carrier targeting murine uPAR for photodynamic therapy and tumor imaging. Acta Biomater. 2015, 23, 116–126. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Compound Names | Structures | Molecular Weight g/mol | Targets |
|---|---|---|---|---|
| 1 | Dabigatran | 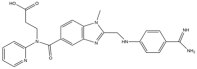 | 471.5 | Thrombin |
| 2 | Ximelagatran | 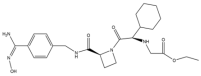 | 473.6 | Thrombin |
| 3 | Melagatran | 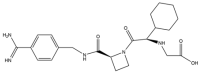 | 429.5 | Thrombin |
| 4 | AZD0837 | 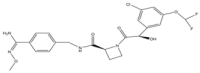 | 496.9 | Thrombin |
| 5 | Apixaban | 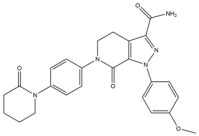 | 459.5 | FXa |
| 6 | Betrixaban | 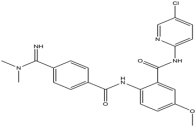 | 451.9 | FXa |
| 7 | Edoxaban | 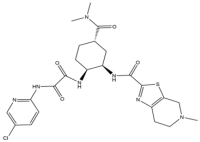 | 548.1 | FXa |
| 8 | Rivaroxaban |  | 435.9 | FXa |
| 9 | Darexaban |  | 476.4 | FXa |
| 10 | Eribaxaban | 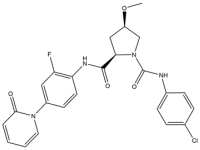 | 484.9 | FXa |
| 11 | Otamixaban |  | 446.5 | FXa |
| 12 | TAK-442 |  | 480 | FXa |
| 13 | LY517717 |  | 459.6 | FXa |
| 14 | Milvexian | 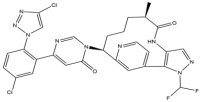 | 626.4 | FXIa |
| 15 | LNP023 | 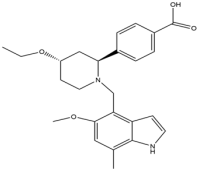 | 422.5 | CFB |
| 16 | Nafamostat | 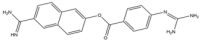 | 347.4 | broad-spectrum |
| 17 | Camostat | 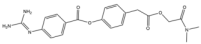 | 398.4 | broad-spectrum |
| 18 | Upamostat | 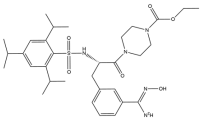 | 629.8 | broad-spectrum |
| 19 | Quercetin | 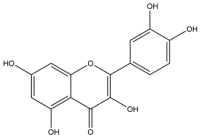 | 302.23 | broad-spectrum |
| 20 | Myricetin |  | 318.23 | broad-spectrum |
| Number | Compound | Therapy | Target | Phase | Indications | Usage Limitations | Solutions |
|---|---|---|---|---|---|---|---|
| 1 | Dabigatran | Small molecule | Thrombin | IV [155] | NVAF [38,156], VTE [157] | Emergency surgery, advanced age [158], pregnant women, severe liver and kidney dysfunction, mechanical heart valve, concomitant administration of drug classes that are strong inhibitors of CYP3A4 and P-glycoprotein [159] | Idarucizumab [160], reduce the dosage, switch to safer medications [158] |
| 2 | Ximelagatran | Small molecule | Thrombin | III | VTE [161] | Liver dysfunction [162] | No findings at this stage |
| 3 | Melagatran | Small molecule | Thrombin | III | VTE [161] | Liver dysfunction [162] | No findings at this stage |
| 4 | AZD0837 | Small molecule | Thrombin | II | AF [45], Chronic ventricular fibrillation [46] | No findings at this stage | No findings at this stage |
| 5 | Apixaban | Small molecule | Thrombin | III | DVT, PE [163] | Dialysis patients, severely impaired liver function, active pathological bleeding [54], pregnant women, severe liver and kidney dysfunction, mechanical heart valve, concomitant administration of drug classes that are strong inhibitors of CYP3A4 and P-glycoprotein [159] | Switch to safer medications |
| 6 | Betrixaban | Small molecule | Thrombin | III | VTE [164] | No findings at this stage | No findings at this stage |
| 7 | Edoxaban | Small molecule | Thrombin | III | AF [165], VTE [166] | Pregnant women, severe liver and kidney dysfunction, mechanical heart valve, concomitant administration of drug classes that are strong inhibitors of CYP3A4 and P-glycoprotein [159] | Anticoagulation with intravenous unfractionated heparin, low molecular weight heparin (LMWH), or warfarin [167] |
| 8 | Rivaroxaban | Small molecule | Thrombin | III | VTE [168] | Pregnant women, severe liver and kidney dysfunction, mechanical heart valve, concomitant administration of drug classes that are strong inhibitors of CYP3A4 and P-glycoprotein [159] | Anticoagulation with intravenous unfractionated heparin, low molecular weight heparin (LMWH), or warfarin [167] |
| 9 | Darexaban | Small molecule | Thrombin | III | ACS, AF, DVT, PE [169] | No findings at this stage | No findings at this stage |
| 10 | Otamixaban | Small molecule | Thrombin | II | ACS, Non-urgent PCI [170] | No findings at this stage | No findings at this stage |
| 11 | TAK-442 | Small molecule | Thrombin | II | ACS [171] | No findings at this stage | No findings at this stage |
| 12 | LY517717 | Small molecule | Thrombin | II | DVT, PE [172] | No findings at this stage | No findings at this stage |
| 13 | Milvexian | Small molecule | FXIa | II | No findings at this stage [63] | No findings at this stage | No findings at this stage |
| 14 | LNP023 | Small molecule | Complement Factor B | III | PNH [173], IgAN [174] | No findings at this stage | No findings at this stage |
| 15 | Nafamostat | Small molecule | broad-spectrum | IV | Acute pancreatitis, AKI, various malignant tumors [65] | No findings at this stage | No findings at this stage |
| 16 | Camostat | Small molecule | broad-spectrum | IV | Chronic pancreatitis, post-operative reflux esophagitis, COVID-19 [175] | No findings at this stage | No findings at this stage |
| 17 | Upamostat | Small molecule | broad-spectrum | II | Pancreatic cancer, breast cancer, COVID-19 [176] | No findings at this stage | No findings at this stage |
| 18 | Aprotinin | Polypeptide | broad-spectrum | IV | Anti-inflammatory and antithrombotic during operation [95] | Sensitive to Aprotinin [177,178] | Switch to safer medications |
| 19 | Antipain | Polypeptide | broad-spectrum | IV | Analgesia [179,180] | No findings at this stage | No findings at this stage |
| 20 | Bacitracin A | Polypeptide | broad-spectrum | IV | Bacterial infection [181] | No findings at this stage | No findings at this stage |
| 21 | leupeptin | Polypeptide | broad-spectrum | IV | COVID-19 [182] | No findings at this stage | No findings at this stage |
| 22 | Desirudin | Polypeptide | Thrombin | III | VTE [44] | Renal dysfunction [183] | Dose adjustment [183] |
| 23 | PEG-Hirudin | Polypeptide | Thrombin | II | No findings at this stage | No findings at this stage | No findings at this stage |
| 24 | MK-0616 | Polypeptide | PCSK9 | II | Hypercholesterolemia [184] | No findings at this stage | No findings at this stage |
| 25 | VX-950 | Polypeptide | NS3/4A protease | I | Chronic hepatitis C [185] | Skin disease [186] | Discontinue and take oral corticosteroids [187] |
| 26 | AZD8233 | ASO | PCSK9 | II | Hypercholesterolemia | No findings at this stage | No findings at this stage |
| 27 | Fesomersen | ASO | FXIa | II | No findings at this stage [188] | No findings at this stage | No findings at this stage |
| 28 | IONIS-FXIRX | ASO | FXIa | II | No findings at this stage [188] | No findings at this stage | No findings at this stage |
| 29 | Inclisiran | SiRNA | PCSK9 | III | Hypercholesterolemia [81,189] | No findings at this stage | No findings at this stage |
| 30 | Sibrotuzumab | Antibody | FAP | I | Fibroblast activation protein-positive cancer [190] | No findings at this stage | No findings at this stage |
| 31 | Evolocumab | Antibody | PCSK9 | III | Hyperlipidemia [147] | Liver function impairment [191] | No findings at this stage |
| 32 | Alirocumab | Antibody | PCSK9 | IV | Hypercholesterolemia [192] | No findings at this stage | No findings at this stage |
| 33 | Narsoplimab | Antibody | MASP-2 | III | IgAN [193] | No findings at this stage | No findings at this stage |
| 34 | XIsomab 3G3 | Antibody | FXIa | II | No findings at this stage [188] | No findings at this stage | No findings at this stage |
| 35 | Osocimab | Antibody | FXIa | II | VTE [194] | No findings at this stage | No findings at this stage |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Huang, M.; Jiang, L. Advancements in Serine Protease Inhibitors: From Mechanistic Insights to Clinical Applications. Catalysts 2024, 14, 787. https://doi.org/10.3390/catal14110787
Wei Y, Huang M, Jiang L. Advancements in Serine Protease Inhibitors: From Mechanistic Insights to Clinical Applications. Catalysts. 2024; 14(11):787. https://doi.org/10.3390/catal14110787
Chicago/Turabian StyleWei, Yang, Mingdong Huang, and Longguang Jiang. 2024. "Advancements in Serine Protease Inhibitors: From Mechanistic Insights to Clinical Applications" Catalysts 14, no. 11: 787. https://doi.org/10.3390/catal14110787
APA StyleWei, Y., Huang, M., & Jiang, L. (2024). Advancements in Serine Protease Inhibitors: From Mechanistic Insights to Clinical Applications. Catalysts, 14(11), 787. https://doi.org/10.3390/catal14110787