Abstract
Several chemical and biocatalytic methods have been described for chiral γ-lactams syntheses. However, only one biocatalytic method has been reported for γ4-lactam resolution, while γ2- and γ3-lactams have not been reported. On the other hand, its resolution through biocatalysts is complicated since enzymes such as ENZA-1 (Rhodococcus equi NCIB 40213) and ENZA-20 (Pseudomonas solanacearum NCIB 40249) are difficult to obtain. Therefore, in this paper, the resolution of γ-lactams 7-9 was carried out through a hydrolysis reaction using the commercially available enzyme CaLB.
1. Introduction
γ-lactams are scaffolds of biologically active chiral compounds and drugs, such as the proteasome inhibitor Lactacystin [1], the antidepressant drug (R)-Rolipram [2], and the respiratory stimulant Doxapram [3] (Figure 1).
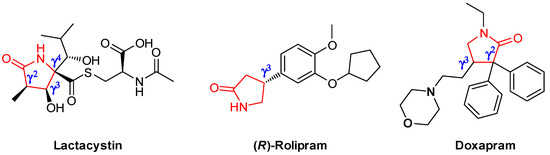
Figure 1.
Examples of biologically active chiral compounds that have a γ-lactam as their base nucleus and different substitution patterns.
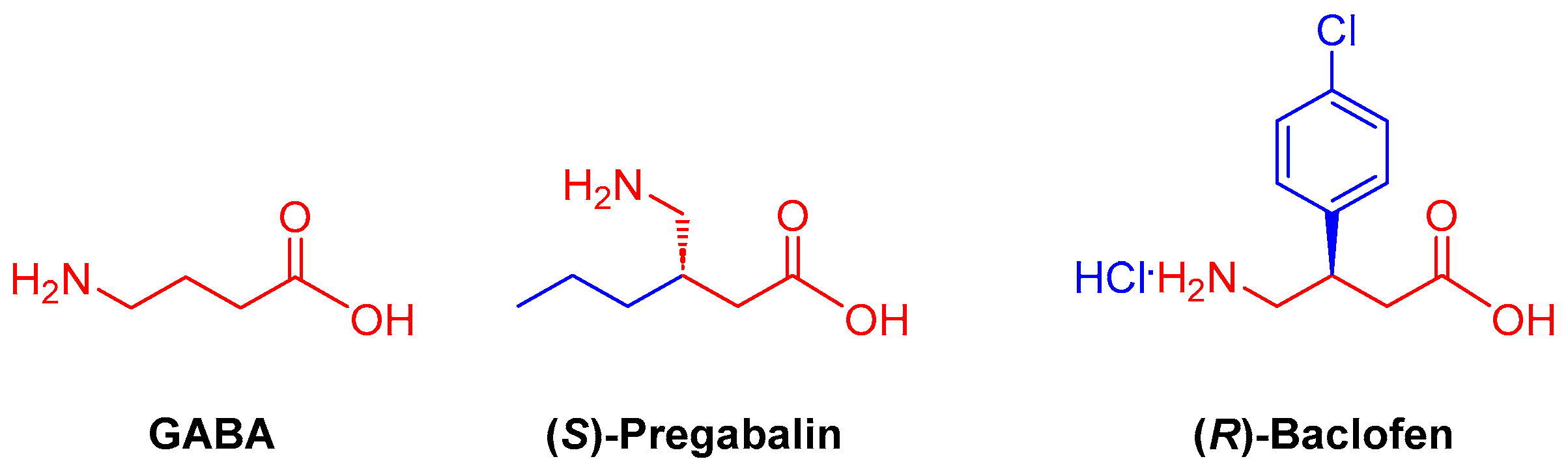
Furthermore, its ring-open derivatives (Figure 2) are necessary for protein synthesis, so its deficiency is associated with diseases [4]. Deficiency of the γ-amino acid (GABA) is associated with several neurological disorders, such as schizophrenia [5], attention-deficit/hyperactivity disorder [6], and major depressive disorder [7]. Likewise, they are key intermediaries to access a variety of natural products and drug molecules; for example, (S)-Pregabalin is an anticonvulsant with anxiolytic and analgesic properties [8], while (R)-Baclofen is a muscle relaxant [9].

Figure 2.
Examples of γ-amino acids with biological properties of great relevance.
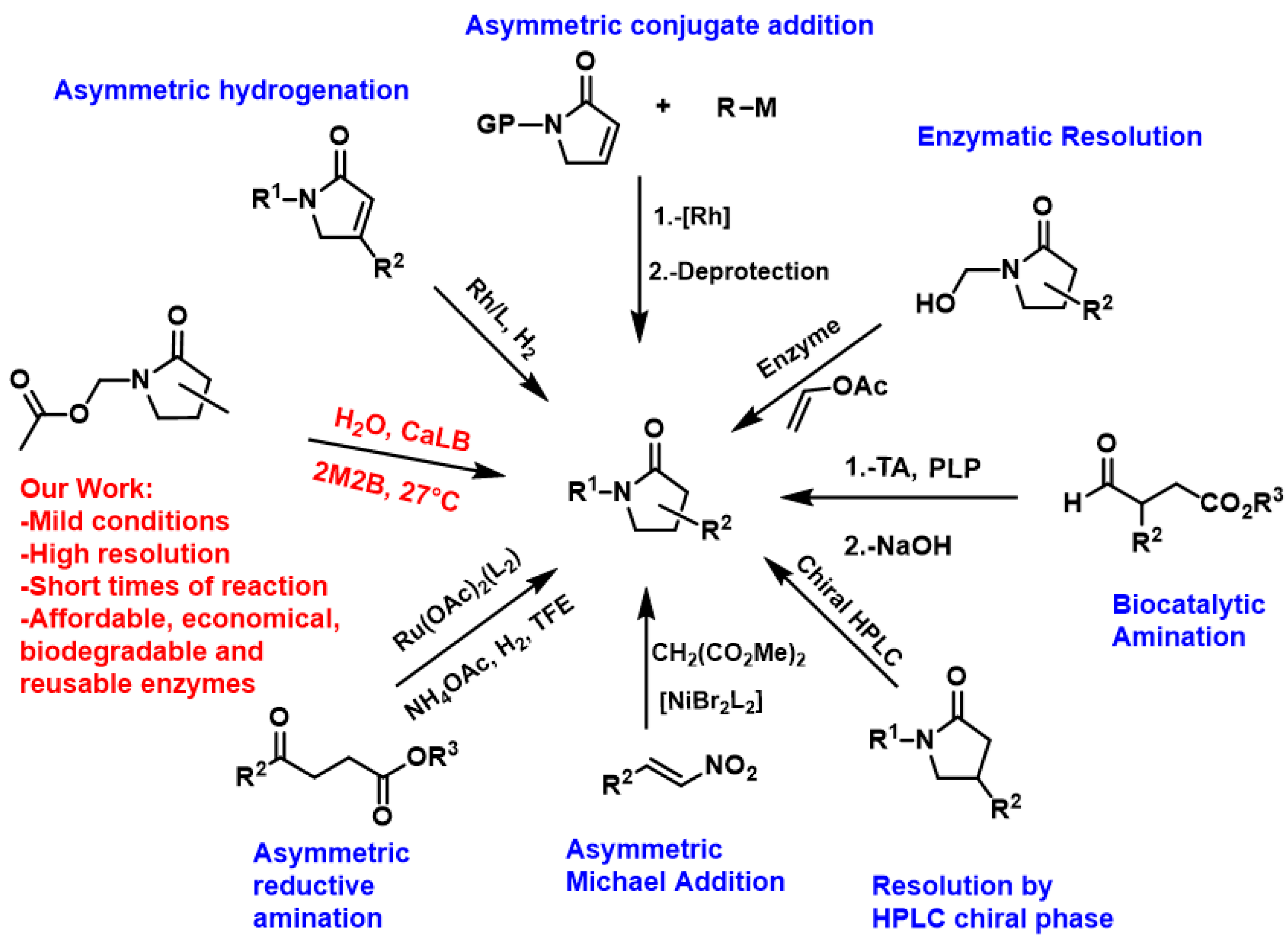
Due to these properties, tremendous efforts have been dedicated to the preparation of chiral γ-lactams, through chemocatalysis as well as biocatalysis, among others [10]. For example, within chemocatalysis, the Pd-catalyzed asymmetric conjugate addition of arylboronic acids to α,β-unsaturated γ-lactams, represents a simple and efficient strategy for obtaining chiral γ-lactams [11]. Zhang et al. [12] used a Rh catalytic system for the asymmetric synthesis of chiral β-substituted γ-lactams through the asymmetric hydrogenation of α,β-substituted β-lactams. Another method is reductive amination of ketoacids/esters followed by intramolecular cyclization, which is considered one of the most attractive approaches to access chiral γ-lactams. Yin et al. [13] carried out an asymmetric reductive amination cascade reaction catalyzed by Ru and its cyclization, providing enantioenriched γ-lactams with up to 97% enantiomeric excess. However, this method employs heavy metals and contaminants such as Rh and Ru, which are not abundant in nature and are also expensive.
On the other hand, the methods for its chemical synthesis are generally based on the formation of C-C bonds through the Michael reaction. In this case, Zlotin et al. [14] carried out the asymmetric Michael addition reaction between nitroalkenes and 1,3-dicarbonyl compounds catalyzed by Ni(II). Another method used is the separation of the racemic mixture by liquid chromatography with a chiral stationary phase (chiral HPLC) [15]. Likewise, Biocatalytic amination is another feasible method for the synthesis of chiral γ-lactams. For example, Kroutil et al. [16] carried out the asymmetric amination of aldehydes with a library of wild-type transaminases (TA), obtaining γ-aminoesters, and subsequently carried out ring closure using basic medium to obtain chiral γ-lactams.
On the other hand, enzymatic resolution is a methodology also used to obtain chiral γ-lactams. Rousseau et al. [17] carried out the enzymatic resolution of γ-butyrolactams, through a transesterification with vinyl acetate catalyzed by porcine pancreatic lipases and Pseudomona cepacia. However, they were only able to resolve the γ4-lactam and not the resolution of the γ2-lactam. Also, Fülop et al. [18] carried out enzymatic resolutions for abacavir intermediate with CaLB through ring cleavage obtaining good enantiomeric selectivities. As described previously, the methods reported in the literature have several disadvantages, such as the use of metal catalysts, which are expensive with low abundance in nature, in addition to polluting the environment and having long reaction times. In the literature so far, only one method for biocatalytic γ4-lactam resolution has been reported, while γ2- and γ3-lactams have not been reported.
Also, the resolution of γ-lactams through biocatalysts is complicated since enzymes such as ENZA-1 (Rhodococcus equi NCIB 40213) and ENZA-20 (Pseudomonas solanacearum NCIB 40249) [19] are difficult to obtain. In this context, Candida antarctica lipase B (CaLB) is commercially available at a low cost, both in its free and immobilized form, with the latter being easy to use and recycle. It has also been the most used lipase in recent years, due to its characteristics such as catalytic promiscuity, thermostability, high activity in organic solvents, as well as its regio-, chemo-, and enantioselectivity [20]. For us, it is of utmost importance to corroborate whether the CaLB enzyme is capable of resolving γ2-, γ3-, and γ4-lactams, which is why in this work the resolution was carried out through a hydrolysis reaction (Figure 3).

Figure 3.
Methods for obtaining chiral γ-lactams: Asymmetric conjugate addition [11], Asymmetric hydrogenation [12], Asymmetric reductive amination [13], Asymmetric Michael Addition [14], Resolution by HPL chiral phase [15], Biocatalytic Amination [16], Enzymatic Resolution [17].
2. Results and Discussion
2.1. γ-Lactams Syntheses
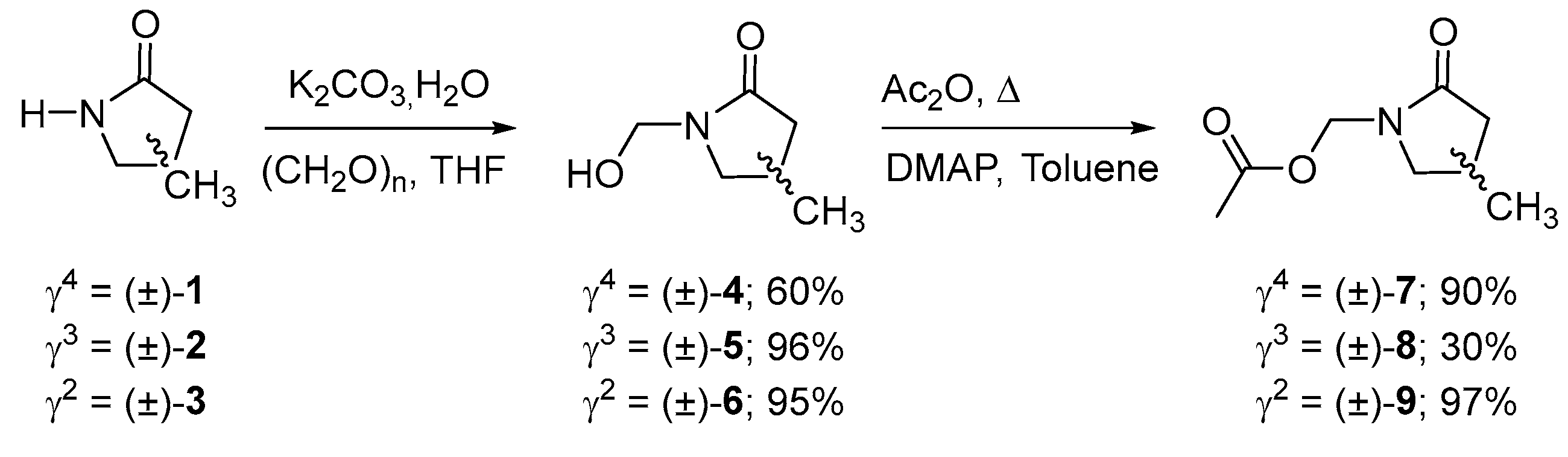
γ2-, γ3, and γ4-lactams (±)-1-3, were obtained using the methodology described by us [21]. Subsequently, a hydroxymethylenation reaction was carried out using the methodology described by Giacomini et al. [22], with 0.04 eq of potassium bicarbonate, 3 eq of paraformaldehyde, and water refluxed in THF over 3 h, obtaining the N-substituted γ-lactams (±)-4-6, with yields of 60, 96, and 95%, respectively. Subsequently, its acylation was carried out using 1.2 eq of acetic anhydride and 1 eq of DMAP in toluene at reflux over 1 h, obtaining the products (±)-7-9 with yields of 90, 30, and 97%, respectively (Scheme 1).
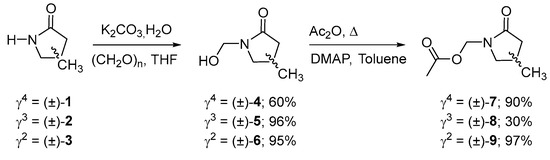
Scheme 1.
Synthesis of the N-substituted γ-lactams (±)-7-9.
2.2. Enzymatic Resolution of γ-Lactams (±)-7-9
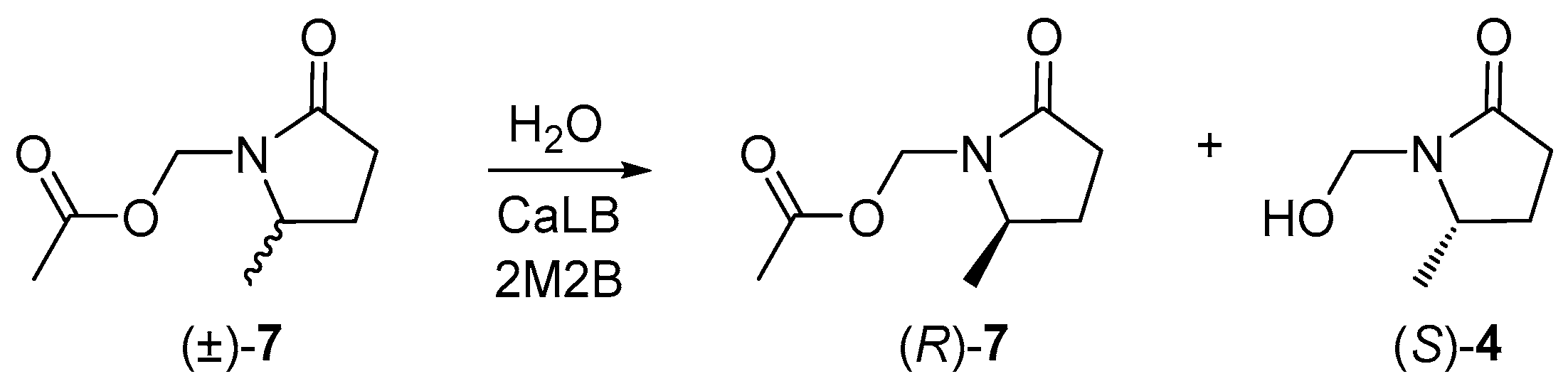
Once γ-lactams (±)-7-9 were synthesized, γ4-lactam (±)-7 was used as a model for enzymatic resolution, because it was expected to show greater recognition by the enzyme as the stereogenic center would be closer to the catalytic site. Therefore, the optimal conditions for its resolution were sought. First, an enzymatic hydrolysis reaction was carried out using the CaLB, water as the nucleophile, and 2-methyl-2-butanol (2M2B) as a solvent (Scheme 2, entry 1, Table 1). Variations in temperature, nucleophile equivalents, as well as enzyme concentration results, are shown in Table 1.

Scheme 2.
CaLB-catalyzed enzymatic resolution of (±)-7.

Table 1.
Optimization of the enzymatic resolution for compound (±)-7.
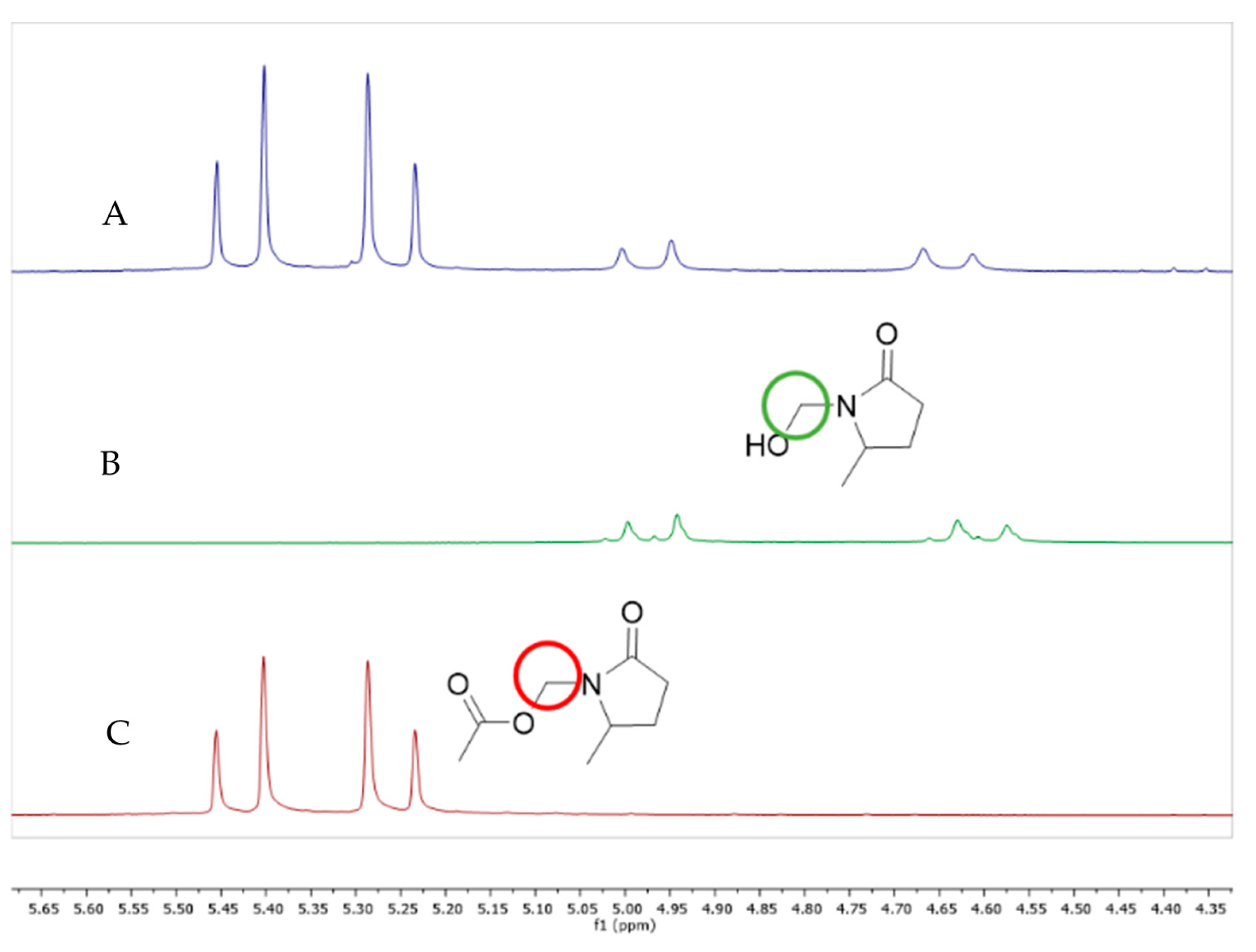
The ratio between the product and starting material was determined by 1H NMR of crude oil (Figure 4). Crude oil was purified by a gradient chromatographic column to obtain the isolated compound. Optical rotation was taken as +7.1°, thus allowing us to correlate it with (S) enantiomer, which is reported in the literature [17]. Likewise, enantiomeric excess was determined by chiral HPLC.

Figure 4.
1H NMR signals for (A) Crude oil, (B) γ4-lactam 7 and (C) γ4-lactam 4.
As can be seen from Table 1, the best conditions to obtain γ4-lactam (S)-4 with high e.e. are those from entry 4, in which the reaction benefits from a longer reaction time and higher water equivalents.
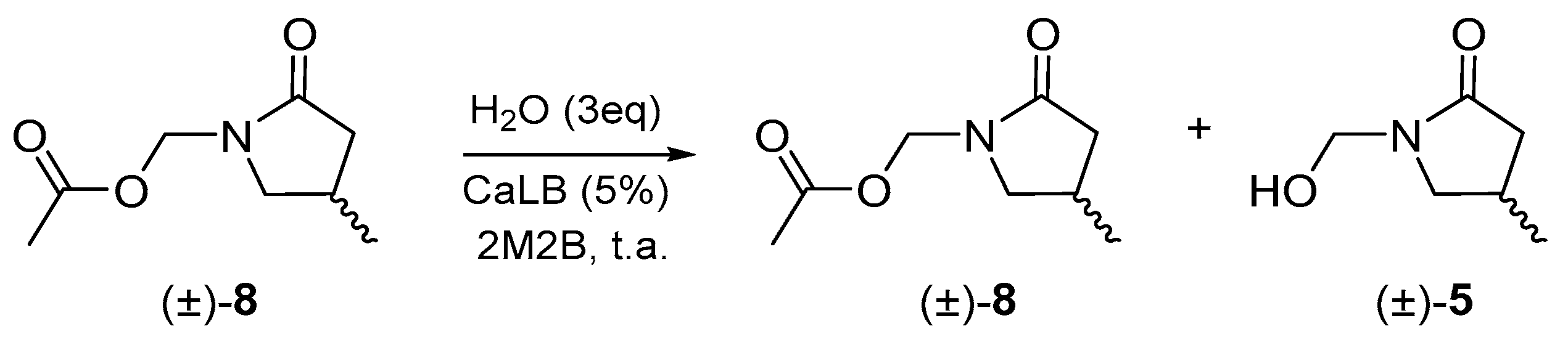
Once the optimal conditions for the resolution of the γ4-lactam (±)-7 were found, they were used to carry out enzymatic resolution on γ3-lactam (±)-8. However, the enzyme had poor selectivity according to the enantiomeric excess calculated (Scheme 3, Table 2).

Scheme 3.
Enzymatic resolution for γ3-lactam (±)-8.
Table 2.
Enzymatic resolution for γ3-lactam (±)-8.
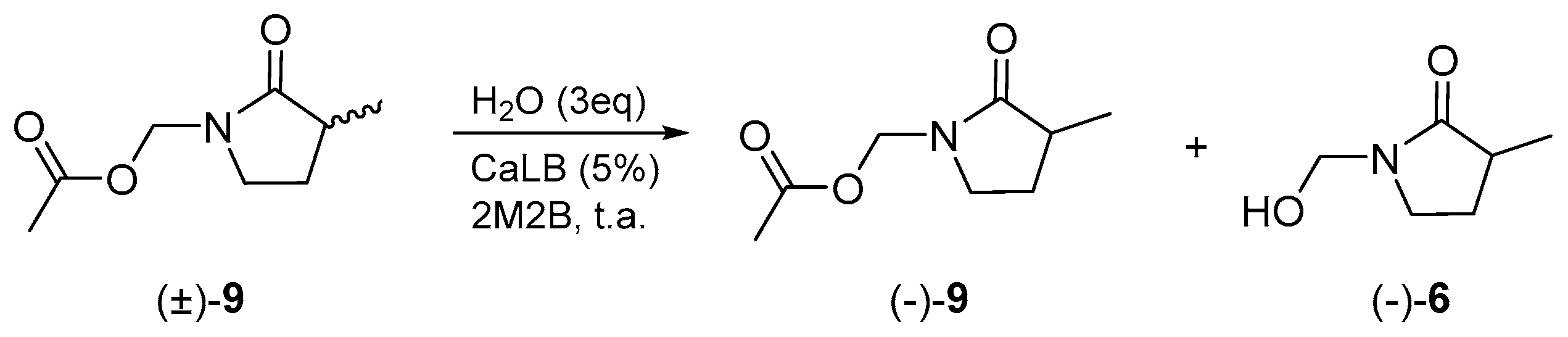
On the other hand, the resolution of γ2-lactam (±)-9 was carried out, observing moderate recognition by the enzyme. The optical rotation was taken as −10.7°, and the enantiomeric excess and enantiomeric ratio (E) were calculated (Scheme 4, Table 3).

Scheme 4.
Enzymatic resolution for γ2-lactam (±)-9.
Table 3.
Enzymatic resolution for compound (±)-9.
Table 4 compares the best results obtained for each N-substituted γ2-, γ3-, and γ4-lactams (±)-7-9. As can be seen, a change in the position of the methyl group is important because it affects enzymatic recognition. Furthermore, these results are of great importance since the resolution by hydrolysis of γ-lactams has not been reported so far.
Table 4.
Enzymatic resolution for γ-lactams (±)-9.
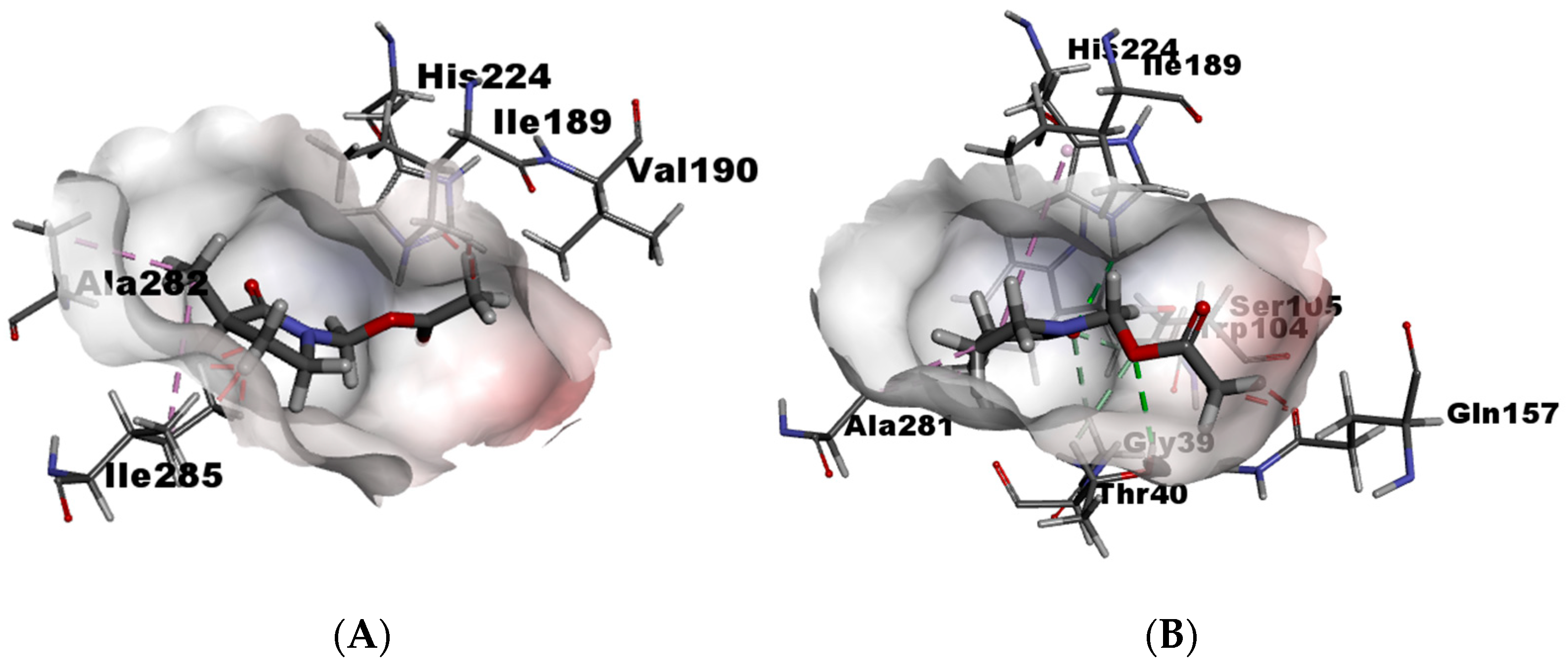
To explain these results, in silico calculations were performed employing Molegro Virtual Docker (See Supplementary Materials). In these calculations, it is possible to know the affinity enzyme-substrate through interactions with amino acid residues, and substrate orientation within the catalytic site. For γ4-lactam (±)-7, enantiomers (S) and (R) were compared. The in silico results show that γ-lactam (R) has unfavorable bumps with Ser105 because of the orientation occupied by the acyl group in the catalytic site (Figure 5A), while γ-lactam (S) does not present unfavorable bumps since the acyl group adopts a better conformation inside the catalytic site (Figure 5B). The orientation of both carbonyl groups seems to play an important role in recognition by the enzyme. In the case of (S), both carbonyls are oriented towards the interior of the active site, generating hydrogen bonds with Gln157, Thr40, Ser105, and His224, which are amino acids of the pocket acyl and the catalytic triad, thus enabling better recognition.
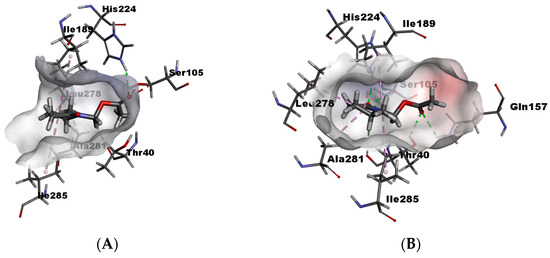
Figure 5.
(A) γ4-lactam-7-(R) within catalytic site and (B) γ4-lactam-7-(S) within catalytic site.
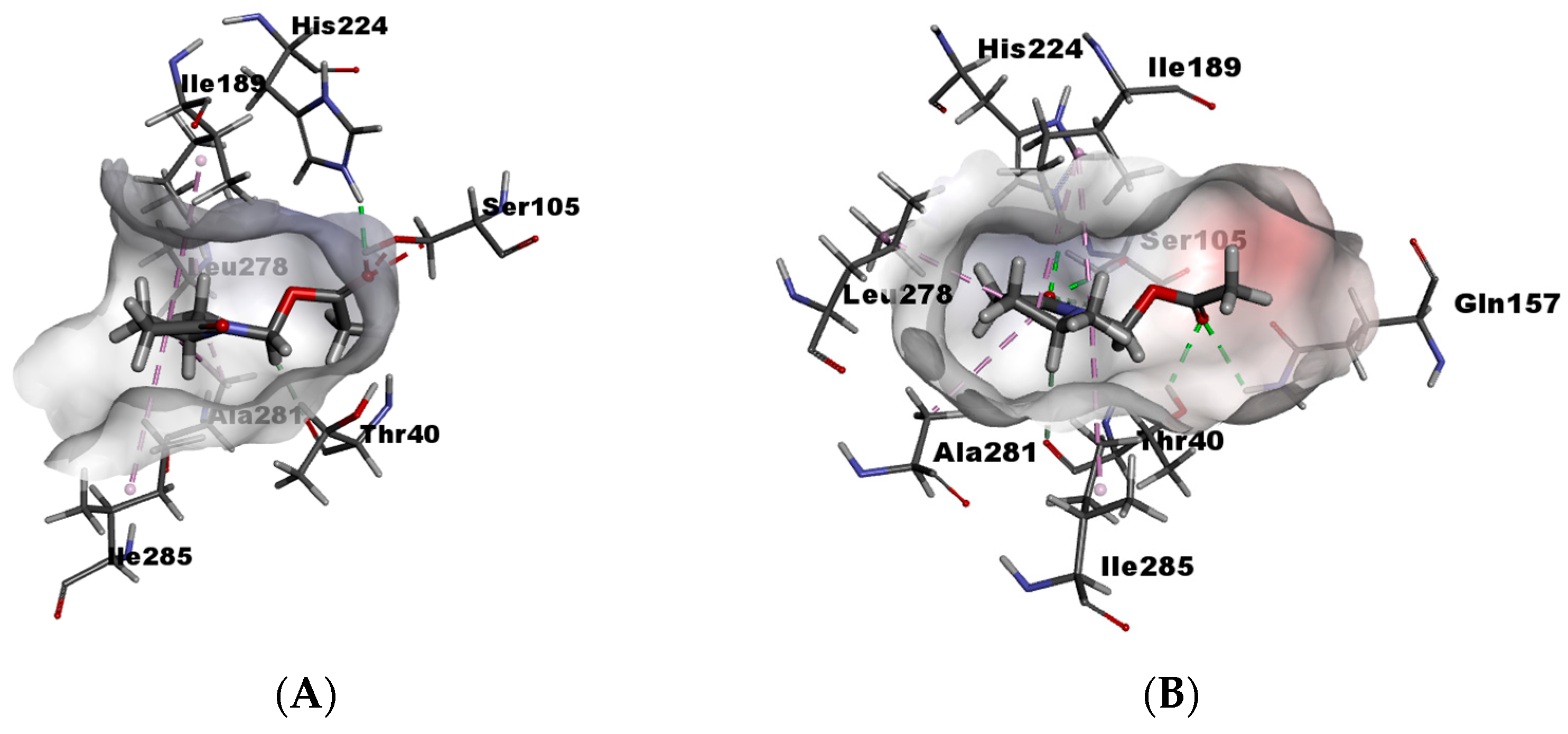
For γ3-lactam (±)-8 enantiomers with the enzyme, it can be seen that the (R) enantiomer has a slightly better recognition because it forms hydrogen bonds with Thr40, His224, and Ser105, and does not have unfavorable bumps (Figure 6A).

Figure 6.
(A) γ3-lactam-8-(R) within catalytic site and (B) γ3-lactam-8-(S) within catalytic site.
In contrast, its (S) enantiomer presents hydrogen bonds with His224, and Ser105 also presents an unfavorable bump with Gln157 (Figure 6B). However, the interaction difference is not very significant, and experimentally, low selectivity is observed.
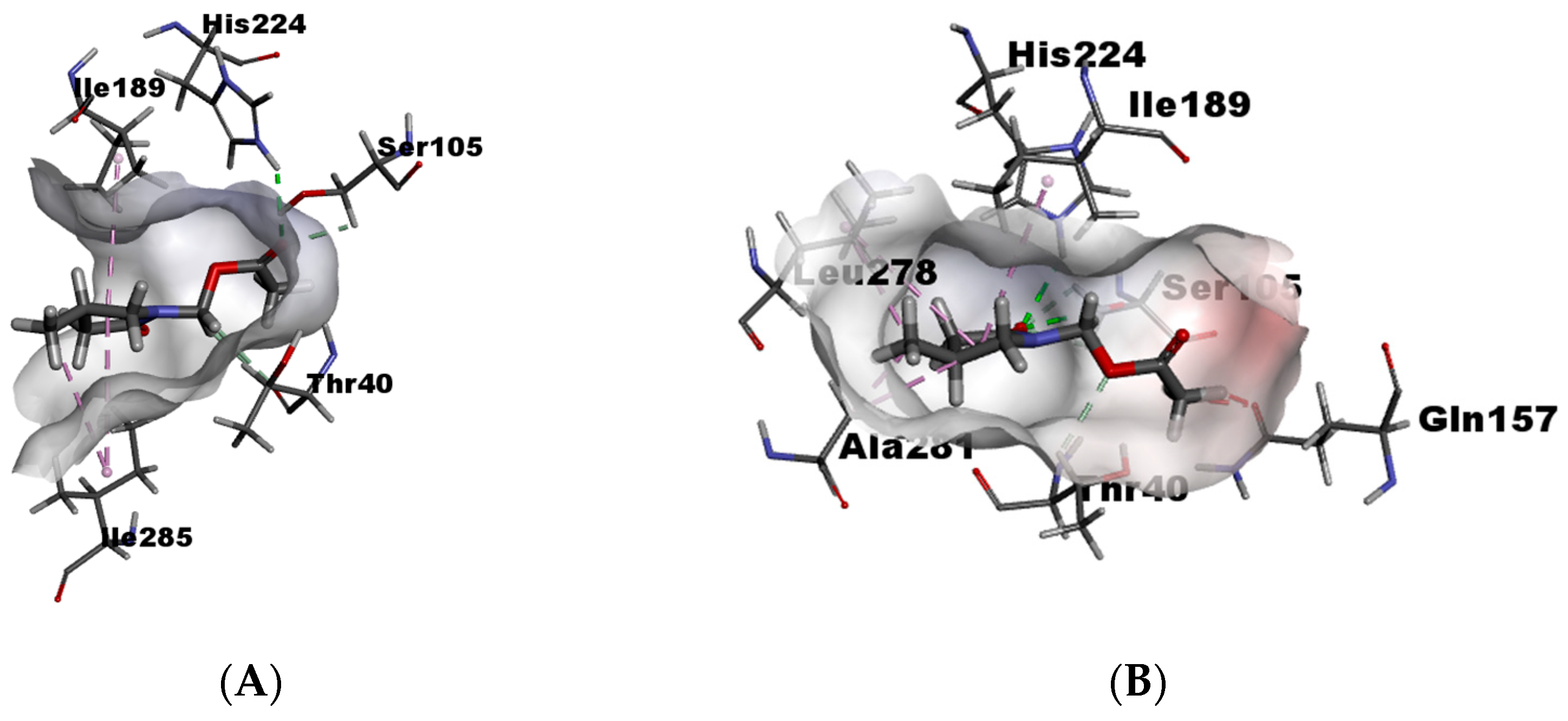
On the other hand, in γ2-lactam (±)-9, the (S) enantiomer has better recognition due to the presence of a greater number of favorable interactions as hydrogen bondings with His224, Ser105, and Thr40 (Figure 7B). In contrast, the (R) enantiomer presents unfavorable bumps with Ile189 and Ile285 and no hydrogen bonding (Figure 7A), which results in better selectivity towards the (S) enantiomer than the (R).

Figure 7.
(A) γ2-lactam-9-(R) within catalytic site and (B) γ2-lactam-9-(S) within catalytic site.
These in silico results are consistent with the experimental results obtained.
3. Materials and Methods
The lipase Novozym® 435 (Novozymes, Mexico City, Mexico) was used as the catalyst for all enzymatic reactions. Novozym® 435 is the commercially available form of the recombinant Candida antarctica lipase B, immobilized by adsorption in a hydrophobic microporous acrylic resin. Reactions were monitored by thin-layer chromatography (TLC) and carried out on 0.25 mm Merck (Rahway, NJ, USA) silica gel plates (60F-254) using UV light as the visualizing agent. Column chromatography was performed on Merck silica gel 60 (0.040–0.063 mm). 1H and 13C NMR spectra were recorded on Varian Gemini 200 MHz equipment. Chemical shift δ values (ppm) are reported relative to the internal tetramethylsilane (TMS) reference (δ = 0.0 ppm) for determinations in CDCl3. Microwave reactions were performed in sealed vessels in a CEM (Matthews, NC, USA) Discover apparatus.
Molecular Docking.
Structures were created in Spartan 14 [23] and then submitted to conformational analysis within molecular mechanics level theory and an MMFF force field. Conformers of minimum energy were selected and optimized within DFT level theory using B3LYP [24,25] and the 6-31G* basis set. Molecular docking calculations were carried out in Molegro Virtual Docker (MVD) [26], using the Cal-B structure PDB: 1LBS [27]. All docking images were made with Discovery Studio Visualizer [28]. See Supplementary Materials for more details.
Syntheses of γ-lactams (±)-1-3; General Procedure 1 (GP1).
γ-lactams (±)-1-3 were obtained by preparing the γ-nitroaliphatic methyl esters under microwave irradiation, as has been reported [20]. Then, the reduction of the nitro group using Pd/C [29] yielded the corresponding γ-lactams (±)-1-3, resulting from a tandem cyclization of the free amines.
γ-Nitroaliphatic methyl esters synthesis.
A solution of α,β-unsaturated ester (methyl methacrylate, methyl crotonate, and methyl acrylate), and the corresponding nitroalkane (nitromethane or nitroethane) in DBU or TMG were added to a 10 mL glass microwave reaction vessel containing a stir bar. The reaction vessel was sealed with a cap and placed into the microwave cavity. The microwave unit was programmed to 70–75 °C with a power of 50 Watts over 5 min. After the reaction was completed and the vessel was cooled to below 50 °C using a compressed air flow, the crude material was purified by column chromatography to give the nitro methyl esters.
Nitro group reduction.
Ammonium formate (8 eq) was added to a solution of nitro methyl esters (1 eq), with Pd/C in MeOH and the mixture was stirred overnight. The solution is then filtered with a vacuum and Pd/C is washed with absolute methanol (10 mL). γ-lactams (±)-1-3 were purified by flash column chromatography.
Synthesis of N-hydroxymethylated γ-lactams (±)-4-6; General Procedure 2 (GP2).
In a 50 mL round flask equipped with a magnetic stirrer, the corresponding γ-lactam dissolved in THF was placed. Subsequently, potassium bicarbonate (0.04 eq), paraformaldehyde (3 eq), and HPLC water (20.1 mmol) were added. The reaction was refluxed over 3 h. After obtaining the corresponding N-hydroxymethylated γ-lactam, it was purified by column chromatography.
Synthesis of N-Acetoxymethyl γ-lactams (±)-7-9; General Procedure 3 (GP3).
In a 50 mL round flask equipped with a magnetic stirrer, the corresponding N-hydroxymethylated γ-lactam dissolved in toluene was placed. Subsequently, acetic anhydride (1.2 eq) and DMAP (1 eq) were added, and the reaction mixture was brought to reflux for 1 h. The corresponding N-substituted γ-lactam was purified by column chromatography.
(rac)-5-Methylpyrrolidin-2-one, (±)-1. Prepared according to GP1, methyl acrylate (20 mmol, 1.81 mL) was mixed with nitroethane (25 mmol, 1.78 mL), DBU (1 mmol, 0.15 mL), and irradiated with microwaves over 5 min at 70 °C and 50 W potency. Crude was purified (Hex:AcOEt 90:10 → 70:30) to obtain a colorless oil, yield: 1.61 g (50%). Methyl nitroester (12.4 mmol, 2 g) was placed in a 250 mL round flask, and MeOH (75 mL), Pd/C (6% p/p, 0.12 g), and ammonium formate (99.2 mmol, 6.25 g) were added. The mixture was stirred overnight, crude was filtered, rotaevaporated, and after purification by flash chromatographic column (Hex:AcOEt 90:10 → 60:40), γ-lactam (±)-1 was isolated as a colorless oil. Yield: 1.03 g (84%). 1H NMR (200 MHz, CDCl3): δ 1.23 (d, J = 6.2 Hz, 3H, CH3), 1.60–1.71 (m, 1H, CH2CH2CHN), 2.18–2.40 (m, 3H, CH2CH2CHN), 3.68–3.91 (m, 1H, CHN). 13C NMR (50 MHz, CDCl3): 178.6, 50.4, 30.8, 29.3, 22.4. Compared with lit [30].
(rac)-4-Methylpyrrolidin-2-one, (±)-2. Prepared according to GP1, methyl crotonate (35 mmol, 3.7 mL) was mixed with nitromethane (87.5 mmol, 4.72 mL) and DBU (1.75 mmol, 0.261 mL) and irradiated with microwaves over 15 min at 75 °C and 50 W potency. Crude was purified (Hex:AcOEt 98:02 → 90:10) to obtain a colorless oil, yield: 2.98 g (53%). Methyl nitroester (12.4 mmol, 2 g) was placed in a 250 mL round flask, MeOH (75 mL), Pd/C (6% p/p, 0.12 g) and ammonium formate (99.2 mmol, 6.25 g) were added. The mixture was stirred overnight, crude was filtered, rotaevaporated, and after purification by flash chromatographic column (Hex:AcOEt 90:10 → 60:40), γ-lactam (±)-2 was isolated as a colorless oil. Yield: 0.92 g (75%). 1H NMR (200 MHz, CDCl3): δ 1.13 (d, J = 6.6 Hz, 3H, CH3),1.94–2.05 (dd, J = 5.0, 15.4 Hz, 1H, CH2CH), 2.4–2.61 (m, 2H, CH2CH), 3.18–3.26 (dd, J = 5.8, 7.5 Hz, 1H, CH2N), 3.69–3.78 (dd, J = 8.2 Hz, 1H, CH2N). 13C NMR (50 MHz, CDCl3): δ 170.1, 56.4, 37.1, 24.3, 20.5. Compared with lit [22].
(rac)-3-Methylpyrrolidin-2-one, (±)-3. Prepared according to GP1, methtyl methacrylate (20 mmol, 2.1 mL), was mixed with nitromethane (25 mmol, 1.35 mL) and TMG (10 mmol, 1.25 mL) and irradiated with microwaves over 20 min at 70 °C and 50 W potency. Crude was purified (Hex:AcOEt 90:10 → 70:30) to obtain a colorless oil, yield: 1.38 g (43%). Methyl nitroester (12.4 mmol, 2 g) was placed in a 250 mL round flask, and MeOH (75 mL), Pd/C (6% p/p, 0.12 g) and ammonium formate (99.2 mmol, 6.25 g) were added. The mixture was stirred overnight, crude was filtered, rotaevaporated, and after purification by flash chromatographic column (Hex:AcOEt 90:10 → 60:40). γ-lactam (±)-3 was isolated as a colorless oil. Yield: 1.1 g (90%). 1H NMR (200 MHz, CDCl3: δ 1.20 (d, J = 6.6 Hz, 3H, CH3), 1.50–1.70 (m, 1H, CH2CH), 2.22–2.37 (m, 3H, CH2CHCH2N), 3.70–3.86 (m, 1H, CH2N). 13C NMR (50 MHz, CDCl3): δ 180.6, 64.5, 31.7, 30.2, 30.2. Compared with lit [22].
(rac)-1-(Hydroxymethyl)-5-methylpyrrolidin-2-one (±)-4. Prepared according to GP2, (±)-1 (5.04 mmol, 500 mg), potassium bicarbonate (0.2 mmol, 27.3 mg), paraformaldehyde (15.13 mmol, 454 mg), and water (20.1 mmol, 0.362 mL) were refluxed in THF (20 mL) over 3 h. After chromatographic column (Hex:AcOEt 90:10 → 0:100), γ-lactam (±)-4 was isolated as a colorless oil. Yield: 0.39 g (60%). 1H NMR (200 MHz, CDCl3): δ 4.99 (dd, J = 10.9, 6.6 Hz, 1H CH2OH), 4.62 (dd, J = 8.5, 10.9 Hz, 1H CH2OH), 3.81 (m, 2H, CHCH3, OH), 2.42 (m, 2H, CH2CO), 2.20 (m, 1H, CH2CH), 1.60 (m, 1H, CH2CH), 1.29 (d, J = 6.3 Hz, 3H, CH3). 13C NMR (50 MHz, CDCl3): δ 176.6, 65.0, 53.2, 30.7, 27.1, 20.4.
(rac)-1-(Hydroxymethyl)-4-methylpyrrolidin-2-one (±)-5. Prepared according to GP2, (±)-2 (11 mmol, 1.09 g) potassium bicarbonate (0.4 mmol, 60.6 mg), paraformaldehyde (33 mmol, 0.99 g), and water (40.2 mmol, 0.788 mL) were refluxed in THF (40 mL) over 3 h. After chromatographic column (Hex:AcOEt 90:10 → 0:100), γ-lactam (±)-5 was isolated as a colorless oil. Yield: 1.24 g (96%). 1H NMR (200 MHz, CDCl3): δ 4.74 (AB, J = 10.6 Hz, 2H, CH2OH), 3.68 (dd, J = 7.7, 9.6 Hz, 1H, CH2NH), 3.16 (dd, J = 5.9, 9.6 Hz, 1H, CH2NH), 2.50 (m, 2H, CHCH3, CH2CO), 2.03 (m, 1H, CH2CO), 1.14 (d, J = 6.6 Hz, 3H, CH3). 13C NMR (50 MHz, CDCl3): δ 175.8, 66.3, 53.4, 37.0, 39.7, 26.5, 19.6.
(rac)-1-(Hydroxymethyl)-3-methylpyrrolidin-2-one (±)-6. Prepared according to GP2, (±)-3 (1 mmol, 100 mg), potassium bicarbonate (0.4 mmol, 5.6 mg), paraformaldehyde (3 mmol, 90.8 mg), and water (72.5 µL) were refluxed in THF (12.5 mL) over 3 h. After chromatographic column (Hex:AcOEt 90:10 → 0:100), γ-lactam (±)-6 was isolated as a colorless oil. Yield: 0.12 g (95%). 1H NMR (200 MHz, CDCl3): δ 4.8 (d, J = 10.7 Hz, 1H CH2OH), 4.68 (d, J = 10.7 Hz, 1H CH2OH), 3.49 (dd, J = 8.6, 1.5 Hz, 1H CH2CH2CH), 3.46 (d, J = 8.5 Hz, 1H CH2CH2CH), 2.46 (m, 1H, CH2CH), 2.23 (m, 1H, CHCH3), 1.62 (m, 1H, CH2CH), 1.16 (d, J = 7.1 Hz, 3H, CH3). 13C NMR (50 MHz, CDCl3): δ 178.8, 66.8, 44.4, 37.3, 27.3, 16.1.
(rac)-1-(Acetoxymethyl)-5-methylpyrrolidin-2-one (±)-7. Prepared according to GP3, (±)-4 (2.5 mmol, 300 mg), acetic anhydride (2.82 mmol, 0.26 mL), and DMAP (2.35 mmol, 287 mg) were refluxed in toluene (15 mL) over 1 h. After chromatographic column (Hex:AcOEt 90:10 → 0:100), γ-lactam (±)-7 was isolated as a colorless oil. Yield: 0.36 g (90%). 1H NMR (200 MHz, CDCl3): δ 5.34 (AB, J = 10.5 Hz, 2H, CH2O), 3.80 (m, 1H, CHCH3), 2.42 (m, 2H, CH2CO), 2.24 (m, 1H, CH2CH), 2.07 (s, 3H, CH3CO), 1.62 (m, 1H, CH2CH), 1.29 (d, J = 6.3 Hz, 3H, CH3CH). 13C NMR (50 MHz, CDCl3): δ 176.26, 170.8, 64.7, 53.1, 29.9, 26.8, 20.8, 19.9.
(rac)-1-(Acetoxymethyl)-4-methylpyrrolidin-2-one (±)-8. Prepared according to GP3, (±)-5 (3.93 mmol, 390 mg), acetic anhydride (11.93 mmol, 1.12 mL), and DMAP (9.9 mmol, 1.21 g) were refluxed in toluene (30 mL) over 1 h. After chromatographic column (Hex:AcOEt 90:10 → 0:100), γ-lactam (±)-8 was isolated as a colorless oil. Yield: 0.15 g (30%). 1H NMR (200 MHz, CDCl3): δ 5.27 (AB, J = 10.0 Hz, 2H, CH2O), 3.62 (dd, J = 9.6, 7.7 Hz, 1H, CH2N), 3.08 (dd, J = 9.6, 6.3 Hz, 1H, CH2N), 2.48 (m, 2H, CHCH3, CH2CO), 2.05 (s, 3H, CH3CO), 1.96 (m, 1H, CH2CO), 1.11 (d, J = 6.6 Hz, 3H, CH3CH). 13C NMR (50 MHz, CDCl3): δ 175.9, 171.0, 66.6, 54.4, 39.1, 26.8, 20.8, 19.48.
(rac)-1-(Acetoxymethyl)-3-methylpyrrolidin-2-one (±)-9. Prepared according to GP3, (±)-6 (0.82 mmol, 106 mg), acetic anhydride (0.986 mmol, 93 µL), and DMAP (0.82 mmol, 100 mg) were refluxed in toluene (12 mL) over 1 h. After chromatographic column (Hex:AcOEt 90:10 → 0:100) γ-lactam (±)-9 was isolated as a colorless oil. Yield: 0.13 g (97%). 1H NMR (200 MHz, CDCl3): δ 5.33 (AB, J = 10.3 Hz, 2H, CH2O), 3.45 (m, 2H, CH2CH2CH), 2.51 (m, 1H, CH2CH), 2.27 (m, 1H, CHCH3), 2.08 (s, 3H, CH3CO), 1.67 (m, 1H, CH2CH), 1.22 (d, J = 7 Hz, 3H, CH3CH). 13C NMR (50 MHz, CDCl3): δ 178.47, 170.9, 66.9, 45.0, 36.5, 27.3, 20.7, 15.8.
4. Conclusions
A synthetic approach to γ2, γ3, and γ4-lactam resolution employing CaL-B has been described. In this methodology, the substituent position is important, as the farther the stereogenic center is from the catalytic site, the lower the recognition is expected. γ4-lactams have good recognition and the product can be obtained enantio-enriched, while γ2-lactams can be obtained at moderated enantiomeric ratios. On the other hand, γ3-lactams do not have good selectivity and are obtained as racemic mixtures. Since resolution is performed under innocuous reagents, this approach is an eco-friendly alternative to access enantio-enriched γ4-lactams. Chemical correlation allowed us to assign the enantiomer recognized by CaLB as (R), which was supported by the in silico results.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal14120861/s1.
Author Contributions
Conceptualization, J.E.; methodology, L.G.H.-V. and G.K.S.-M.; validation, L.G.H.-V. and J.E.; formal analysis, L.G.H.-V. and G.K.S.-M.; investigation, L.G.H.-V. and G.K.S.-M.; resources, J.E.; writing—original draft preparation, L.G.H.-V. and G.K.S.-M.; writing—review and editing, L.G.H.-V. and J.E.; supervision, J.E.; project administration, J.E.; funding acquisition, J.E. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by CONAHCYT, project number CB2019/610262.
Data Availability Statement
There is no server where data can be saved for consultation but, if needed, data can be shared by contacting jaime@uaem.mx.
Acknowledgments
G.K.S.-M. thanks CONAHCYT (731743) for the doctoral fellowship. L.G.H.-V. thanks CONAHCYT (299370) for the post-doctoral fellowship.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Fenteany, G.; Standaert, R.F.; Lane, W.S.; Choi, S.; Corey, E.J.; Schreiber, S.L. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 1995, 268, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Mix, E.; Winblad, B. The antidepressant and antiinflammatory effects of rolipram in the central nervous system. CNS Drug Rev. 2001, 7, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Lunsford, C.D.; Cale, A.D.; Ward, J.W.; Franko, B.V.; Jenkins, H. 4-(β-Substituted ethyl)-3,3-diphenyl-2-pyrrolidinones. A New Series of CNS Stimulants. J. Med. Chem. 1964, 7, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Akram, M.; Asif, H.M.; Uzair, M.; Akhtar, N.; Madni, A.; Ali-Shah, S.M.; Hasan, Z.; Ullah, A. Amino acids: A review article. J. Med. Plants Res. 2011, 5, 3997–4000. [Google Scholar]
- Kozhuharova, P.; Diaconescu, A.O.; Allen, P. Reduced cortical GABA and glutamate in high schizotypy. Psychopharmacology 2021, 238, 2459–2470. [Google Scholar] [CrossRef]
- Edden, R.A.E.; Crocetti, D.; Zhu, H.; Gilbert, D.L.; Mostofsky, S.H. Reduced GABA Concentration in Attention-Deficit/Hyperactivity Disorder. Arch. Gen. Psychiatry 2012, 69, 750–753. [Google Scholar] [CrossRef]
- Sarawagi, A.; Soni, N.D.; Patel, A.B. Glutamate and GABA homeostasis and neurometabolism in major depressive disorder. Front. Psychiatry 2021, 12, 637863. [Google Scholar] [CrossRef]
- Lauria-Horner, B.A.; Pohl, R.B. Pregabalin: A new anxiolytic. Expert Opin. Investig. Drugs 2003, 12, 663–672. [Google Scholar] [CrossRef]
- Ramesh, P.; Suman, D.; Reddy, K.S.N. Asymmetric Synthetic Strategies of (R)-Baclofen: An Antispastic Drug. Synthesis 2018, 50, 211–226. [Google Scholar] [CrossRef]
- Rivas, F.; Ling, T. Advanceds toward the Synthesis of Functionalized y-Lactams. Org. Prep. Proced. Int. 2016, 48, 254–295. [Google Scholar] [CrossRef]
- Zhang, T.; Nishiura, Y.; Cusumano, A.Q.; Stoltz, B.M. Palladium-Catalyzed Asymmetric Conjugate Addition of Arylboronic Acids to α,β-Unsaturated Lactams: Enantioselective Construction of All-Carbon Quaternary Stereocenters in Saturated Nitrogen-Containing Heterocycles. Org. Lett. 2023, 23, 6479–6484. [Google Scholar] [CrossRef] [PubMed]
- Lang, Q.; Gu, G.; Cheng, Y.; Yin, Q.; Zhang, X. Highly Enantioselective Synthesis of Chiral γ-Lactams by Rh-Catalyzed Asymmetric Hydrogenation. ACS Catal. 2018, 8, 4824–4828. [Google Scholar] [CrossRef]
- Shi, Y.; Tan, X.; Gao, S.; Zhang, Y.; Wang, J.; Zhang, X.; Yin, Q. Direct Synthesis of Chiral NH Lactams via Ru-Catalyzed Asymmetric Reductive Amination/Cyclization Cascade of Keto Acids/Esters. Org. Lett. 2020, 22, 2707–2713. [Google Scholar] [CrossRef]
- Sukhanova, A.A.; Nelyubina, Y.V.; Zlotin, S.G. Asymmetric synthesis of 3-prenyl-substituted pyrrolidin-2-ones. Mendeleev Commun. 2016, 26, 471–473. [Google Scholar] [CrossRef]
- Ates, C.; Lurquin, F.; Quesnel, Y.; Schule, A. 4-Substituted Pyrrolidin-2-ones and Their Use. WO 2007/031263 A1, 22 March 2007. [Google Scholar]
- Fuchs, C.S.; Farnberger, J.E.; Steinkellner, G.; Sattler, J.H.; Pickl, M.; Simon, R.C.; Zepeck, F.; Gruber, K.; Kroutil, W. Asymmetric Amination of α-Chiral Aliphatic Aldehydes via Dynamic Kinetic Resolution to Acces Stereocomplementary Brivaracetam and Pregabalin Precursors. Adv. Synth. Catal. 2018, 360, 768–778. [Google Scholar] [CrossRef]
- Jouglet, B.; Rousseau, G. Enzymic resolution of N-Hydroxymethyl γ-Butyrolactams. An Access to optically active γ-Butyrolactams. Tetrahedron Lett. 1993, 34, 2307–2310. [Google Scholar] [CrossRef]
- Galla, Z.; Forró, E.; Fülöp, F. Enhanced enzymatic synthesis of the enantiopure intermediate for the blockbuster drug intermediate abacavir through a two-step enzymatic cascade reaction. Tetrahedron Asymm. 2016, 27, 729–731. [Google Scholar] [CrossRef]
- Evans, C.; McCague, R.; Roberts, S.M.; Sutherland, A.G. Synthesis of Either Enantiomer of cis-3-Aminocyclopebtanecarboxylic Acid from Both Enantiomers of Racemic 2-Azabicyclo[2.2.1]hept-5-en-3-one. J. Chem. Soc. Perkin Trans. 1 1991, 656–657. [Google Scholar] [CrossRef]
- Ortiz, C.; Ferreira, M.L.; Barbosa, O.; dos Santos, J.C.; Rodrigues, R.C.; Berenguer-Murcia, A.; Briand, L.E.; Fernandez-Lafuente, R. Novozym 435: The “perfect” lipase immobilized biocatalyst? Catal. Sci. Technol. 2019, 9, 2380–2420. [Google Scholar] [CrossRef]
- Escalante, J.; Díaz-Coutiño, F.D. Synthesis of γ-Nitro Aliphatic Methyl Esters Via Michael Additions Promoted by Microwave Irradiation. Molecules 2009, 14, 1595–1604. [Google Scholar] [CrossRef]
- Martelli, G.; Cirillo, M.; Giraldi, V.; Giacomini, D. Chemoenzymatic enantioselective route to get (+) and (−) 4-acetoxy-azetidin-2-one by lipase-catalysed kinetic resolution and their applications. Bioorg. Chem. 2022, 120, 105580. [Google Scholar] [CrossRef]
- Spartan’14; Wavefunction, Inc.: Irvine, CA, USA, 2014.
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.; Christensen, M.H. MolDock: A new Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Uppenberg, J.; Öhrner, N.; Norin, M.; Hult, K.; Kleywegt, G.J.; Patkar, S.; Waagen, V.; Anthonsen, T.; Jones, T.A. Crystallographic and Molecular-Modeling Studies of Lipase B from Candida antarctica Reveal a Stereospecificity Pocket for Secondary Alcohols. Biochemistry 1995, 34, 16838–16851. [Google Scholar] [CrossRef]
- BIOVIA, Dassault Systèmes. Discovery Studio Visualizer, Discovery Studio 2019 Client; Dassault Systèmes: San Diego, CA, USA, 2019. [Google Scholar]
- Díaz-Coutiño, F.D.; Escalante, J. Efficient ‘One Pot’ Nitro Reduction-Protection of γ-Nitro Aliphatic Methyl Esters. J. Mex. Chem. Soc. 2009, 53, 93–95. [Google Scholar] [CrossRef]
- Brenner, M.; Seebach, D. Enantioselective Preparation of γ-Amino Acids and γ-Lactams from Nitro Olefins and Carboxylic Acids, with the Valine-Derived 4-Isopropyl-5,5-diphenyl-1,3-oxazolidin-2-one as an Auxiliary. Helv. Chim. Acta 1999, 82, 2365–2379. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).