
Using a Single-Atom FeN4 Catalyst on Defective Graphene for the Efficient Reduction of NO to Alanine: A Computational Study


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
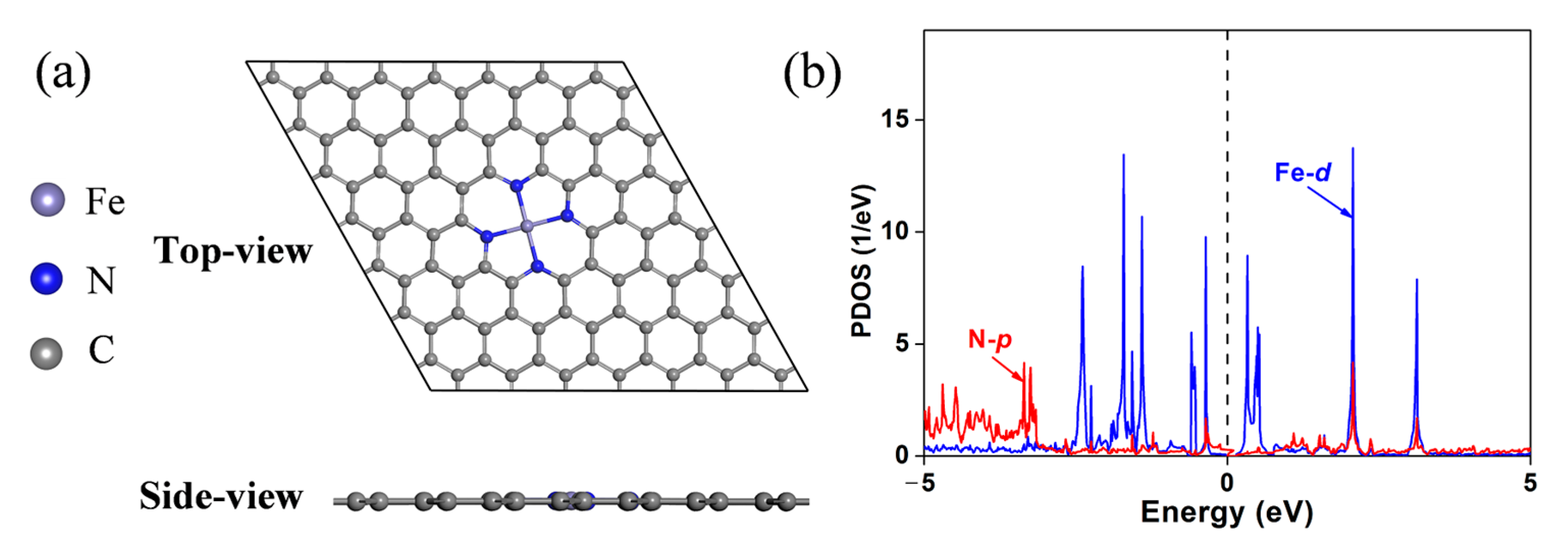
2.1. The Structure and Stability of Fe-NG
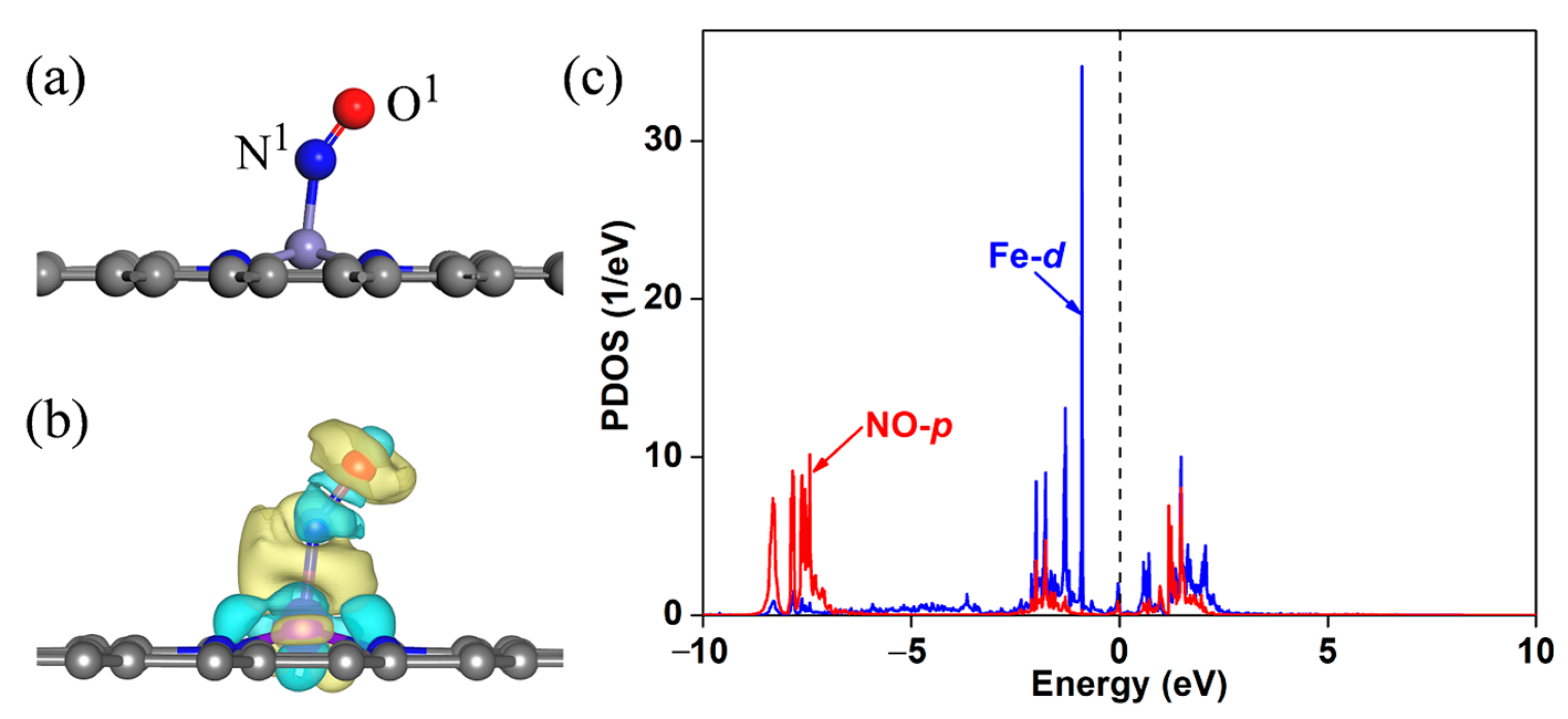
2.2. NO Adsorption on Fe-NG
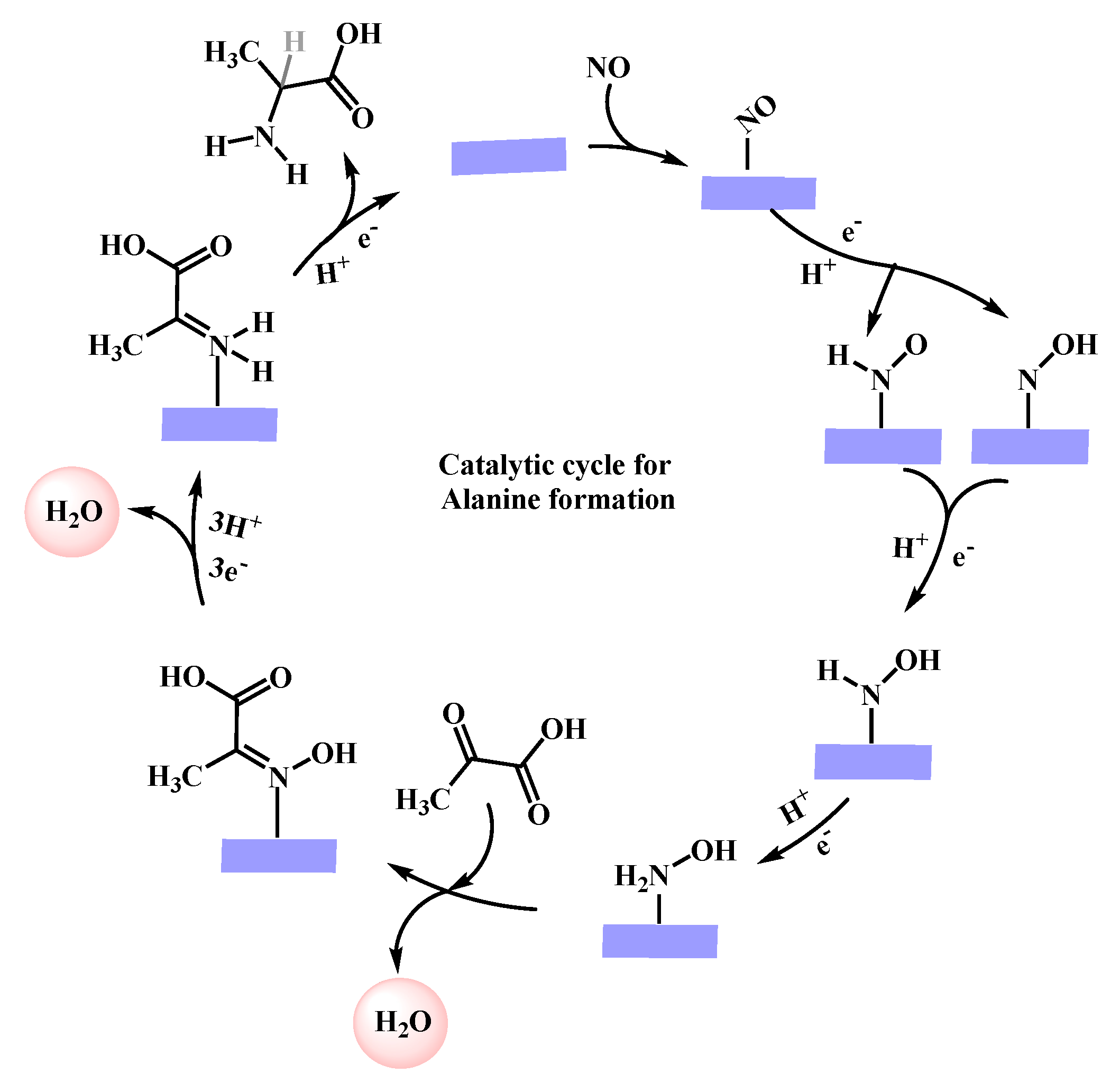
2.3. Catalytic Cycle for Alanine Formation on Fe-NG
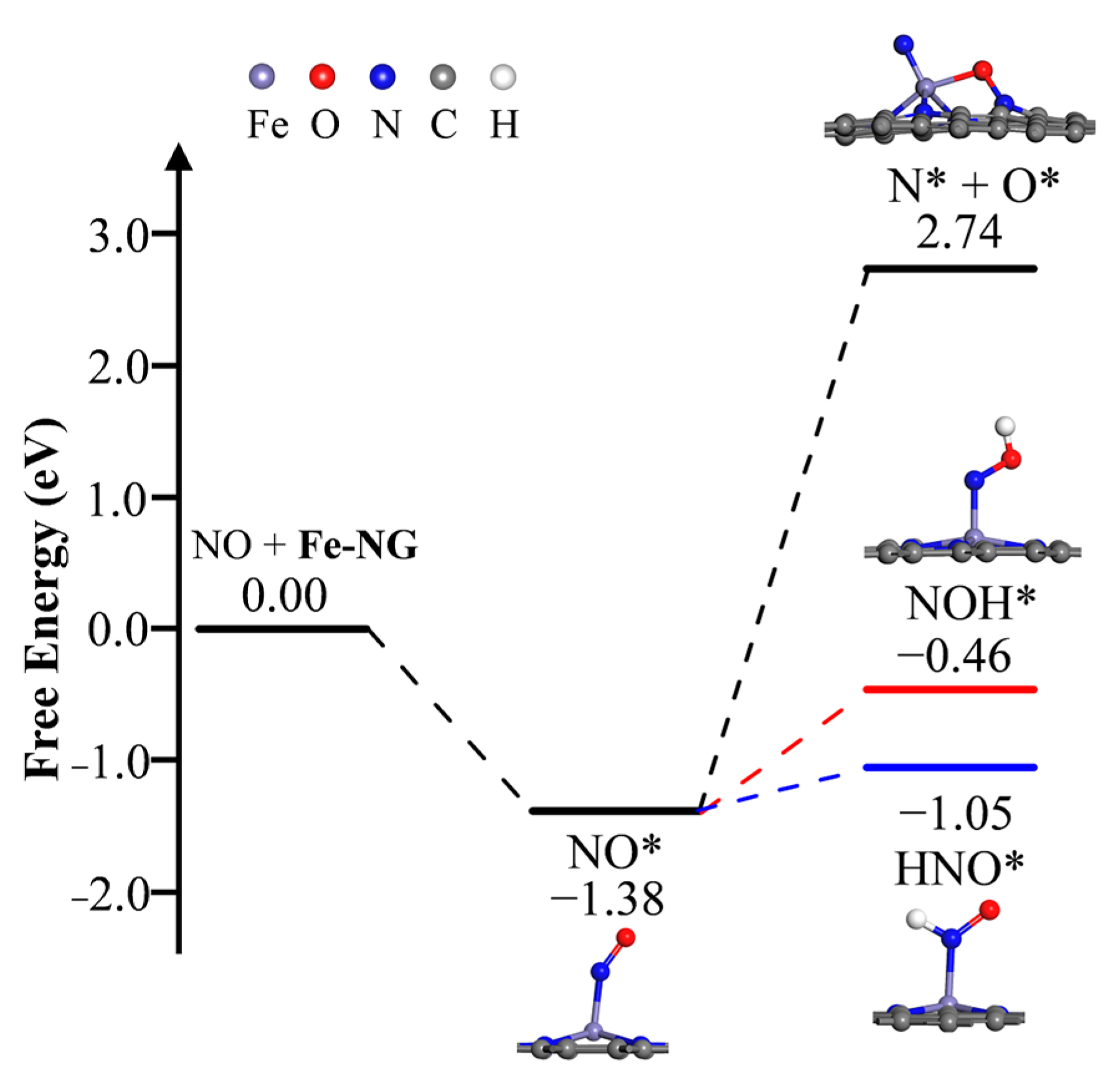
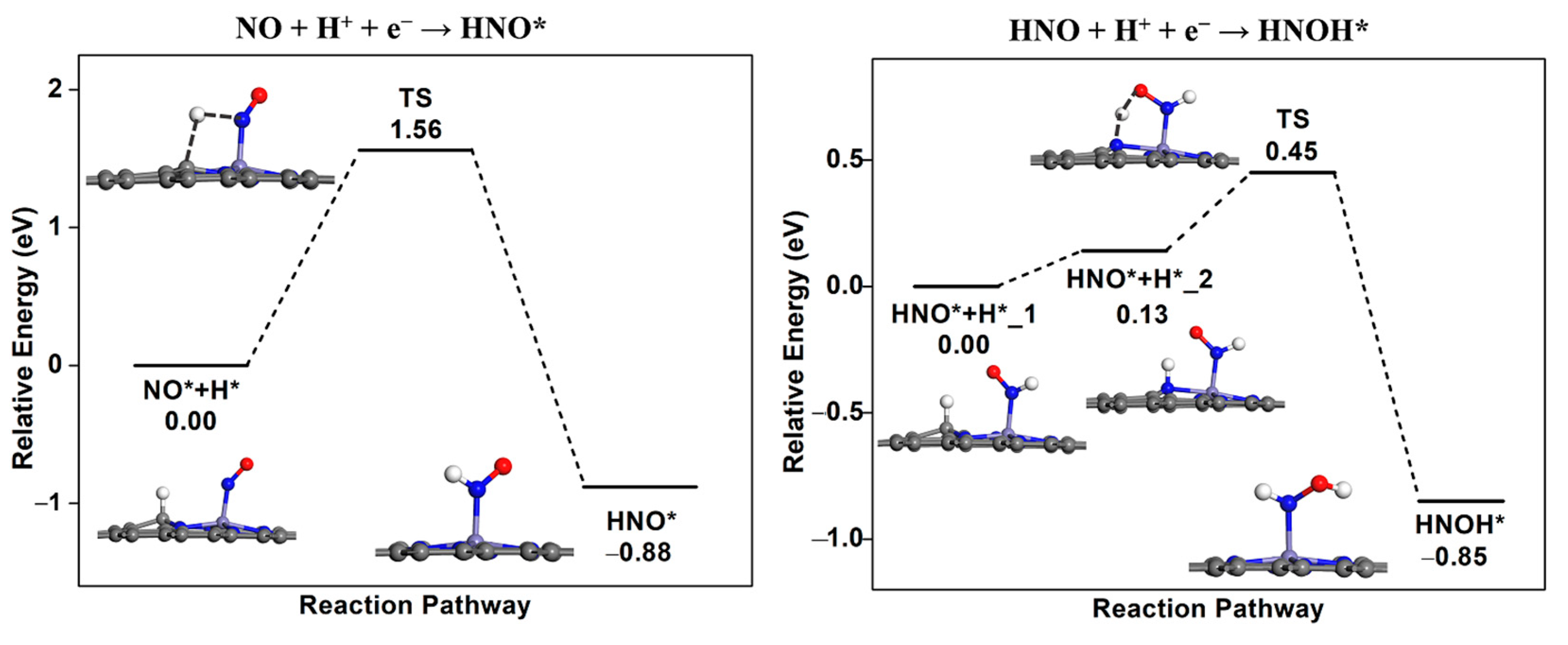
2.3.1. Hydrogenation and Splitting of NO* Species
2.3.2. Alanine Formation on Fe-NG
2.4. Side Reaction Analyses
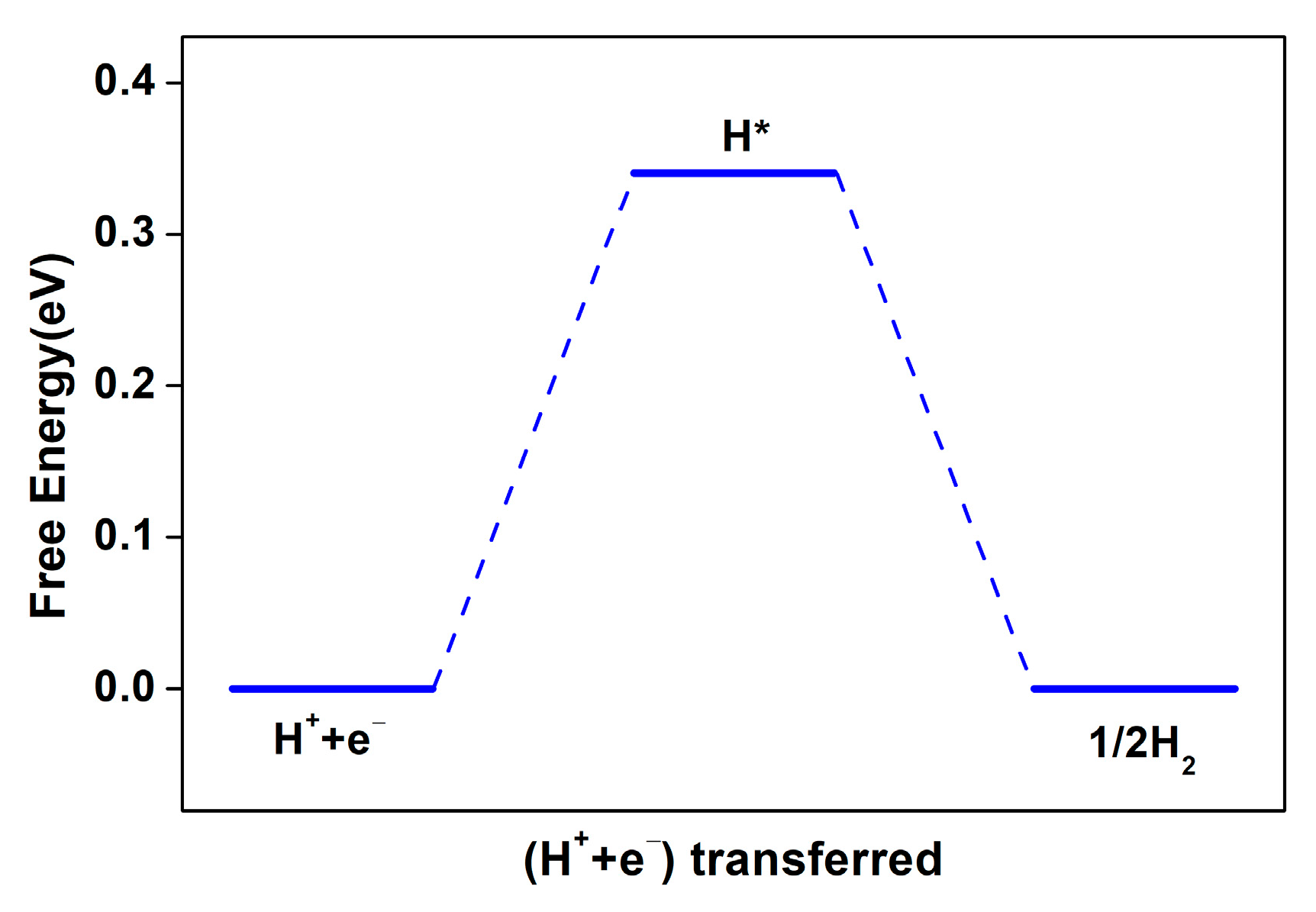
2.4.1. The Hydrogen Evolution Reaction on Fe-NG
2.4.2. NH3 Formation on Fe-NG
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Postgate, J.R. Nitrogen Fixation; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Cui, C.; Jia, Y.; Zhang, H.; Geng, L.; Luo, Z. Plasma-assisted chain reactions of Rh3+ clusters with dinitrogen: N≡N bond dissociation. J. Phys. Chem. Lett. 2020, 11, 8222–8230. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liu, D.; Cao, H.; Hou, F.; Liang, J.; Dou, S.X. Nitrogen reduction to ammonia on atomic-scale active sites under mild conditions. Small Methods 2019, 3, 1800501. [Google Scholar] [CrossRef]
- Liang, J.; Liu, Q.; Alshehri, A.A.; Sun, X. Recent advances in nanostructured heterogeneous catalysts for N-cycle electrocatalysis. Nano Res. Energy 2022, 1, 9120010. [Google Scholar] [CrossRef]
- Tong, D.; Zhang, Q.; Davis, S.J.; Liu, F.; Zheng, B.; Geng, G.; Xue, T.; Li, M.; Hong, C.; Lu, Z.; et al. Targeted emission reductions from global super-polluting power plant units. Nat. Sustain. 2018, 1, 59–68. [Google Scholar] [CrossRef]
- Mohan, S.; Dinesha, P.; Kumar, S. NOx reduction behaviour in copper zeolite catalysts for ammonia SCR systems: A review. Chem. Eng. J. 2020, 384, 123253. [Google Scholar] [CrossRef]
- Zha, Z.; Li, F.; Ge, Z.; Lu, Q.; Ma, Y.; Zeng, M.; Wu, Y.; Hou, Z.; Zhang, H. Reactivity and kinetics of biomass pyrolysis products for in-situ reduction of NOx in a bubbling fluidized bed. Chem. Eng. J. 2024, 483, 149138. [Google Scholar] [CrossRef]
- Liao, P.; Kang, J.; Xiang, R.; Wang, S.; Li, G. Electrocatalytic systems for NOx valorization in organonitrogen synthesis. Angew. Chem. Int. Ed. 2024, 63, e202311752. [Google Scholar] [CrossRef]
- Shimomura, S.; Higuchi, M.; Matsuda, R.; Yoneda, K.; Hijikata, Y.; Kubota, Y.; Mita, Y.; Kim, J.; Takata, M.; Kitagawa, S. Selective sorption of oxygen and nitric oxide by an electron-donating flexible porous coordination polymer. Nat. Chem. 2010, 2, 633–637. [Google Scholar] [CrossRef]
- Zhang, L.; Chen, Z.; Liu, Z.; Bu, J.; Ma, W.; Yan, C.; Bai, R.; Lin, J.; Zhang, Q.; Liu, J.; et al. Efficient electrocatalytic acetylene semihydrogenation by electron–rich metal sites in N–heterocyclic carbene metal complexes. Nat. Commun. 2021, 12, 6574. [Google Scholar] [CrossRef] [PubMed]
- Muhammad Farhan, S.; Pan, W.; Zhijian, C.; JianJun, Y. Innovative catalysts for the selective catalytic reduction of NOx with H2: A systematic review. Fuel 2024, 355, 129364. [Google Scholar] [CrossRef]
- Sawabe, K.; Hiro, T.; Shimizu, K.-I.; Satsuma, A. Density functional theory calculation on the promotion effect of H2 in the selective catalytic reduction of NOx over Ag–MFI zeolite. Catal. Today 2010, 153, 90–94. [Google Scholar] [CrossRef]
- Sung, C.-Y.; Broadbelt, L.J.; Snurr, R.Q. A DFT study of adsorption of intermediates in the NOx reduction pathway over BaNaY zeolites. Catal. Today 2008, 136, 64–75. [Google Scholar] [CrossRef]
- Suryanto, B.H.R.; Du, H.-L.; Wang, D.; Chen, J.; Simonov, A.N.; MacFarlane, D.R. Challenges and prospects in the catalysis of electroreduction of nitrogen to ammonia. Nat. Catal. 2019, 2, 290–296. [Google Scholar] [CrossRef]
- Qin, L.; Sun, F.; Gong, Z.; Ma, G.; Chen, Y.; Tang, Q.; Qiao, L.; Wang, R.; Liu, Z.-Q.; Tang, Z. Electrochemical NO3− reduction catalyzed by atomically precise Ag30Pd4 bimetallic nanocluster: Synergistic catalysis or tandem catalysis? ACS Nano 2023, 17, 12747–12758. [Google Scholar] [CrossRef]
- Wang, D.; Lu, X.F.; Luan, D.; Lou, X.W. Selective electrocatalytic conversion of nitric oxide to high value-added chemicals. Adv. Mater. 2024, 36, 2312645. [Google Scholar] [CrossRef]
- Jia, S.; Wu, L.; Liu, H.; Wang, R.; Sun, X.; Han, B. Nitrogenous intermediates in NOx-involved electrocatalytic reactions. Angew. Chem. Int. Ed. 2024, 63, e202400033. [Google Scholar] [CrossRef]
- Clayborne, A.; Chun, H.-J.; Rankin, R.B.; Greeley, J. Elucidation of pathways for NO electroreduction on Pt(111) from first principles. Angew. Chem. Int. Ed. 2015, 54, 8255–8258. [Google Scholar] [CrossRef]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Kyriakou, G.; Boucher, M.B.; Jewell, A.D.; Lewis, E.A.; Lawton, T.J.; Baber, A.E.; Tierney, H.L.; Flytzani-Stephanopoulos, M.; Sykes, E.C.H. Isolated metal atom geometries as a strategy for selective heterogeneous hydrogenations. Science 2012, 335, 1209–1212. [Google Scholar] [CrossRef]
- Liu, P.; Zhao, Y.; Qin, R.; Mo, S.; Chen, G.; Gu, L.; Chevrier, D.M.; Zhang, P.; Guo, Q.; Zang, D.; et al. Photochemical route for synthesizing atomically dispersed palladium catalysts. Science 2016, 352, 797–800. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, Z. Single Mo atom supported on defective boron nitride monolayer as an efficient electrocatalyst for nitrogen fixation: A computational study. J. Am. Chem. Soc. 2017, 139, 12480–12487. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-H.; Yang, L.-M.; Ganz, E. Efficient and selective electroreduction of CO2 by single-atom catalyst two-dimensional TM–Pc monolayers. ACS Sustain. Chem. Eng. 2018, 6, 15494–15502. [Google Scholar] [CrossRef]
- Chen, Z.; Zhao, J.; Cabrera, C.R.; Chen, Z. Computational screening of efficient single-atom catalysts based on graphitic carbon nitride (g-C3N4) for nitrogen electroreduction. Small Methods 2019, 3, 1800368. [Google Scholar] [CrossRef]
- Jouny, M.; Lv, J.-J.; Cheng, T.; Ko, B.H.; Zhu, J.-J.; Goddard, W.A.; Jiao, F. Formation of carbon–nitrogen bonds in carbon monoxide electrolysis. Nat. Chem. 2019, 11, 846–851. [Google Scholar] [CrossRef]
- Udayasurian, S.R.; Li, T. Recent research progress on building C–N bonds via electrochemical NOx reduction. Nanoscale 2024, 16, 2805–2819. [Google Scholar] [CrossRef]
- Li, Y.; Verma, V.; Su, H.; Zhang, X.; Zhou, S.; Lawson, T.; Li, J.; Amal, R.; Hou, Y.; Dai, L. Rationally designed carbon-based catalysts for electrochemical C-N coupling. Adv. Energy Mater. 2024, 14, 2401341. [Google Scholar] [CrossRef]
- Guo, C.; Zhou, W.; Lan, X.; Wang, Y.; Li, T.; Han, S.; Yu, Y.; Zhang, B. Electrochemical upgrading of formic acid to formamide via coupling nitrite Co-reduction. J. Am. Chem. Soc. 2022, 144, 16006–16011. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Wen, X.; Liu, Y.; Chen, C.; Xie, C.; Wang, D.; Qiu, M.; He, N.; Zhou, P.; Chen, W.; et al. Oxygen vacancy-mediated selective C–N coupling toward electrocatalytic urea synthesis. J. Am. Chem. Soc. 2022, 144, 11530–11535. [Google Scholar] [CrossRef]
- Jiao, D.; Dong, Y.; Cui, X.; Cai, Q.; Cabrera, C.R.; Zhao, J.; Chen, Z. Boosting the efficiency of urea synthesis via cooperative electroreduction of N2 and CO2 on MoP. J. Mater. Chem. A 2023, 11, 232–240. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; Klika, K.D.; Izawa, K.; Sato, T.; Meanwell, N.A.; Soloshonok, V.A. Applications of fluorine-containing amino acids for drug design. Eur. J. Med. Chem. 2020, 186, 111826. [Google Scholar] [CrossRef]
- Aguilar Troyano, F.J.; Merkens, K.; Anwar, K.; Gómez-Suárez, A. Radical-based synthesis and modification of amino acids. Angew. Chem. Int. Ed. 2021, 60, 1098–1115. [Google Scholar] [CrossRef] [PubMed]
- Hug, J.J.; Krug, D.; Müller, R. Bacteria as genetically programmable producers of bioactive natural products. Nat. Rev. Chem. 2020, 4, 172–193. [Google Scholar] [CrossRef] [PubMed]
- Noisier, A.F.M.; Brimble, M.A. C–H functionalization in the synthesis of amino acids and peptides. Chem. Rev. 2014, 114, 8775–8806. [Google Scholar] [CrossRef]
- Zhang, X.; Jantama, K.; Moore, J.C.; Shanmugam, K.T.; Ingram, L.O. Production of L-alanine by metabolically engineered escherichia coli. Appl. Microbiol. Biotechnol. 2007, 77, 355–366. [Google Scholar] [CrossRef]
- Breuer, M.; Ditrich, K.; Habicher, T.; Hauer, B.; Keßeler, M.; Stürmer, R.; Zelinski, T. Industrial methods for the production of optically active intermediates. Angew. Chem. Int. Ed. 2004, 43, 788–824. [Google Scholar] [CrossRef]
- Xian, J.; Li, S.; Su, H.; Liao, P.; Wang, S.; Xiang, R.; Zhang, Y.; Liu, Q.; Li, G. Electrosynthesis of α-amino acids from NO and other NOx species over cofe alloy-decorated self-standing carbon fiber membranes. Angew. Chem. Int. Ed. 2023, 62, e202306726. [Google Scholar] [CrossRef]
- Li, M.; Wu, Y.; Zhao, B.-H.; Cheng, C.; Zhao, J.; Liu, C.; Zhang, B. Electrosynthesis of amino acids from NO and α-keto acids using two decoupled flow reactors. Nat. Catal. 2023, 6, 906–915. [Google Scholar] [CrossRef]
- Xian, J.; Li, S.; Su, H.; Liao, P.; Wang, S.; Zhang, Y.; Yang, W.; Yang, J.; Sun, Y.; Jia, Y.; et al. Electrocatalytic synthesis of essential amino acids from nitric oxide using atomically dispersed Fe on N-doped carbon. Angew. Chem. Int. Ed. 2023, 62, e202304007. [Google Scholar] [CrossRef]
- Bedford, R.B. How low does iron go? Chasing the active species in Fe-catalyzed cross-coupling reactions. Acc. Chem. Res. 2015, 48, 1485–1493. [Google Scholar] [CrossRef]
- Zhao, T.; Tian, Y.; Wang, Y.; Yan, L.; Su, Z. Mechanistic insight into electroreduction of carbon dioxide on FeNx (x = 0–4) embedded graphene. Phys. Chem. Chem. Phys. 2019, 21, 23638–23644. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Peterson, A.A.; Abild-Pedersen, F.; Studt, F.; Rossmeisl, J.; Nørskov, J.K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 2010, 3, 1311–1315. [Google Scholar] [CrossRef]
- Delley, B. An all–electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Johnson, R. NIST 101. Computational Chemistry Comparison and Benchmark Database, CCCBDBD Computational Chemistry Comparison and Benchmark Database, 1999.
- Govind, N.; Petersen, M.; Fitzgerald, G.; King–Smith, D.; Andzelm, J. A generalized synchronous transit method for transition state location. Comp. Mater. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Yuan, X.; Guo, Z.; Liu, J.; Zhao, T.; Su, Z. Using a Single-Atom FeN4 Catalyst on Defective Graphene for the Efficient Reduction of NO to Alanine: A Computational Study. Catalysts 2024, 14, 876. https://doi.org/10.3390/catal14120876
Tian Y, Yuan X, Guo Z, Liu J, Zhao T, Su Z. Using a Single-Atom FeN4 Catalyst on Defective Graphene for the Efficient Reduction of NO to Alanine: A Computational Study. Catalysts. 2024; 14(12):876. https://doi.org/10.3390/catal14120876
Chicago/Turabian StyleTian, Yu, Xiaoxi Yuan, Zexuan Guo, Jingyao Liu, Tingting Zhao, and Zhongmin Su. 2024. "Using a Single-Atom FeN4 Catalyst on Defective Graphene for the Efficient Reduction of NO to Alanine: A Computational Study" Catalysts 14, no. 12: 876. https://doi.org/10.3390/catal14120876
APA StyleTian, Y., Yuan, X., Guo, Z., Liu, J., Zhao, T., & Su, Z. (2024). Using a Single-Atom FeN4 Catalyst on Defective Graphene for the Efficient Reduction of NO to Alanine: A Computational Study. Catalysts, 14(12), 876. https://doi.org/10.3390/catal14120876