
Regulating the Hydrodeoxygenation Activity of Molybdenum Carbide with Different Diamines as Carbon Sources

Abstract
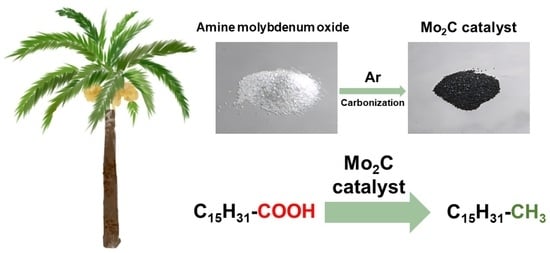
:
1. Introduction
2. Results and Discussion
2.1. Characterizations of Catalyst
2.1.1. XRD Results
2.1.2. TGA-DSC Results
2.1.3. SEM, TEM, and HRTEM Results
2.1.4. BET and BJH Results
2.1.5. XPS Result
2.1.6. Raman Result
2.2. The Conversion of Palmitic Acid over Molybdenum Carbide Catalysts
2.3. The Structure–Activity Relationship of Mo2C Catalysts
3. Materials and Methods
3.1. Materials
3.2. Catalysts Preparation
3.3. Catalytic Activity Test
3.4. Catalysts Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shindell, D.; Smith, C.J. Climate and air-quality benefits of a realistic phase-out of fossil fuels. Nature 2019, 573, 408–411. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Davis, S.J.; Creutzig, F.; Fuss, S.; Minx, J.; Gabrielle, B.; Kato, E.; Jackson, R.B.; Cowie, A.; Kriegler, E.; et al. Biophysical and economic limits to negative CO2 emissions. Nat. Clim. Chang. 2016, 6, 42–50. [Google Scholar] [CrossRef]
- Degirmenci, H.; Uludag, A.; Ekici, S.; Hikmet Karakoc, T. Analyzing the hydrogen supply chain for airports: Evaluating environmental impact, cost, sustainability, viability, and safety in various scenarios for implementation. Energy Convers. Manag. 2023, 293, 117537. [Google Scholar] [CrossRef]
- Liu, Y.; Hajj, M.; Bao, Y. Review of robot-based damage assessment for offshore wind turbines. Renew. Sustain. Energy Rev. 2022, 158, 112187. [Google Scholar] [CrossRef]
- Berga, L. The Role of Hydropower in Climate Change Mitigation and Adaptation: A Review. Engineering 2016, 2, 313–318. [Google Scholar] [CrossRef]
- Ayachi, S.; He, X.; Yoon, H.J. Solar Thermoelectricity for Power Generation. Adv. Energy Mater. 2023, 13, 2300937. [Google Scholar] [CrossRef]
- Chen, W.-H.; Lee, K.T.; Ong, H.C. Biofuel and Bioenergy Technology. Energies 2019, 12, 290. [Google Scholar] [CrossRef]
- Mukhtar, A.; Saqib, S.; Mubashir, M.; Ullah, S.; Inayat, A.; Mahmood, A.; Ibrahim, M.; Show, P.L. Mitigation of CO2 emissions by transforming to biofuels: Optimization of biofuels production processes. Renew. Sustain. Energy Rev. 2021, 150, 111487. [Google Scholar] [CrossRef]
- Li, D.; Xin, H.; Du, X.; Hao, X.; Liu, Q.; Hu, C. Recent advances for the production of hydrocarbon biofuel via deoxygenation progress. Sci. Bull. 2015, 60, 2096–2106. [Google Scholar] [CrossRef]
- Xu, J.; Jiang, J.; Zhao, J. Thermochemical conversion of triglycerides for production of drop-in liquid fuels. Renew. Sustain. Energy Rev. 2016, 58, 331–340. [Google Scholar] [CrossRef]
- Ding, S.; Parlett, C.M.A.; Fan, X. Recent developments in multifunctional catalysts for fatty acid hydrodeoxygenation as a route towards biofuels. Mol. Catal. 2022, 523, 111492. [Google Scholar] [CrossRef]
- Kon, K.; Toyao, T.; Onodera, W.; Siddiki, S.M.A.H.; Shimizu, K.-i. Hydrodeoxygenation of Fatty Acids, Triglycerides, and Ketones to Liquid Alkanes by a Pt–MoOx/TiO2 Catalyst. ChemCatChem 2017, 9, 2822–2827. [Google Scholar] [CrossRef]
- Phan, D.-P.; Pham, T.M.; Lee, H.; Tran, M.H.; Park, E.D.; Kim, J.; Lee, E.Y. Hydrodeoxygenation of stearic acid over zeolite–MOF composite-supported Pt catalysts. J. Ind. Eng. Chem. 2023, 127, 590–599. [Google Scholar] [CrossRef]
- Zhang, J.; Huo, X.; Li, Y.; Strathmann, T.J. Catalytic Hydrothermal Decarboxylation and Cracking of Fatty Acids and Lipids over Ru/C. ACS Sustain. Chem. Eng. 2019, 7, 14400–14410. [Google Scholar] [CrossRef]
- Deng, Y.; Ge, Y.; Xu, M.; Yu, Q.; Xiao, D.; Yao, S.; Ma, D. Molybdenum Carbide: Controlling the Geometric and Electronic Structure of Noble Metals for the Activation of O–H and C–H Bonds. Acc. Chem. Res. 2019, 52, 3372–3383. [Google Scholar] [CrossRef]
- Du, X.; Liu, J.; Li, D.; Xin, H.; Lei, X.; Zhang, R.; Zhou, L.; Yang, H.; Zeng, Y.; Zhang, H.; et al. Structural and electronic effects boosting Ni-doped Mo2C catalyst toward high-efficiency C-O/C-C bonds cleavage. J. Energy Chem. 2022, 75, 109–116. [Google Scholar] [CrossRef]
- Lin, Z.; Wan, W.; Yao, S.; Chen, J.G. Cobalt-modified molybdenum carbide as a selective catalyst for hydrodeoxygenation of furfural. Appl. Catal. B Environ. 2018, 233, 160–166. [Google Scholar] [CrossRef]
- Sosa, L.F.; de Souza, P.M.; Rafael, R.A.; Wojcieszak, R.; Briois, V.; Francisco, L.R.; Rabelo-Neto, R.C.; Marceau, E.; Paul, S.; Toniolo, F.S.; et al. Study of the performance of SiO2-supported Mo2C and metal-promoted Mo2C catalysts for the hydrodeoxygenation of m-cresol. Appl. Catal. B Environ. 2023, 331, 122720. [Google Scholar] [CrossRef]
- Chang, Z.; Duan, P.; Xu, Y. Catalytic hydropyrolysis of microalgae: Influence of operating variables on the formation and composition of bio-oil. Bioresour. Technol. 2015, 184, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; York, A.P.E.; Coleman, K.S.; Claridge, J.B.; Sloan, J.; Charnock, J.; Green, M.L.H. Effect of carburising agent on the structure of molybdenum carbides. J. Mater. Chem. 2001, 11, 3094–3098. [Google Scholar] [CrossRef]
- Galashev, A.Y. Greenhouse effect of clusterization of CO2 and CH4 with atmospheric moisture. Environ. Chem. Lett. 2011, 9, 37–41. [Google Scholar] [CrossRef]
- Deng, S.; Shi, X.; Zhao, Y.; Wang, C.; Wu, J.; Yao, X. Catalytic Mo2C decorated N-doped honeycomb-like carbon network for high stable lithium-sulfur batteries. Chem. Eng. J. 2022, 433, 133683. [Google Scholar] [CrossRef]
- Wan, C.; Regmi, Y.N.; Leonard, B.M. Multiple Phases of Molybdenum Carbide as Electrocatalysts for the Hydrogen Evolution Reaction. Angew. Chem. Int. Ed. 2014, 53, 6407–6410. [Google Scholar] [CrossRef]
- Wan, C.; Knight, N.A.; Leonard, B.M. Crystal structure and morphology control of molybdenum carbide nanomaterials synthesized from an amine–metal oxide composite. Chem. Commun. 2013, 49, 10409–10411. [Google Scholar] [CrossRef]
- Lin, H.; Shi, Z.; He, S.; Yu, X.; Wang, S.; Gao, Q.; Tang, Y. Heteronanowires of MoC–Mo2C as efficient electrocatalysts for hydrogen evolution reaction. Chem. Sci. 2016, 7, 3399–3405. [Google Scholar] [CrossRef]
- Šljukić, B.; Vujković, M.; Amaral, L.; Santos, D.M.F.; Rocha, R.P.; Sequeira, C.A.C.; Figueiredo, J.L. Carbon-supported Mo2C electrocatalysts for hydrogen evolution reaction. J. Mater. Chem. A 2015, 3, 15505–15512. [Google Scholar] [CrossRef]
- Gao, Q.; Zhang, C.; Xie, S.; Hua, W.; Zhang, Y.; Ren, N.; Xu, H.; Tang, Y. Synthesis of Nanoporous Molybdenum Carbide Nanowires Based on Organic−Inorganic Hybrid Nanocomposites with Sub-Nanometer Periodic Structures. Chem. Mater. 2009, 21, 5560–5562. [Google Scholar] [CrossRef]
- Sarr, B.; Mbaye, A.; Diop, C.A.K.; Melin, F.; Hellwig, P.; Sidibé, M.; Rousselin, Y. Crystal structure of bis (diisopropylammonium) molybdate. Acta Crystallogr. Sect. E 2018, 74, 1682–1685. [Google Scholar] [CrossRef] [PubMed]
- Upreti, S.; Ramanan, A. Structure-Directing Role of Hydrogen-Bonded Dimers of Phenylenediammonium Cations: Supramolecular Assemblies of Octamolybdate-Based Organic−Inorganic Hybrids. Cryst. Growth Des. 2005, 5, 1837–1843. [Google Scholar] [CrossRef]
- Hubbard, D.J.; Johnston, A.R.; Casalongue, H.S.; Sarjeant, A.N.; Norquist, A.J. Synthetic Approaches for Noncentrosymmetric Molybdates. Inorg. Chem. 2008, 47, 8518–8525. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, C.; Wu, C.; Liu, Y.; Zhang, X.; Li, X.; Li, Y.; Sun, J.; Su, Z. Defined organic-octamolybdate crystalline superstructures derived Mo2C@C as efficient hydrogen evolution electrocatalysts. Chin. Chem. Lett. 2023, volume, 108782. [Google Scholar] [CrossRef]
- Ren, X.; Zhao, J.; Wei, Q.; Ma, Y.; Guo, H.; Liu, Q.; Wang, Y.; Cui, G.; Asiri, A.M.; Li, B.; et al. High-Performance N2-to-NH3 Conversion Electrocatalyzed by Mo2C Nanorod. ACS Cent. Sci. 2019, 5, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-Y.; Zhang, Y.; Jiang, W.-J.; Zhang, X.; Dai, Z.; Wan, L.-J.; Hu, J.-S. Pomegranate-like N,P-Doped Mo2C@C Nanospheres as Highly Active Electrocatalysts for Alkaline Hydrogen Evolution. ACS Nano 2016, 10, 8851–8860. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Liu, Y.; Peng, Z.; Zhang, Z.; Zhou, H.; Zhang, X.; Yakobson, B.I.; Goddard, W.A., III; Guo, X.; Hauge, R.H.; et al. Atomic H-Induced Mo2C Hybrid as an Active and Stable Bifunctional Electrocatalyst. ACS Nano 2017, 11, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Murugappan, K.; Anderson, E.M.; Teschner, D.; Jones, T.E.; Skorupska, K.; Román-Leshkov, Y. Operando NAP-XPS unveils differences in MoO3 and Mo2C during hydrodeoxygenation. Nat. Catal. 2018, 1, 960–967. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Ji, L.; Czioska, S.; Guo, L.; Chen, Z. Highly Dispersed Mo2C Nanoparticles Embedded in Ordered Mesoporous Carbon for Efficient Hydrogen Evolution. ACS Appl. Energy Mater. 2018, 1, 736–743. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, W.; Jiang, J.; Xu, J.; Zhai, Q.; Wei, L.; Long, F.; Liu, C.; Liu, P.; Tan, W.; et al. Nitrogen-rich carbon-supported ultrafine MoC nanoparticles for the hydrotreatment of oleic acid into diesel-like hydrocarbons. Chem. Eng. J. 2020, 382, 122464. [Google Scholar] [CrossRef]
- Du, X.; Zhou, K.; Zhou, L.; Lei, X.; Yang, H.; Li, D.; Hu, C. Efficient catalytic conversion of jatropha oil to high grade biofuel on Ni-Mo2C/MCM-41 catalysts with tuned surface properties. J. Energy Chem. 2021, 61, 425–435. [Google Scholar] [CrossRef]
- Nguyen, T.P.; Choi, K.S.; Kim, S.Y.; Lee, T.H.; Jang, H.W.; Van Le, Q.; Kim, I.T. Strategy for controlling the morphology and work function of W2C/WS2 nanoflowers. J. Alloys Compd. 2020, 829, 154582. [Google Scholar] [CrossRef]
- Žula, M.; Grilc, M.; Likozar, B. Mechanistic reaction micro-kinetics-based structure–activity relationships for palmitic acid hydrodeoxygenation over NiMoSx/Al2O3 catalysts. Chem. Eng. J. 2023, 467, 143425. [Google Scholar] [CrossRef]
- Hollak, S.A.W.; Gosselink, R.W.; van Es, D.S.; Bitter, J.H. Comparison of Tungsten and Molybdenum Carbide Catalysts for the Hydrodeoxygenation of Oleic Acid. ACS Catal. 2013, 3, 2837–2844. [Google Scholar] [CrossRef]
- Peroni, M.; Lee, I.; Huang, X.; Baráth, E.; Gutiérrez, O.Y.; Lercher, J.A. Deoxygenation of Palmitic Acid on Unsupported Transition-Metal Phosphides. ACS Catal. 2017, 7, 6331–6341. [Google Scholar] [CrossRef]
- Kubičková, I.; Snåre, M.; Eränen, K.; Mäki-Arvela, P.; Murzin, D.Y. Hydrocarbons for diesel fuel via decarboxylation of vegetable oils. Catal. Today 2005, 106, 197–200. [Google Scholar] [CrossRef]
- Chang, H.; Abdulkareem-Alsultan, G.; Taufiq-Yap, Y.H.; Mohd Izham, S.; Sivasangar, S. Development of porous MIL-101 derived catalyst application for green diesel production. Fuel 2024, 355, 129459. [Google Scholar] [CrossRef]
- Shamanaev, I.V.; Deliy, I.V.; Gerasimov, E.Y.; Pakharukova, V.P.; Kodenev, E.G.; Aleksandrov, P.V.; Bukhtiyarova, G.A. Synergetic Effect of Ni2P/SiO2 and γ-Al2O3 Physical Mixture in Hydrodeoxygenation of Methyl Palmitate. Catalysts 2017, 7, 329. [Google Scholar] [CrossRef]
- Zeng, Y.; Wang, H.; Yang, H.; Juan, C.; Li, D.; Wen, X.; Zhang, F.; Zou, J.-J.; Peng, C.; Hu, C. Ni nanoparticle coupled surface oxygen vacancies for efficient synergistic conversion of palmitic acid into alkanes. Chin. J. Catal. 2023, 47, 229–242. [Google Scholar] [CrossRef]
- Lee, C.-W.; Lin, P.-Y.; Chen, B.-H.; Kukushkin, R.G.; Yakovlev, V.A. Hydrodeoxygenation of palmitic acid over zeolite-supported nickel catalysts. Catal. Today 2021, 379, 124–131. [Google Scholar] [CrossRef]
- Liang, J.; Chen, T.; Liu, J.; Zhang, Q.; Peng, W.; Li, Y.; Zhang, F.; Fan, X. Chemoselective hydrodeoxygenation of palmitic acid to diesel-like hydrocarbons over Ni/MoO2@Mo2CTx catalyst with extraordinary synergic effect. Chem. Eng. J. 2020, 391, 123472. [Google Scholar] [CrossRef]
- Chen, X.; Chen, X.; Li, C.; Liang, C. Engineering the structural formula of N-doped molybdenum carbide nanowires for the deoxygenation of palmitic acid. Sustain. Energy Fuels 2020, 4, 2370–2379. [Google Scholar] [CrossRef]
- Du, X.; Lei, X.; Zhou, L.; Peng, Y.; Zeng, Y.; Yang, H.; Li, D.; Hu, C.; Garcia, H. Bimetallic Ni and Mo Nitride as an Efficient Catalyst for Hydrodeoxygenation of Palmitic Acid. ACS Catal. 2022, 12, 4333–4343. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y.; Yao, L.; Hu, C. Temperature hysteresis in dry reforming of methane on Ni/SBA-15 catalyst: Low temperature activity originated from the synergy of Ni0 and carbon nanotubes. Appl. Catal. B Environ. Energy 2023, 333, 122756. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, K.; Yang, Y.; Liu, H.; Qiao, Z.; Luo, H. Synthesis of mesoporous Al2O3 with large surface area and large pore diameter by improved precipitation method. Microporous Mesoporous Mater. 2014, 193, 47–53. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | SBET a (m2/g) | VBJH b (cm3/g) | DP c (nm) | Mo d (wt%) | C e (wt%) | N e (wt%) | Mo/C f |
|---|---|---|---|---|---|---|---|
| Mo2C-6 | 90 | 0.062 | 3.1 | 79.0 | 6.6 | 0.2 | 1.5 |
| Mo2C-8 | 57 | 0.058 | 4.4 | 82.7 | 7.2 | 0.2 | 1.4 |
| Mo2C-10 | 53 | 0.035 | 7.5 | 77.1 | 9.3 | 0.2 | 1.0 |
| Mo2C-12 | 31 | 0.039 | 14.7 | 78.1 | 9.5 | 0.2 | 1.0 |
| Catalysts | Raw Material | Reaction Conditions | Activity Performance | Ref. |
|---|---|---|---|---|
| Pd/C | Stearic acid | Batch reactor, T = 360 °C, P = 1.7 MPa (5% H2/He), t = 6 h Rea/Cat(wt/wt) = 20 | 60% conversion, 90% alkanes selectivity | [43] |
| Ru/C | Stearic acid | Batch reactor, T = 330 °C, P = 1 MPa N2, t = 12.5 h, Rea/Cat(wt/wt) = 10 | 100% conversion, 90% alkanes selectivity | [14] |
| Ni/P-MIL-101 | Palmitic acid | Batch reactor, T = 400 °C, P = 3 MPa H2, t = 3 h, Rea/Cat(wt/wt) = 20 | 100% conversion, 100% alkanes selectivity | [44] |
| Ni2P/SiO2–γ-Al2O3 | Methyl palmitate | Trickle-bed reactor, T = 290 °C, P = 3 MPa H2, H2/feed = 600 Nm3/m3, WHSV = 5 h−1 | 100% conversion, 99% pentadecane selectivity | [45] |
| Ni/CeO2 | Palmitic acid | Batch reactor, T = 270 °C, P = 2 MPa H2, t = 10 h, Rea/Cat(wt/wt) = 5 | 100% conversion, 94.8% pentadecane selectivity | [46] |
| Ni-Mo(R)/ZSM-5 | Palmitic acid | Batch reactor, T = 300 °C, P = 3.5 MPa H2, t = 4 h, Rea/Cat(wt/wt) = 4 | 100% conversion, 99% alkanes selectivity | [47] |
| Ni/MoO2@Mo2CTx | Palmitic acid | Batch reactor, T = 280 °C, P = 4 MPa H2, t = 4 h, Rea/Cat(wt/wt) = 5 | 100% conversion, 97.09 alkanes selectivity | [48] |
| Mo2.56CN0.50 | Palmitic acid | Trickle-bed reactor, T = 300 °C, P = 4 MPa H2, H2/feed = 600 Nm3/m3, WHSV = 50.84 h−1 | 99.9% conversion, 81.0% pentadecane selectivity | [49] |
| Ni3Mo3N@600 | Palmitic acid | Batch reactor, T = 270 °C, P = 2 MPa H2, t = 10 h, Rea/Cat(wt/wt) = 10 | 100% conversion, 94.3% alkanes selectivity | [50] |
| Mo2C-12 | Palmitic acid | Batch reactor, T = 275 °C, P = 2 MPa H2, t = 8 h, Rea/Cat(wt/wt) = 5 | 100% conversion, 96.7% alkanes selectivity | This work |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, L.; Yang, H.; Du, X.; Hu, C. Regulating the Hydrodeoxygenation Activity of Molybdenum Carbide with Different Diamines as Carbon Sources. Catalysts 2024, 14, 138. https://doi.org/10.3390/catal14020138
Zhou L, Yang H, Du X, Hu C. Regulating the Hydrodeoxygenation Activity of Molybdenum Carbide with Different Diamines as Carbon Sources. Catalysts. 2024; 14(2):138. https://doi.org/10.3390/catal14020138
Chicago/Turabian StyleZhou, Linyuan, Huiru Yang, Xiangze Du, and Changwei Hu. 2024. "Regulating the Hydrodeoxygenation Activity of Molybdenum Carbide with Different Diamines as Carbon Sources" Catalysts 14, no. 2: 138. https://doi.org/10.3390/catal14020138
APA StyleZhou, L., Yang, H., Du, X., & Hu, C. (2024). Regulating the Hydrodeoxygenation Activity of Molybdenum Carbide with Different Diamines as Carbon Sources. Catalysts, 14(2), 138. https://doi.org/10.3390/catal14020138