
The Effect of Precursor Concentration on the Crystallite Size of CeO2 to Enhance the Sulfur Resistance of Pt/CeO2 for Water Gas Shift
 and
and
Abstract
:
1. Introduction
2. Results
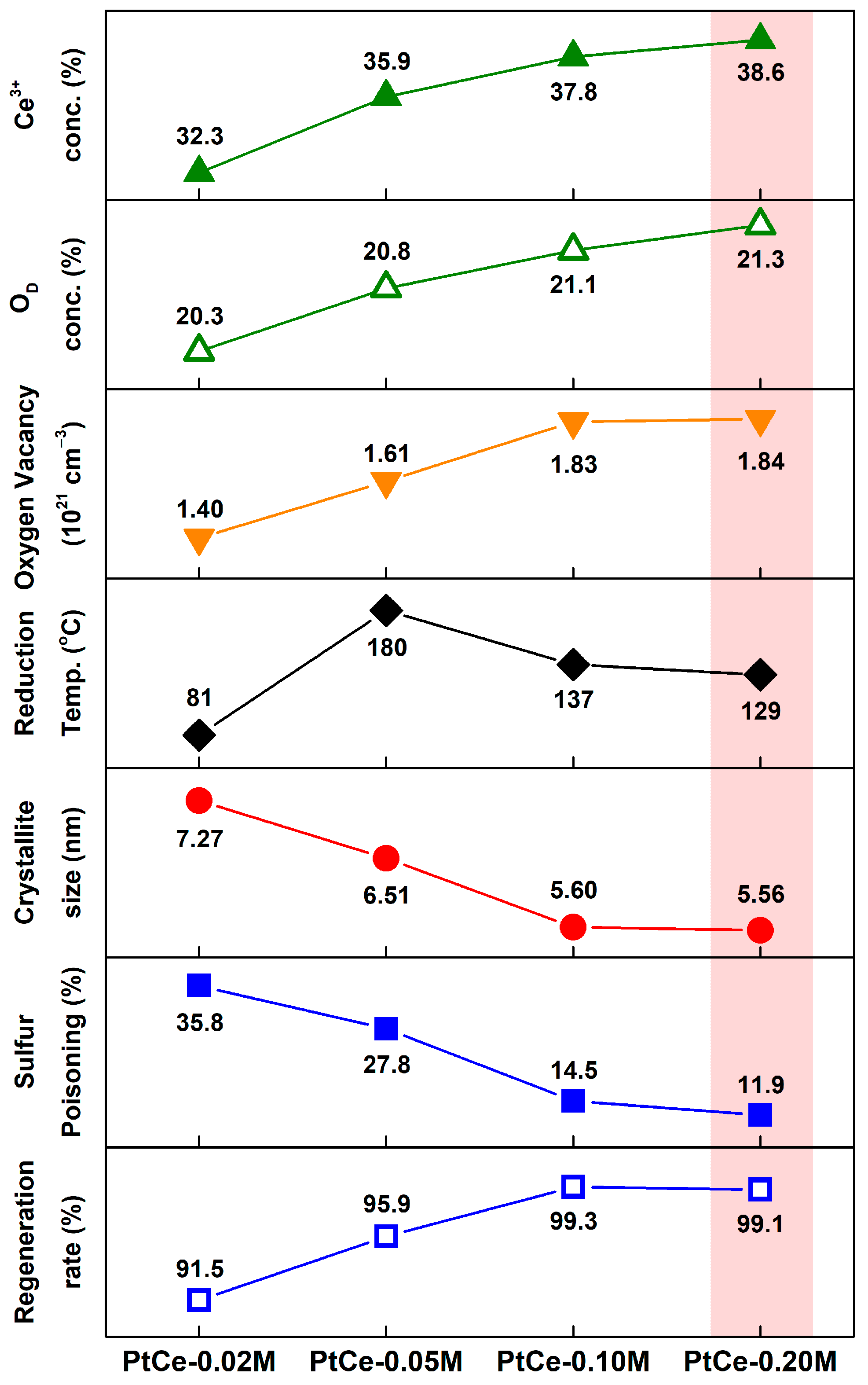
2.1. Catalytic Activity
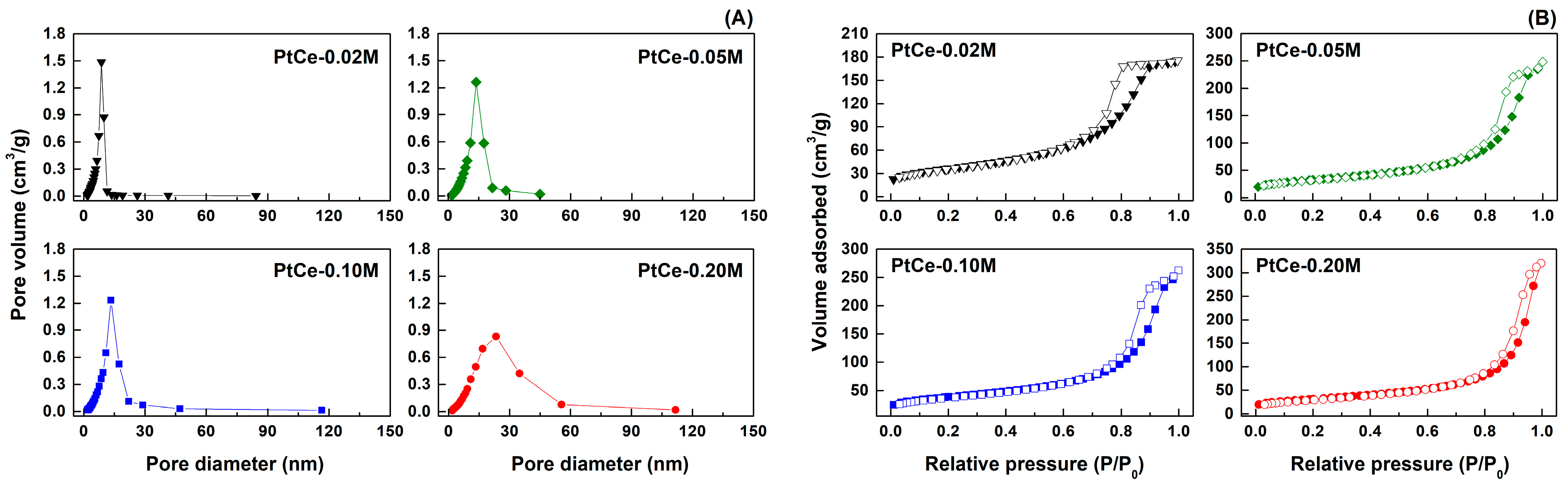
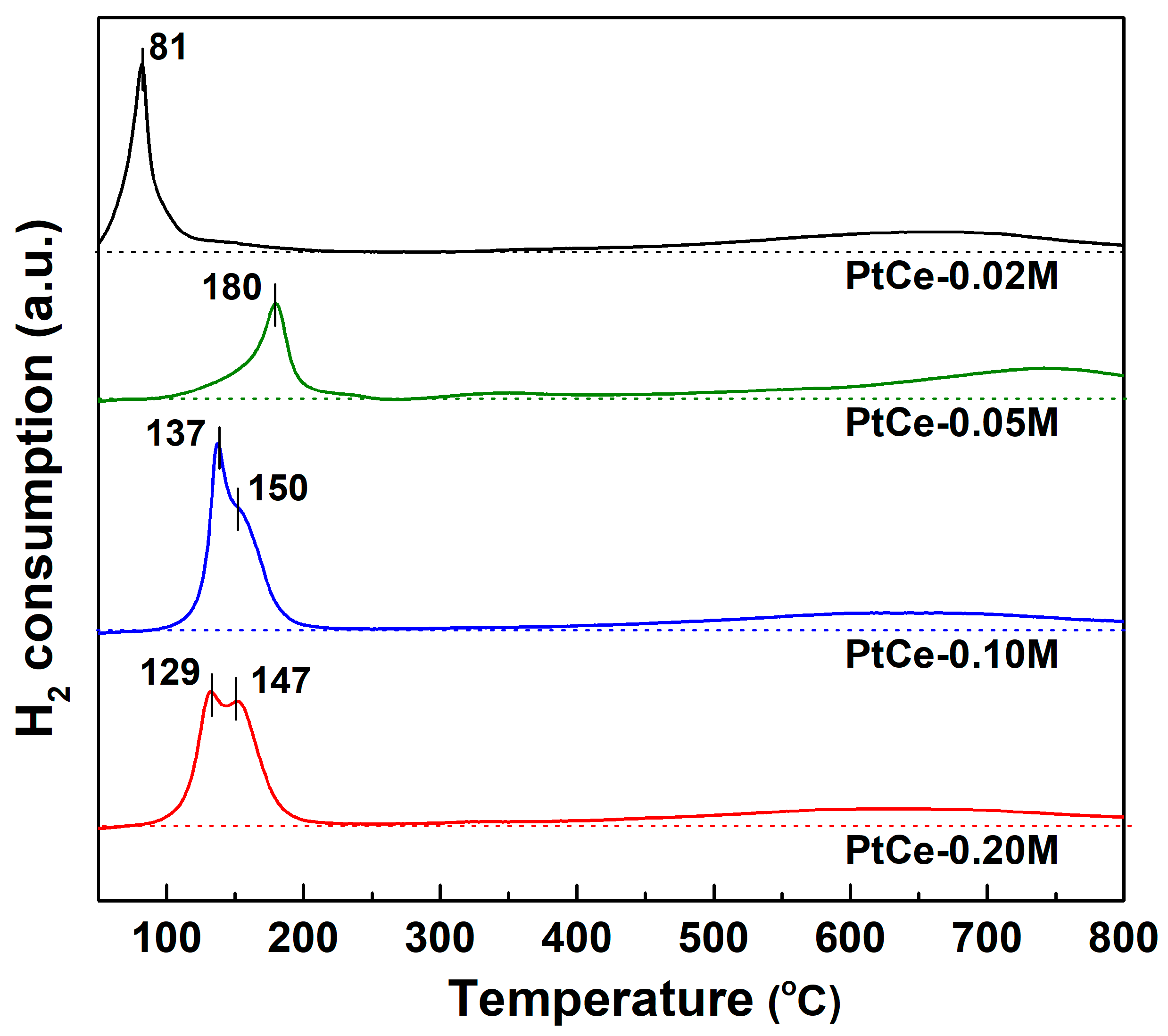
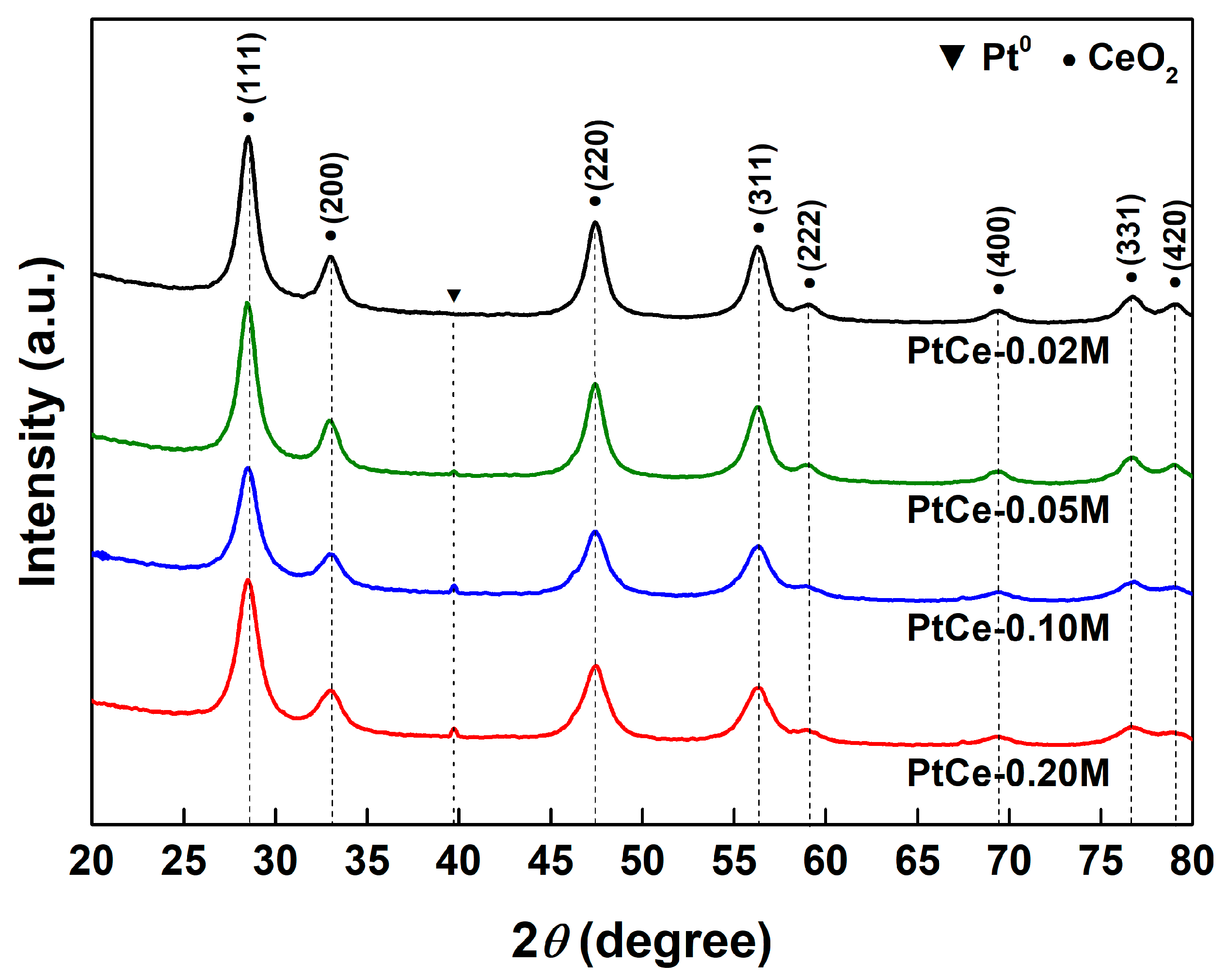
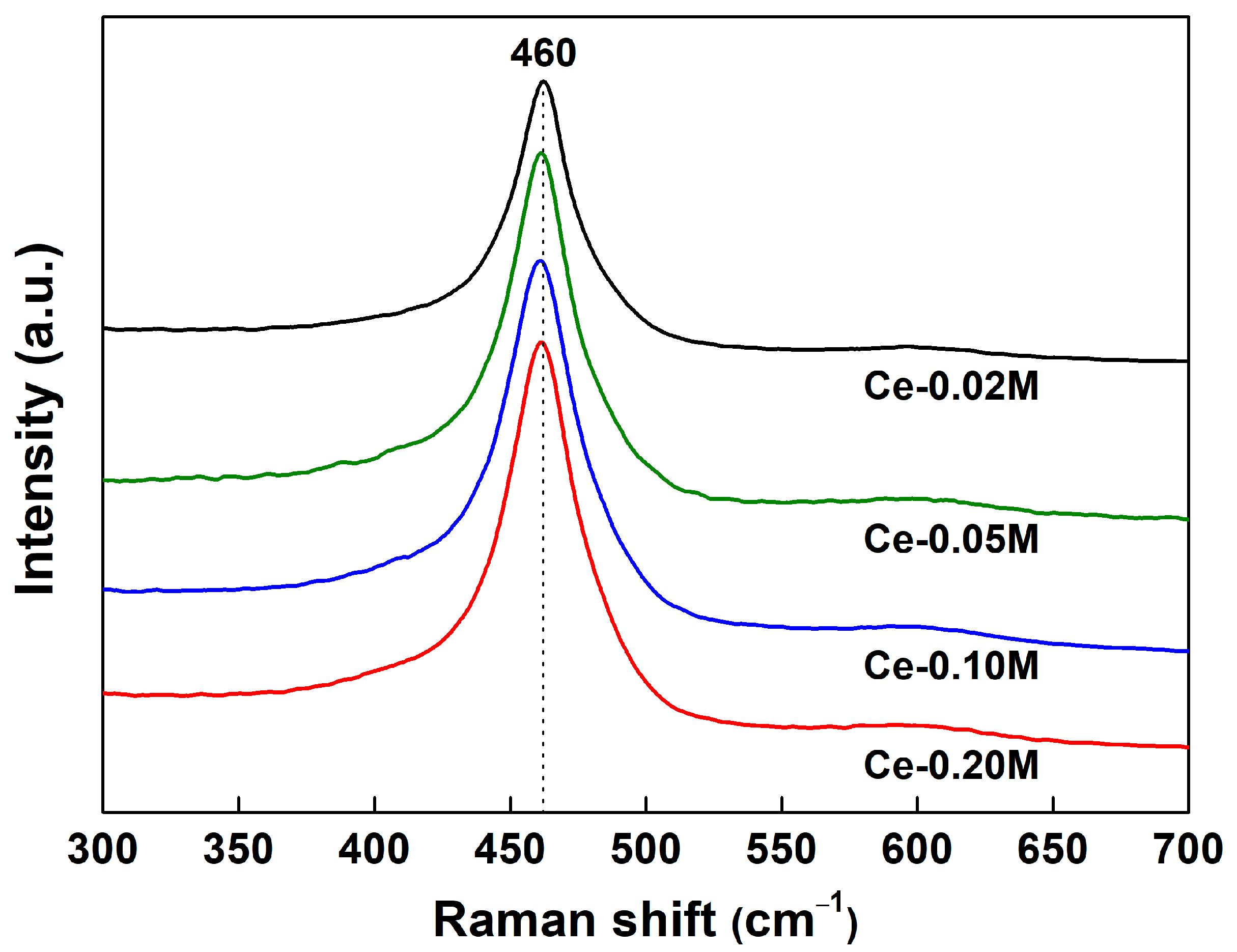
2.2. Catalyst Characterization Results
3. Discussion
4. Materials and Methods
4.1. Catalyst Synthesis
4.2. Characterization
4.3. Catalytic Reaction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lee, Y.L.; Kim, K.J.; Hong, G.R.; Roh, H.S. Target-Oriented Water–Gas Shift Reactions with Customized Reaction Conditions and Catalysts. Chem. Eng. J. 2023, 458, 141422. [Google Scholar] [CrossRef]
- Nandhini, R.; Berslin, D.; Sivaprakash, B.; Rajamohan, N.; Vo, D.V.N. Thermochemical Conversion of Municipal Solid Waste into Energy and Hydrogen: A Review. Environ. Chem. Lett. 2022, 20, 1645–1669. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Kim, K.J.; Kim, B.J.; Hong, G.R.; Jang, W.J.; Bae, J.W.; Park, Y.K.; Jeon, B.H.; Roh, H.S. From Gray to Blue Hydrogen: Trends and Forecasts of Catalysts and Sorbents for Unit Process. Renew. Sustain. Energy Rev. 2023, 186, 113635. [Google Scholar] [CrossRef]
- Sajid, M.; Raheem, A.; Ullah, N.; Asim, M.; Ur Rehman, M.S.; Ali, N. Gasification of Municipal Solid Waste: Progress, Challenges, and Prospects. Renew. Sustain. Energy Rev. 2022, 168, 112815. [Google Scholar] [CrossRef]
- Alaedini, A.H.; Tourani, H.K.; Saidi, M. A Review of Waste-to-Hydrogen Conversion Technologies for Solid Oxide Fuel Cell (SOFC) Applications: Aspect of Gasification Process and Catalyst Development. J. Environ. Manag. 2023, 329, 117077. [Google Scholar] [CrossRef]
- Rasul, M.G.; Hazrat, M.A.; Sattar, M.A.; Jahirul, M.I.; Shearer, M.J. The Future of Hydrogen: Challenges on Production, Storage and Applications. Energy Convers. Manag. 2022, 272, 116326. [Google Scholar] [CrossRef]
- Lee, Y.L.; Lee, K.; Hyun Ko, C.; Roh, H.S. Optimization of Nano-Catalysts for Application in Compact Reformers. Chem. Eng. J. 2022, 431, 134299. [Google Scholar] [CrossRef]
- Hakandai, C.; Sidik Pramono, H.; Aziz, M. Conversion of Municipal Solid Waste to Hydrogen and Its Storage to Methanol. Sustain. Energy Technol. Assess. 2022, 51, 101968. [Google Scholar] [CrossRef]
- Saravanakumar, A.; Chen, W.H.; Arunachalam, K.D.; Park, Y.K.; Chyuan Ong, H. Pilot-Scale Study on Downdraft Gasification of Municipal Solid Waste with Mass and Energy Balance Analysis. Fuel 2022, 315, 123287. [Google Scholar] [CrossRef]
- Lee, Y.L.; Kim, K.J.; Hong, G.R.; Ahn, S.Y.; Kim, B.J.; Shim, J.O.; Roh, H.S. Highly Sulfur Tolerant and Regenerable Pt/CeO2 Catalyst for Waste to Energy. Renew. Energy 2021, 178, 334–343. [Google Scholar] [CrossRef]
- Lee, Y.L.; Kim, K.J.; Hong, G.R.; Ahn, S.Y.; Kim, B.J.; Park, H.R.; Yun, S.J.; Bae, J.W.; Jeon, B.H.; Roh, H.S. Sulfur-Tolerant Pt/CeO2Catalyst with Enhanced Oxygen Storage Capacity by Controlling the Pt Content for the Waste-to-Hydrogen Processes. ACS Sustain. Chem. Eng. 2021, 9, 15287–15293. [Google Scholar] [CrossRef]
- Kim, K.J.; Jeon, K.W.; Hong, G.R.; Jeon, B.H.; Wook Bae, J.; Jang, W.J.; Lee, Y.L.; Roh, H.S. Elucidating the Effect of Ce/Zr Ratio on High Temperature Shift Activity with Sulfur Poisoning. J. Ind. Eng. Chem. 2022, 115, 537–543. [Google Scholar] [CrossRef]
- Hong, G.R.; Kim, K.J.; Ahn, S.Y.; Kim, B.J.; Park, H.R.; Lee, Y.L.; Lee, S.S.; Jeon, Y.; Roh, H.S. Sulfur-Resistant CeO2-Supported Pt Catalyst for Waste-to-Hydrogen: Effect of Catalyst Synthesis Method. Catalysts 2022, 12, 1670. [Google Scholar] [CrossRef]
- Kim, K.J.; Hong, G.R.; Ahn, S.Y.; Kim, B.J.; Lee, Y.L.; Roh, H.S. Effect of Precipitation Variables on the Performance of CeO2-Based Catalysts for Waste-to-Hydrogen. Catal. Today 2024, 425, 114336. [Google Scholar] [CrossRef]
- Kim, K.J.; Lee, Y.L.; Hong, G.R.; Ahn, S.Y.; Kim, B.J.; Lee, S.S.; Jeon, Y.; Roh, H.S. A Study on the Activity Recovery Behavior of Noble Metal Catalysts against Sulfur Poisoning. Catal. Today 2024, 425, 114361. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Jang, W.J.; Shim, J.O.; Jeon, B.H.; Roh, H.S. CeO2-Based Oxygen Storage Capacity Materials in Environmental and Energy Catalysis for Carbon Neutrality: Extended Application and Key Catalytic Properties. Catal. Rev. Sci. Eng. 2023, 1–84. [Google Scholar] [CrossRef]
- Ouyang, J.; Pei, J.; Kuang, Q.; Xie, Z.; Zheng, L. Supersaturation-Controlled Shape Evolution of α-Fe2O 3 Nanocrystals and Their Facet-Dependent Catalytic and Sensing Properties. ACS Appl. Mater. Interfaces 2014, 6, 12505–12514. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Li, S.; Xiong, L.; Jiao, Y.; Yuan, S.; Wang, J.; Chen, Y. Preparation of Nanostructured CeO2-ZrO2-Based Materials with Stabilized Surface Area and Their Catalysis in Soot Oxidation. Appl. Surf. Sci. 2020, 505, 144301. [Google Scholar] [CrossRef]
- Ramachandran, M.; Subadevi, R.; Sivakumar, M. Role of PH on Synthesis and Characterization of Cerium Oxide (CeO2) Nano Particles by Modified Co-Precipitation Method. Vacuum 2019, 161, 220–224. [Google Scholar] [CrossRef]
- Thanh, N.T.K.; Maclean, N.; Mahiddine, S. Mechanisms of Nucleation and Growth of Nanoparticles in Solution. Chem. Rev. 2014, 114, 7610–7630. [Google Scholar] [CrossRef] [PubMed]
- Roelands, C.P.M.; Ter Horst, J.H.; Kramer, H.J.M.; Jansens, P.J. Analysis of Nucleation Rate Measurements in Precipitation Processes. Cryst. Growth Des. 2006, 6, 1380–1392. [Google Scholar] [CrossRef]
- Papavasiliou, A.; Oliani, B.; Scanferla, M.; Sakellis, E.; Glisenti, A.; Canu, P.; Katsaros, F.K. Highly Homogeneous Multicomponent Mesoporous Catalysts: Defective Amorphous vs. Nanocrystalline CeO2 Structure towards CO-PROX Reaction. Int. J. Hydrogen Energy 2023, 48, 25044–25064. [Google Scholar] [CrossRef]
- Nematollahi, B.; Rezaei, M.; Lay, E.N. Preparation of Highly Active and Stable NiO-CeO2 Nanocatalysts for CO Selective Methanation. Int. J. Hydrogen Energy 2015, 40, 8539–8547. [Google Scholar] [CrossRef]
- Li, F.; Zou, L.; He, J.; Wu, Y.; Yang, L.; Liu, Q.; Wu, Q.; Yang, X. On the Correlation between Structure and Catalytic Activity of Mesoporous Ceria Nanoparticles. J. Catal. 2021, 402, 300–309. [Google Scholar] [CrossRef]
- Tanabe, T.; Nagai, Y.; Hirabayashi, T.; Takagi, N.; Dohmae, K.; Takahashi, N.; Matsumoto, S.; Shinjoh, H.; Kondo, J.N.; Schouten, J.C.; et al. Low Temperature CO Pulse Adsorption for the Determination of Pt Particle Size in a Pt/Cerium-Based Oxide Catalyst. Appl. Catal. A Gen. 2009, 370, 108–113. [Google Scholar] [CrossRef]
- Takeguchi, T.; Manabe, S.; Kikuchi, R.; Eguchi, K.; Kanazawa, T.; Matsumoto, S.; Ueda, W. Determination of Dispersion of Precious Metals on CeO2- Containing Supports. Appl. Catal. A Gen. 2005, 293, 91–96. [Google Scholar] [CrossRef]
- Lee, J.; Ryou, Y.; Kim, J.; Chan, X.; Kim, T.J.; Kim, D.H. Influence of the Defect Concentration of Ceria on the Pt Dispersion and the CO Oxidation Activity of Pt/CeO2. J. Phys. Chem. C 2018, 122, 4972–4983. [Google Scholar] [CrossRef]
- Baraj, E.; Ciahotný, K.; Hlinčík, T. The Water Gas Shift Reaction: Catalysts and Reaction Mechanism. Fuel 2021, 288, 119817. [Google Scholar] [CrossRef]
- Kalamaras, C.M.; Americanou, S.; Efstathiou, A.M. “Redox” vs “Associative Formate with -OH Group Regeneration” WGS Reaction Mechanism on Pt/CeO2: Effect of Platinum Particle Size. J. Catal. 2011, 279, 287–300. [Google Scholar] [CrossRef]
- Vecchietti, J.; Bonivardi, A.; Xu, W.; Stacchiola, D.; Delgado, J.J.; Calatayud, M.; Collins, S.E. Understanding the Role of Oxygen Vacancies in the Water Gas Shift Reaction on Ceria-Supported Platinum Catalysts. ACS Catal. 2014, 4, 2088–2096. [Google Scholar] [CrossRef]
- Wang, B.; Li, X.; Sun, Y.; Xiao, H.; Fu, M.; Li, S.; Liang, H.; Qiao, Z.; Ye, D. Unravelling the Correlation of Dielectric Barrier Discharge Power and Performance of Pt/CeO2 Catalysts for Toluene Oxidation. Catal. Sci. Technol. 2022, 13, 389–399. [Google Scholar] [CrossRef]
- Lee, J.; Ryou, Y.; Chan, X.; Kim, T.J.; Kim, D.H. How Pt Interacts with CeO2 under the Reducing and Oxidizing Environments at Elevated Temperature: The Origin of Improved Thermal Stability of Pt/CeO2 Compared to CeO2. J. Phys. Chem. C 2016, 120, 25870–25879. [Google Scholar] [CrossRef]
- Peng, R.; Li, S.; Sun, X.; Ren, Q.; Chen, L.; Fu, M.; Wu, J.; Ye, D. Size Effect of Pt Nanoparticles on the Catalytic Oxidation of Toluene over Pt/CeO2 Catalysts. Appl. Catal. B 2018, 220, 462–470. [Google Scholar] [CrossRef]
- Zhou, A.; Wang, J.; Wang, H.; Li, H.; Wang, J.; Shen, M. Effect of Active Oxygen on the Performance of Pt/CeO2 Catalysts for CO Oxidation. J. Rare Earths 2018, 36, 257–264. [Google Scholar] [CrossRef]
- Happel, M.; Mysliveček, J.; Johánek, V.; Dvořák, F.; Stetsovych, O.; Lykhach, Y.; Matolín, V.; Libuda, J. Adsorption Sites, Metal-Support Interactions, and Oxygen Spillover Identified by Vibrational Spectroscopy of Adsorbed CO: A Model Study on Pt/Ceria Catalysts. J. Catal. 2012, 289, 118–126. [Google Scholar] [CrossRef]
- Kuan, W.F.; Chung, C.H.; Lin, M.M.; Tu, F.Y.; Chen, Y.H.; Yu, W.Y. Activation of Carbon Dioxide with Surface Oxygen Vacancy of Ceria Catalyst: An Insight from in-Situ X-Ray Absorption near Edge Structure Analysis. Mater. Today Sustain. 2023, 23, 100425. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, C.; Wang, Z.; Ji, J.; Wan, H.; Zou, W.; Tong, Q.; Sun, J.; Dong, L.; Chen, Y.W. Enhanced Low-Temperature Catalytic Performance for Toluene Combustion of CeO2-Supported Pt-Ir Alloy Catalysts. Appl. Surf. Sci. 2022, 580, 152278. [Google Scholar] [CrossRef]
- Tan, W.; Xie, S.; Le, D.; Diao, W.; Wang, M.; Low, K.B.; Austin, D.; Hong, S.; Gao, F.; Dong, L.; et al. Fine-Tuned Local Coordination Environment of Pt Single Atoms on Ceria Controls Catalytic Reactivity. Nat. Commun. 2022, 13, 7070. [Google Scholar] [CrossRef]
- Rocha, L.S.R.; Aparecido Ciola Amoresi, R.; Duarte, T.M.; Marana, N.L.; Sambrano, J.R.; Aldao, C.M.; Simões, A.Z.; Ponce, M.A.; Longo, E. Experimental and Theoretical Interpretation of the Order/Disorder Clusters in CeO2:La. Appl. Surf. Sci. 2020, 510, 145216. [Google Scholar] [CrossRef]
- Soni, S.; Kumar, S.; Dalela, B.; Kumar, S.; Alvi, P.A.; Dalela, S. Defects and Oxygen Vacancies Tailored Structural and Optical Properties in CeO2 Nanoparticles Doped with Sm3+ Cation. J. Alloys. Compd. 2018, 752, 520–531. [Google Scholar] [CrossRef]
- Bindu, P.; Thomas, S. Estimation of Lattice Strain in ZnO Nanoparticles: X-ray Peak Profile Analysis. J. Theor. Appl. Phys. 2014, 8, 123–134. [Google Scholar] [CrossRef]
- Ma, D.; Lu, Z.; Tang, Y.; Li, T.; Tang, Z.; Yang, Z. Effect of Lattice Strain on the Oxygen Vacancy Formation and Hydrogen Adsorption at CeO2(111) Surface. Phys. Lett. A 2014, 378, 2570–2575. [Google Scholar] [CrossRef]
- Lee, Y.L.; Mnoyan, A.; Na, H.S.; Ahn, S.Y.; Kim, K.J.; Shim, J.O.; Lee, K.; Roh, H.S. Comparison of the Effects of the Catalyst Preparation Method and CeO2 morphology on the Catalytic Activity of Pt/CeO2 catalysts for the Water-Gas Shift Reaction. Catal. Sci. Technol. 2020, 10, 6299–6308. [Google Scholar] [CrossRef]
- Liu, H.; Dong, M.; Xiong, J.; Yang, J.; Ning, J.; Liang, Y.; Lu, J. CO2 Laser Promoted Oxygen Vacancy-Active Oxygen Cycle in DRM on Ni/CeO2. Energy Convers. Manag. 2023, 286, 117081. [Google Scholar] [CrossRef]
- Hernández, W.Y.; Laguna, O.H.; Centeno, M.A.; Odriozola, J.A. Structural and Catalytic Properties of Lanthanide (La, Eu, Gd) Doped Ceria. J. Solid State Chem. 2011, 184, 3014–3020. [Google Scholar] [CrossRef]
- Trogadas, P.; Parrondo, J.; Ramani, V. CeO2 Surface Oxygen Vacancy Concentration Governs in Situ Free Radical Scavenging Efficacy in Polymer Electrolytes. ACS Appl. Mater. Interfaces 2012, 4, 5098–5102. [Google Scholar] [CrossRef]
- Rahimi-Nasrabadi, M.; Pourmortazavi, S.M.; Davoudi-Dehaghani, A.A.; Hajimirsadeghi, S.S.; Zahedi, M.M. Synthesis and Characterization of Copper Oxalate and Copper Oxide Nanoparticles by Statistically Optimized Controlled Precipitation and Calcination of Precursor. CrystEngComm 2013, 15, 4077–4086. [Google Scholar] [CrossRef]
- Zhang, J.; Li, H.; Kuang, Q.; Xie, Z. Toward Rationally Designing Surface Structures of Micro- and Nanocrystallites: Role of Supersaturation. ACC Chem. Res. 2018, 51, 2880–2887. [Google Scholar] [CrossRef]
- Shittu, T.; Altarawneh, M. Investigative Properties of CeO2 doped with Niobium: A Combined Characterization and DFT Studies. Nanotechnol. Rev. 2021, 11, 191–203. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | BET S.A. (m2/g) a | Pore Volume (cm3/g) a | Pt0 Dispersion (%) b | Crystallite Size (nm) c | Lattice Parameter (Å) c | Lattice Strain (10−2) c |
|---|---|---|---|---|---|---|
| PtCe–0.02 M | 130 | 0.272 | 59.0 | 7.27 | 5.420 | 1.938 |
| PtCe–0.05 M | 118 | 0.384 | 62.5 | 6.51 | 5.423 | 2.163 |
| PtCe–0.10 M | 138 | 0.406 | 59.0 | 5.60 | 5.420 | 2.514 |
| PtCe–0.20 M | 112 | 0.495 | 60.0 | 5.56 | 5.421 | 2.532 |
| Catalyst | Ce3+ (%) a | OV (%) b | Support | Concentration of Oxygen Vacancies (1021 cm−3) c |
|---|---|---|---|---|
| PtCe–0.02 M | 32.3 | 20.3 | Ce–0.02 M | 1.40 |
| PtCe–0.05 M | 35.9 | 20.8 | Ce–0.05 M | 1.61 |
| PtCe–0.10 M | 37.8 | 21.1 | Ce–0.10 M | 1.83 |
| PtCe–0.20 M | 38.6 | 21.3 | Ce–0.20 M | 1.84 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, G.-R.; Kim, K.-J.; Ahn, S.-Y.; Kim, B.-J.; Shin, B.-G.; Oh, M.-J.; Lee, Y.-L.; Roh, H.-S. The Effect of Precursor Concentration on the Crystallite Size of CeO2 to Enhance the Sulfur Resistance of Pt/CeO2 for Water Gas Shift. Catalysts 2024, 14, 99. https://doi.org/10.3390/catal14020099
Hong G-R, Kim K-J, Ahn S-Y, Kim B-J, Shin B-G, Oh M-J, Lee Y-L, Roh H-S. The Effect of Precursor Concentration on the Crystallite Size of CeO2 to Enhance the Sulfur Resistance of Pt/CeO2 for Water Gas Shift. Catalysts. 2024; 14(2):99. https://doi.org/10.3390/catal14020099
Chicago/Turabian StyleHong, Ga-Ram, Kyoung-Jin Kim, Seon-Yong Ahn, Beom-Jun Kim, Bong-Gyeong Shin, Min-Ju Oh, Yeol-Lim Lee, and Hyun-Seog Roh. 2024. "The Effect of Precursor Concentration on the Crystallite Size of CeO2 to Enhance the Sulfur Resistance of Pt/CeO2 for Water Gas Shift" Catalysts 14, no. 2: 99. https://doi.org/10.3390/catal14020099