
Enhancing Stability of γ-Al2O3-Supported NiCu Catalysts by Impregnating Basic Oxides in the Hydrodeoxygenation of Anisole




Abstract
:1. Introduction
2. Results and Discussion
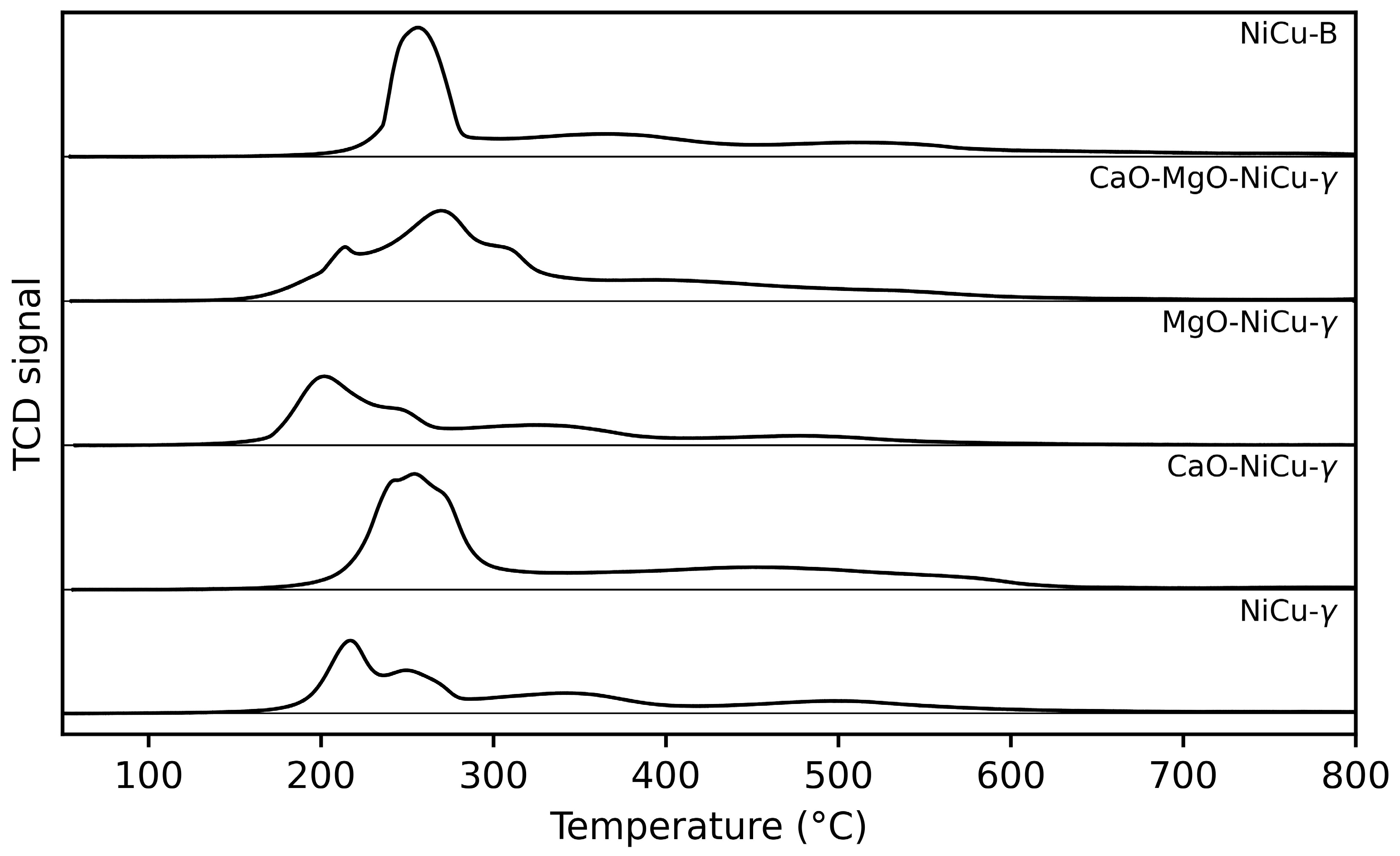
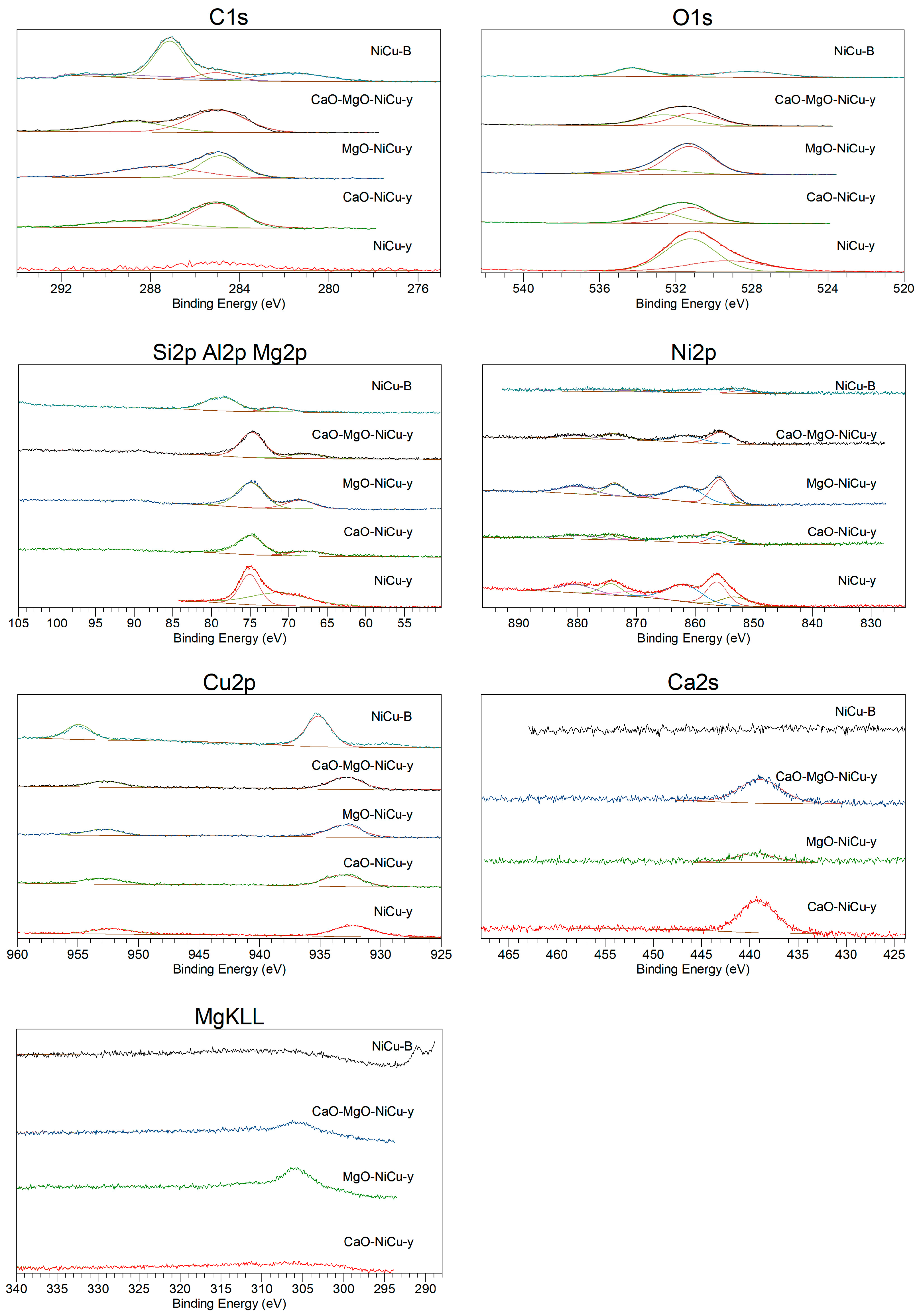
2.1. Catalyst Characterization
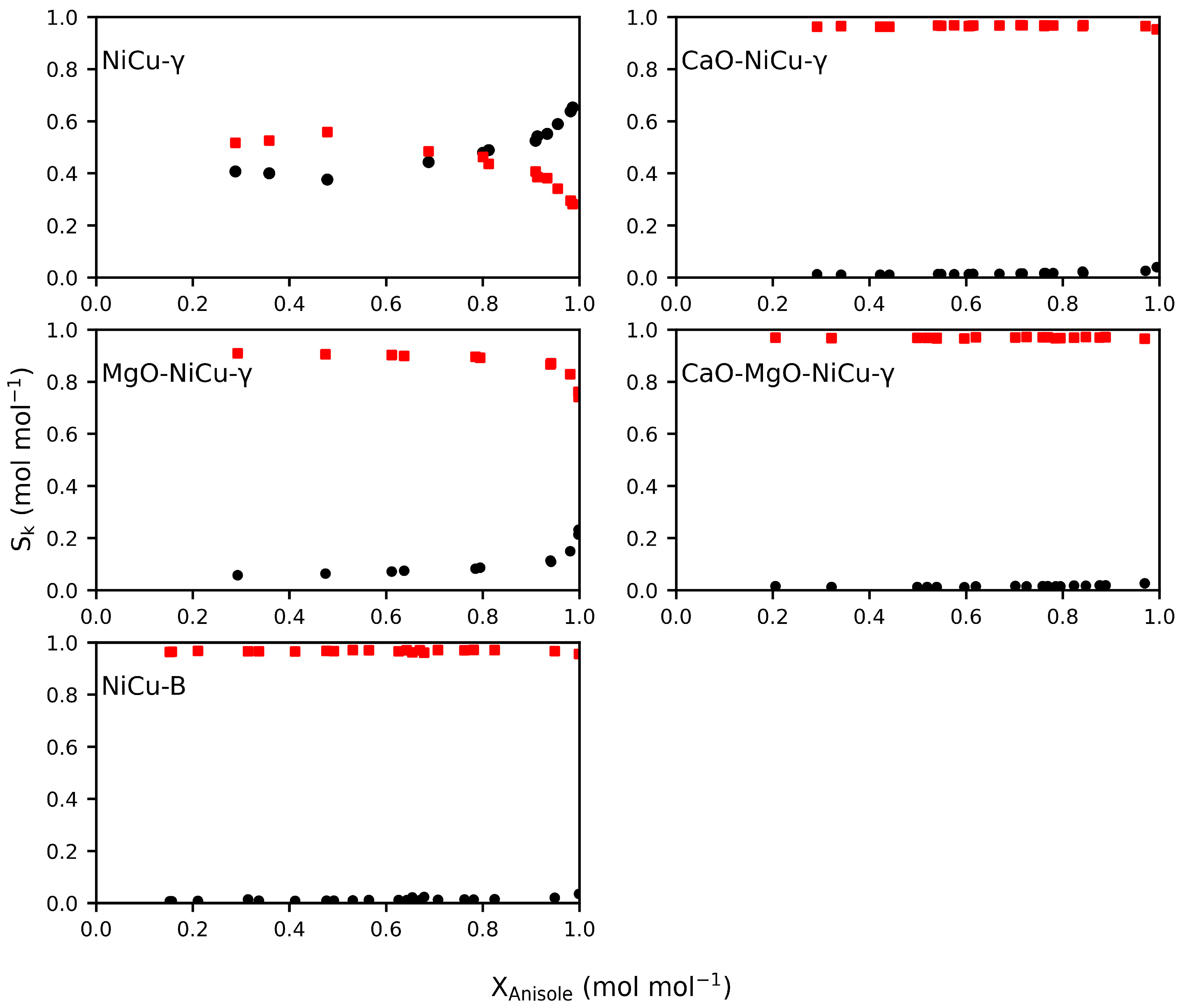
2.2. Catalyst Performance
3. Materials and Methods
3.1. Catalyst Preparation
3.2. Catalyst Characterization
3.3. Catalyst Performance Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Londo, M.; van Stralen, J.; Uslu, A.; Mozaffarian, H.; Kraan, C. Lignocellulosic biomass for chemicals and energy: An integrated assessment of future EU market sizes, feedstock availability impacts, synergy and competition effects, and path dependencies. Biofuels Bioprod. Biorefining 2018, 12, 1065–1081. [Google Scholar] [CrossRef]
- Okolie, J.A.; Mukherjee, A.; Nanda, S.; Dalai, A.K.; Kozinski, J.A. Next-generation biofuels and platform biochemicals from lignocellulosic biomass. Int. J. Energy Res. 2021, 45, 14145–14169. [Google Scholar] [CrossRef]
- de Reviere, A.; Gunst, D.; Sabbe, M.K.; Reyniers, M.-F.; Verberckmoes, A. Dehydration of butanol towards butenes over MFI, FAU and MOR: Influence of zeolite topology. Catal. Sci. Technol. 2021, 11, 2540–2559. [Google Scholar] [CrossRef]
- Serrano-Ruiz, J.C.; Dumesic, J.A. Catalytic routes for the conversion of biomass into liquid hydrocarbon transportation fuels. Energy Environ. Sci. 2011, 4, 83–99. [Google Scholar] [CrossRef]
- Jacobs, B.; Yao, Y.; Van Nieuwenhove, I.; Sharma, D.; Graulus, G.-J.; Bernaerts, K.; Verberckmoes, A. Sustainable lignin modifications and processing methods: Green chemistry as the way forward. Green Chem. 2023, 25, 2042–2086. [Google Scholar] [CrossRef]
- Garcia-Perez, M.; Wang, X.S.; Shen, J.; Rhodes, M.J.; Tian, F.; Lee, W.-J.; Wu, H.; Li, C.-Z. Fast Pyrolysis of Oil Mallee Woody Biomass: Effect of Temperature on the Yield and Quality of Pyrolysis Products. Ind. Eng. Chem. Res. 2008, 47, 1846–1854. [Google Scholar] [CrossRef]
- Bond, J.Q.; Upadhye, A.A.; Olcay, H.; Tompsett, G.A.; Jae, J.; Xing, R.; Alonso, D.M.; Wang, D.; Zhang, T.; Kumar, R.; et al. Production of renewable jet fuel range alkanes and commodity chemicals from integrated catalytic processing of biomass. Energy Environ. Sci. 2014, 7, 1500–1523. [Google Scholar] [CrossRef]
- Wang, H.; Male, J.; Wang, Y. Recent Advances in Hydrotreating of Pyrolysis Bio-Oil and Its Oxygen-Containing Model Compounds. ACS Catal. 2013, 3, 1047–1070. [Google Scholar] [CrossRef]
- Ranga, C.; Alexiadis, V.I.; Lauwaert, J.; Lødeng, R.; Thybaut, J.W. Effect of Co incorporation and support selection on deoxygenation selectivity and stability of (Co)Mo catalysts in anisole HDO. Appl. Catal. A Gen. 2019, 571, 61–70. [Google Scholar] [CrossRef]
- Ardiyanti, A.R.; Bykova, M.V.; Khromova, S.A.; Yin, W.; Venderbosch, R.H.; Yakovlev, V.A.; Heeres, H.J. Ni-Based Catalysts for the Hydrotreatment of Fast Pyrolysis Oil. Energy Fuels 2016, 30, 1544–1554. [Google Scholar] [CrossRef]
- Vandevyvere, T.; Sabbe, M.K.; Mendes, P.S.F.; Thybaut, J.W.; Lauwaert, J. NiCu-based catalysts for the low-temperature hydrodeoxygenation of anisole: Effect of the metal ratio on SiO2 and γ-Al2O3 supports. Green Carbon 2023, 1, 170–184. [Google Scholar] [CrossRef]
- Ardiyanti, A.R.; Khromova, S.A.; Venderbosch, R.H.; Yakovlev, V.A.; Heeres, H.J. Catalytic hydrotreatment of fast-pyrolysis oil using non-sulfided bimetallic Ni-Cu catalysts on a δ-Al2O3 support. Appl. Catal. B Environ. 2012, 117–118, 105–117. [Google Scholar] [CrossRef]
- Sangnikul, P.; Phanpa, C.; Xiao, R.; Zhang, H.; Reubroycharoen, P.; Kuchonthara, P.; Vitidsant, T.; Pattiya, A.; Hinchiranan, N. Role of copper- or cerium-promoters on NiMo/γ-Al2O3 catalysts in hydrodeoxygenation of guaiacol and bio-oil. Appl. Catal. A Gen. 2019, 574, 151–160. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, C.; Liu, Y.; Hou, X.; Zhang, R.; Tang, X. Coke Deposition on Ni/HZSM-5 in Bio-oil Hydrodeoxygenation Processing. Energy Fuels 2015, 29, 1722–1728. [Google Scholar] [CrossRef]
- Ameen, M.; Azizan, M.T.; Ramli, A.; Yusup, S.; Abdullah, B. The effect of metal loading over Ni/γ-Al2O3 and Mo/γ-Al2O3 catalysts on reaction routes of hydrodeoxygenation of rubber seed oil for green diesel production. Catal. Today 2020, 355, 51–64. [Google Scholar] [CrossRef]
- Poissonnier, J.; Ranga, C.; Lødeng, R.; Thybaut, J.W. Oxygen functionality and chain length effects in HDO: Impact of competitive adsorption on reactivity. Fuel 2022, 308, 121940. [Google Scholar] [CrossRef]
- He, Z.; Wang, X. Hydrodeoxygenation of model compounds and catalytic systems for pyrolysis bio-oils upgrading. Catal. Sustain. Energy 2012, 1, 28–52. [Google Scholar] [CrossRef]
- Sitthisa, S.; An, W.; Resasco, D.E. Selective conversion of furfural to methylfuran over silica-supported NiFe bimetallic catalysts. J. Catal. 2011, 284, 90–101. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U., Jr.; Steele, P.H. Pyrolysis of Wood/Biomass for Bio-oil: A Critical Review. Energy Fuels 2006, 20, 848–889. [Google Scholar] [CrossRef]
- Prabhudesai, V.S.; Gurrala, L.; Vinu, R. Catalytic Hydrodeoxygenation of Lignin-Derived Oxygenates: Catalysis, Mechanism, and Effect of Process Conditions. Energy Fuels 2022, 36, 1155–1188. [Google Scholar] [CrossRef]
- Zhang, J.; Fidalgo, B.; Kolios, A.; Shen, D.; Gu, S. Mechanism of deoxygenation in anisole decomposition over single-metal loaded HZSM-5: Experimental study. Chem. Eng. J. 2018, 336, 211–222. [Google Scholar] [CrossRef]
- Grange, P.; Laurent, E.; Maggi, R.; Centeno, A.; Delmon, B. Hydrotreatment of pyrolysis oils from biomass: Reactivity of the various categories of oxygenated compounds and preliminary techno-economical study. Catal. Today 1996, 29, 297–301. [Google Scholar] [CrossRef]
- Dieuzeide, M.L.; Jobbagy, M.; Amadeo, N. Glycerol steam reforming over Ni/γ-Al2O3 catalysts, modified with Mg(II). Effect of Mg (II) content. Catal. Today 2013, 213, 50–57. [Google Scholar] [CrossRef]
- Bouriakova, A.; Mendes, P.S.F.; Katryniok, B.; De Clercq, J.; Thybaut, J.W. Co-metal induced stabilization of alumina-supported copper: Impact on the hydrogenolysis of glycerol to 1,2-propanediol. Catal. Commun. 2020, 146, 106134. [Google Scholar] [CrossRef]
- Vandevyvere, T.; Sabbe, M.K.; Bouriakova, A.; Saravanamurugan, S.; Thybaut, J.W.; Lauwaert, J. Impact of the incipient wetness impregnation sequence during the preparation of La or Ce promoted NiCu-Al2O3 on low-temperature hydrodeoxygenation. Catal. Commun. 2023, 181, 106734. [Google Scholar] [CrossRef]
- Yu, J.; Ge, Q.; Fang, W.; Xu, H. Influences of calcination temperature on the efficiency of CaO promotion over CaO modified Pt/γ-Al2O3 catalyst. Appl. Catal. A Gen. 2011, 395, 114–119. [Google Scholar] [CrossRef]
- Lemonidou, A.A.; Vasalos, I.A. Carbon dioxide reforming of methane over 5 wt.% Ni/CaO-Al2O3 catalyst. Appl. Catal. A Gen. 2002, 228, 227–235. [Google Scholar] [CrossRef]
- Papageridis, K.N.; Charisiou, N.D.; Douvartzides, S.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; AlKhoori, S.; Polychronopoulou, K.; Goula, M.A. Promoting effect of CaO-MgO mixed oxide on Ni/γ-Al2O3 catalyst for selective catalytic deoxygenation of palm oil. Renew. Energy 2020, 162, 1793–1810. [Google Scholar] [CrossRef]
- Charisiou, N.D.; Papageridis, K.N.; Tzounis, L.; Sebastian, V.; Hinder, S.J.; Baker, M.A.; AlKetbi, M.; Polychronopoulou, K.; Goula, M.A. Ni supported on CaO-MgO-Al2O3 as a highly selective and stable catalyst for H2 production via the glycerol steam reforming reaction. Int. J. Hydrogen Energy 2019, 44, 256–273. [Google Scholar] [CrossRef]
- Lisboa, J.d.S.; Santos, D.C.R.M.; Passos, F.B.; Noronha, F.B. Influence of the addition of promoters to steam reforming catalysts. Catal. Today 2005, 101, 15–21. [Google Scholar] [CrossRef]
- Boscagli, C.; Raffelt, K.; Zevaco, T.A.; Olbrich, W.; Otto, T.N.; Sauer, J.; Grunwaldt, J.-D. Mild hydrotreatment of the light fraction of fast-pyrolysis oil produced from straw over nickel-based catalysts. Biomass Bioenergy 2015, 83, 525–538. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Yurdakal, S.; Garlisi, C.; Özcan, L.; Bellardita, M.; Palmisano, G. Chapter 4—(Photo)catalyst Characterization Techniques: Adsorption Isotherms and BET, SEM, FTIR, UV–Vis, Photoluminescence, and Electrochemical Characterizations. In Heterogeneous Photocatalysis; Marcì, G., Palmisano, L., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 87–152. [Google Scholar]
- Li, B.; Su, W.; Wang, X.; Wang, X. Alumina supported Ni and Co catalysts modified by Y2O3 via different impregnation strategies: Comparative analysis on structural properties and catalytic performance in methane reforming with CO2. Int. J. Hydrogen Energy 2016, 41, 14732–14746. [Google Scholar] [CrossRef]
- Longo, A.; Theofanidis, S.A.; Cavallari, C.; Srinath, N.V.; Hu, J.; Poelman, H.; Sabbe, M.K.; Sahle, C.J.; Marin, G.B.; Galvita, V.V. What Makes Fe-Modified MgAl2O4 an Active Catalyst Support? Insight from X-ray Raman Scattering. ACS Catal. 2020, 10, 6613–6622. [Google Scholar] [CrossRef]
- de Reviere, A.; Vandevyvere, T.; Sabbe, M.K.; Verberckmoes, A. Renewable Butene Production through Dehydration Reactions over Nano-HZSM-5/γ-Al2O3 Hybrid Catalysts. Catalysts 2020, 10, 879. [Google Scholar] [CrossRef]
- Gorte, R.J. Temperature-programmed desorption for the characterization of oxide catalysts. Catal. Today 1996, 28, 405–414. [Google Scholar] [CrossRef]
- Gorte, R.J. What do we know about the acidity of solid acids? Catal. Lett. 1999, 62, 1–13. [Google Scholar] [CrossRef]
- Alreshaidan, S.B.; Ibrahim, A.A.; Fakeeha, A.H.; Almutlaq, A.M.; Ali, F.A.; Al-Fatesh, A.S. Effect of Modified Alumina Support on the Performance of Ni-Based Catalysts for CO2 Reforming of Methane. Catalysts 2022, 12, 1066. [Google Scholar] [CrossRef]
- Schreiter, N.; Kirchner, J.; Kureti, S. A DRIFTS and TPD study on the methanation of CO2 on Ni/Al2O3 catalyst. Catal. Commun. 2020, 140, 105988. [Google Scholar] [CrossRef]
- Marinković, D.M.; Avramović, J.M.; Stanković, M.V.; Stamenković, O.S.; Jovanović, D.M.; Veljković, V.B. Synthesis and characterization of spherically-shaped CaO/γ-Al2O3 catalyst and its application in biodiesel production. Energy Convers. Manag. 2017, 144, 399–413. [Google Scholar] [CrossRef]
- Scirè, S.; Crisafulli, C.; Maggiore, R.; Minicò, S.; Galvagno, S. Effect of the acid–base properties of Pd–Ca/Al2O3 catalysts on the selective hydrogenation of phenol to cyclohexanone: FT-IR and TPD characterization. Appl. Surf. Sci. 1998, 136, 311–320. [Google Scholar] [CrossRef]
- Taufiq-Yap, Y.H.; Lee, H.V.; Yunus, R.; Juan, J.C. Transesterification of non-edible Jatropha curcas oil to biodiesel using binary Ca–Mg mixed oxide catalyst: Effect of stoichiometric composition. Chem. Eng. J. 2011, 178, 342–347. [Google Scholar] [CrossRef]
- Khromova, S.A.; Bykova, M.V.; Bulavchenko, O.A.; Ermakov, D.Y.; Saraev, A.A.; Kaichev, V.V.; Venderbosch, R.H.; Yakovlev, V.A. Furfural Hydrogenation to Furfuryl Alcohol over Bimetallic Ni–Cu Sol–Gel Catalyst: A Model Reaction for Conversion of Oxygenates in Pyrolysis Liquids. Top. Catal. 2016, 59, 1413–1423. [Google Scholar] [CrossRef]
- Silva, G.C.R.; Qian, D.; Pace, R.; Heintz, O.; Caboche, G.; Santillan-Jimenez, E.; Crocker, M. Promotional Effect of Cu, Fe and Pt on the Performance of Ni/Al2O3 in the Deoxygenation of Used Cooking Oil to Fuel-Like Hydrocarbons. Catalysts 2020, 10, 91. [Google Scholar] [CrossRef]
- Lin, J.-H.; Biswas, P.; Guliants, V.V.; Misture, S. Hydrogen production by water–gas shift reaction over bimetallic Cu–Ni catalysts supported on La-doped mesoporous ceria. Appl. Catal. A Gen. 2010, 387, 87–94. [Google Scholar] [CrossRef]
- De Rogatis, L.; Montini, T.; Cognigni, A.; Olivi, L.; Fornasiero, P. Methane partial oxidation on NiCu-based catalysts. Catal. Today 2009, 145, 176–185. [Google Scholar] [CrossRef]
- Boudaira, B.; Harabi, A.; Bouzerara, F.; Zenikheri, F.; Foughali, L.; Guechi, A. Preparation and characterization of membrane supports for microfiltration and ultrafiltration using kaolin (DD2) and CaCO3. Desalination Water Treat. 2016, 57, 5258–5265. [Google Scholar] [CrossRef]
- Yoshida, R.; Sun, D.; Yamada, Y.; Sato, S.; Hutchings, G.J. Vapor-phase hydrogenation of levulinic acid to γ-valerolactone over Cu-Ni bimetallic catalysts. Catal. Commun. 2017, 97, 79–82. [Google Scholar] [CrossRef]
- Upare, P.P.; Jeong, M.-G.; Hwang, Y.K.; Kim, D.H.; Kim, Y.D.; Hwang, D.W.; Lee, U.H.; Chang, J.-S. Nickel-promoted copper–silica nanocomposite catalysts for hydrogenation of levulinic acid to lactones using formic acid as a hydrogen feeder. Appl. Catal. A Gen. 2015, 491, 127–135. [Google Scholar] [CrossRef]
- Naghash, A.R.; Etsell, T.H.; Xu, S. XRD and XPS Study of Cu−Ni Interactions on Reduced Copper−Nickel−Aluminum Oxide Solid Solution Catalysts. Chem. Mater. 2006, 18, 2480–2488. [Google Scholar] [CrossRef]
- Albuquerque, M.C.G.; Santamaría-González, J.; Mérida-Robles, J.M.; Moreno-Tost, R.; Rodríguez-Castellón, E.; Jiménez-López, A.; Azevedo, D.C.S.; Cavalcante, C.L.; Maireles-Torres, P. MgM (M = Al and Ca) oxides as basic catalysts in transesterification processes. Appl. Catal. A Gen. 2008, 347, 162–168. [Google Scholar] [CrossRef]
- Hou, Z.; Yokota, O.; Tanaka, T.; Yashima, T. Characterization of Ca-promoted Ni/α-Al2O3 catalyst for CH4 reforming with CO2. Appl. Catal. A Gen. 2003, 253, 381–387. [Google Scholar] [CrossRef]
- Wu, J.; Shen, Y.; Liu, C.; Wang, H.; Geng, C.; Zhang, Z. Vapor phase hydrogenation of furfural to furfuryl alcohol over environmentally friendly Cu–Ca/SiO2 catalyst. Catal. Commun. 2005, 6, 633–637. [Google Scholar] [CrossRef]
- Scirè, S.; Minicò, S.; Crisafulli, C. Selective hydrogenation of phenol to cyclohexanone over supported Pd and Pd-Ca catalysts: An investigation on the influence of different supports and Pd precursors. Appl. Catal. A Gen. 2002, 235, 21–31. [Google Scholar] [CrossRef]
- Zhang, Q.; Feng, X.; Liu, J.; Zhao, L.; Song, X.; Zhang, P.; Gao, L. Hollow hierarchical Ni/MgO-SiO2 catalyst with high activity, thermal stability and coking resistance for catalytic dry reforming of methane. Int. J. Hydrogen Energy 2018, 43, 11056–11068. [Google Scholar] [CrossRef]
- Huang, B.; Li, X.; Ji, S.; Lang, B.; Habimana, F.; Li, C. Effect of MgO promoter on Ni-based SBA-15 catalysts for combined steam and carbon dioxide reforming of methane. J. Nat. Gas Chem. 2008, 17, 225–231. [Google Scholar] [CrossRef]
- Li, H.; Tian, H.; Chen, S.; Sun, Z.; Liu, T.; Liu, R.; Assabumrungrat, S.; Saupsor, J.; Mu, R.; Pei, C.; et al. Sorption enhanced steam reforming of methanol for high-purity hydrogen production over Cu-MgO/Al2O3 bifunctional catalysts. Appl. Catal. B Environ. 2020, 276, 119052. [Google Scholar] [CrossRef]
- He, C.; Ruan, T.; Ouyang, X.; Ma, Y.; Qian, Y.; Qiu, X. Selective hydrodeoxygenation of monophenolics from lignin bio-oil for preparing cyclohexanol and its derivatives over Ni-Co/Al2O3-MgO catalyst. Ind. Crops Prod. 2023, 202, 117045. [Google Scholar] [CrossRef]
- Shen, J.; Lochhead, M.J.; Bray, K.L.; Chen, Y.; Dumesic, J.A. Structural and Acid/Base Properties of Supported Europium Oxides. J. Phys. Chem. 1995, 99, 2384–2392. [Google Scholar] [CrossRef]
- Saraeian, A.; Burkhow, S.J.; Jing, D.; Smith, E.A.; Shanks, B.H. Catalyst Property Effects on Product Distribution during the Hydrodeoxygenation of Lignin Pyrolysis Vapors over MoO3/γ-Al2O3. ACS Sustain. Chem. Eng. 2021, 9, 6685–6696. [Google Scholar] [CrossRef]
- Wang, X.; Arai, M.; Wu, Q.; Zhang, C.; Zhao, F. Hydrodeoxygenation of lignin-derived phenolics—A review on the active sites of supported metal catalysts. Green Chem. 2020, 22, 8140–8168. [Google Scholar] [CrossRef]
- Daniell, W.; Schubert, U.; Glöckler, R.; Meyer, A.; Noweck, K.; Knözinger, H. Enhanced surface acidity in mixed alumina–silicas: A low-temperature FTIR study. Appl. Catal. A Gen. 2000, 196, 247–260. [Google Scholar] [CrossRef]
- Sokolov, S.; Stoyanova, M.; Rodemerck, U.; Linke, D.; Kondratenko, E.V. Effect of support on selectivity and on-stream stability of surface VOx species in non-oxidative propane dehydrogenation. Catal. Sci. Technol. 2014, 4, 1323–1332. [Google Scholar] [CrossRef]
- De Waele, J.; Galvita, V.V.; Poelman, H.; Gabrovska, M.; Nikolova, D.; Damyanova, S.; Thybaut, J.W. Ethanol dehydrogenation over Cu catalysts promoted with Ni: Stability control. Appl. Catal. A Gen. 2020, 591, 117401. [Google Scholar] [CrossRef]
- Van der Borght, K.; Toch, K.; Galvita, V.V.; Thybaut, J.W.; Marin, G.B. Information-Driven Catalyst Design Based on High-Throughput Intrinsic Kinetics. Catalysts 2015, 5, 1948–1968. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | SBET (m2 g−1) | Vp (cm3 g−1) | Cacid (µmol NH3 g−1) | Cbasic (µmol CO2 g−1) | Activity (molanisole s−1 kgcat−1) | SCyclohexane (%) |
|---|---|---|---|---|---|---|
| γ-Al2O3 | 240 ± 37 | 0.60 ± 0.10 | 427 | 36 | / | / |
| B-Al2O3 | 219 ± 21 | 0.65 ± 0.11 | 280 | 87 | / | / |
| NiCu-γ | 172 ± 49 | 0.38 ± 0.11 | 90 | 17 | 0.062 ± 0.011 | 49 ± 6 |
| CaO-NiCu-γ | 121 ± 24 | 0.26 ± 0.04 | 33 | 63 | 0.072 ± 0.012 | 1.6 ± 0.3 |
| MgO-NiCu-γ | 141 ± 42 | 0.33 ± 0.09 | 51 | 57 | 0.127 ± 0.034 | 8 ± 2 |
| CaO-MgO-NiCu-γ | 117 ± 20 | 0.28 ± 0.07 | 54 | 58 | 0.093 ± 0.024 | 1.5 ± 0.1 |
| NiCu-B | 155 ± 12 | 0.45 ± 0.02 | 50 | 56 | 0.069 ± 0.017 | 1.3 ± 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vandevyvere, T.; Sabbe, M.K.; Thybaut, J.W.; Lauwaert, J. Enhancing Stability of γ-Al2O3-Supported NiCu Catalysts by Impregnating Basic Oxides in the Hydrodeoxygenation of Anisole. Catalysts 2024, 14, 166. https://doi.org/10.3390/catal14030166
Vandevyvere T, Sabbe MK, Thybaut JW, Lauwaert J. Enhancing Stability of γ-Al2O3-Supported NiCu Catalysts by Impregnating Basic Oxides in the Hydrodeoxygenation of Anisole. Catalysts. 2024; 14(3):166. https://doi.org/10.3390/catal14030166
Chicago/Turabian StyleVandevyvere, Tom, Maarten K. Sabbe, Joris W. Thybaut, and Jeroen Lauwaert. 2024. "Enhancing Stability of γ-Al2O3-Supported NiCu Catalysts by Impregnating Basic Oxides in the Hydrodeoxygenation of Anisole" Catalysts 14, no. 3: 166. https://doi.org/10.3390/catal14030166